The Effect of Oligomerization on A Solid-Binding Peptide Binding to Silica-Based Materials

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Production and Purification of Truncated Derivatives
2.3. Construction, Production, and Purification of a Synthetic Linker Derivative
2.4. Silica Binding Assay
2.5. Circular Dichroism (CD) Spectroscopy
2.6. Circular Dichroism Secondary Structure Analysis
2.7. Fluorescence Spectroscopy
2.8. Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D) Interaction Analyses
3. Results and Discussion
3.1. Effect of Linker Oligomerization on the Secondary Structure of PG
3.2. Effect of Linker Oligomerization Chemical Stability of PG
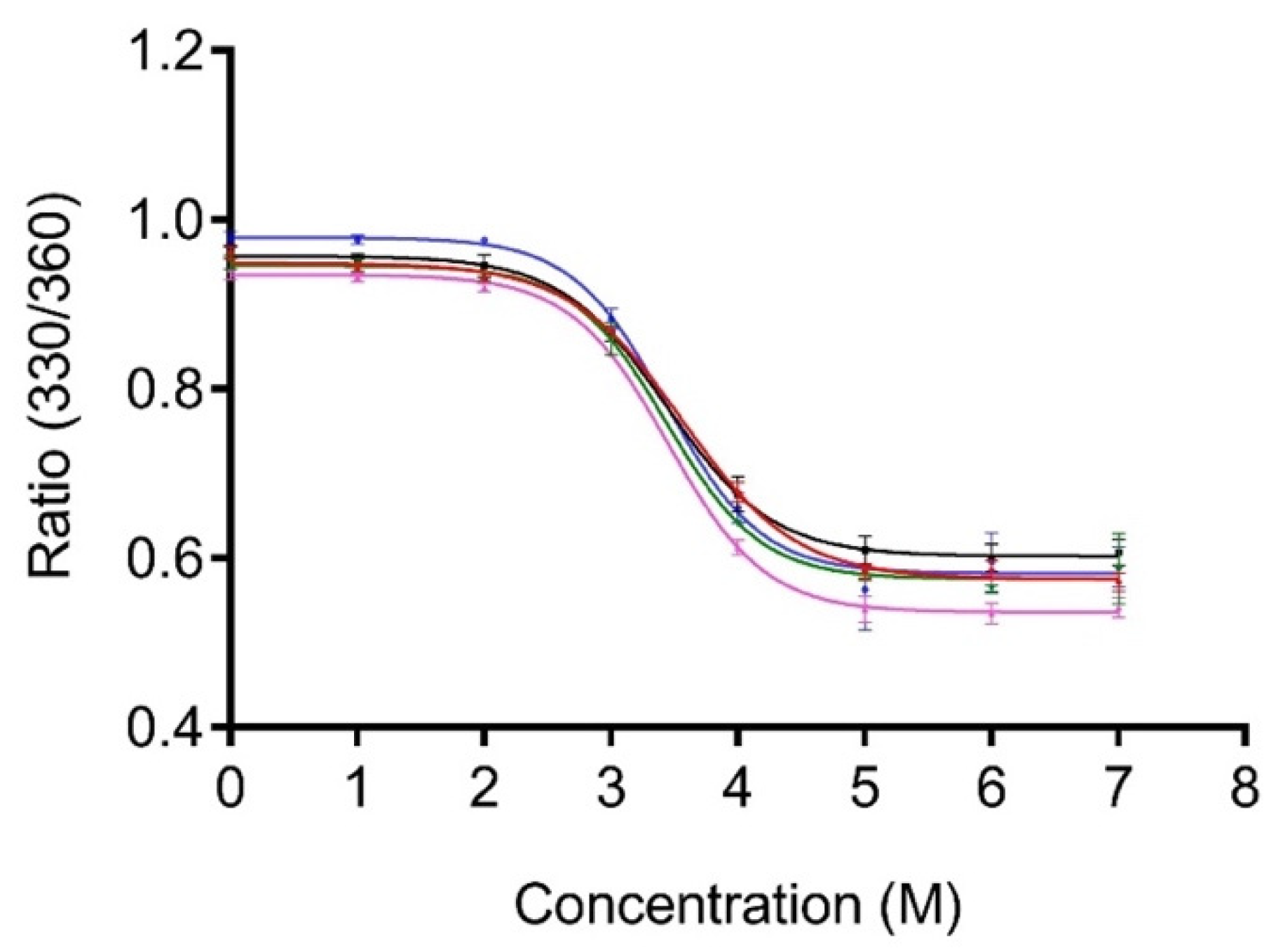
3.3. Minimal Number of Repeats Required for Silica Binding
3.4. Binding Orientation of Truncated Derivative 3 × LPG
3.5. Physiochemical Properties of 4 × LPG Derivatives and Linker Repeats
3.6. Introduction of a (GGGGS)n Linker
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sunna, A.; Chi, F.; Bergquist, P.L. A linker peptide with high affinity towards silica-containing materials. New Biotechnol. 2013, 30, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Care, A.; Bergquist, P.L.; Sunna, A. Solid-binding peptides: Smart tools for nanobiotechnology. Trends Biotechnol. 2015, 33, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Rothenstein, D.; Claasen, B.; Omiecienski, B.; Lammel, P.; Bill, J. Isolation of ZnO-Binding 12-mer Peptides and Determination of Their Binding Epitopes by NMR Spectroscopy. J. Am. Chem. Soc. 2012, 134, 12547–12556. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Farmer, B.L.; Pandey, R.B.; Slocik, J.M.; Patnaik, S.S.; Pachter, R.; Naik, R.R. Nature of Molecular Interactions of Peptides with Gold, Palladium, and Pd−Au Bimetal Surfaces in Aqueous Solution. J. Am. Chem. Soc. 2009, 131, 9704–9714. [Google Scholar] [CrossRef] [PubMed]
- Kumada, Y.; Tokunaga, Y.; Imanaka, H.; Imamura, K.; Sakiyama, T.; Katoh, S.; Nakanishi, K. Screening and Characterization of Affinity Peptide Tags Specific to Polystyrene Supports for the Orientated Immobilization of Proteins. Biotechnol. Prog. 2006, 22, 401–405. [Google Scholar] [CrossRef]
- Care, A.; Chi, F.; Bergquist, P.L.; Sunna, A. Biofunctionalization of silica-coated magnetic particles mediated by a peptide. J. Nanopart. Res. 2014, 16, 2543. [Google Scholar] [CrossRef]
- Tang, Z.; Palafox-Hernandez, J.P.; Law, W.-C.; Hughes, Z.E.; Swihart, M.T.; Prasad, P.; Knecht, M.R.; Walsh, T.R. Biomolecular Recognition Principles for Bionanocombinatorics: An Integrated Approach to Elucidate Enthalpic and Entropic Factors. ACS Nano 2013, 7, 9632–9646. [Google Scholar] [CrossRef]
- Rimola, A.; Sodupe, M.; Ugliengo, P. Affinity Scale for the Interaction of Amino Acids with Silica Surfaces. J. Phys. Chem. C 2009, 113, 5741–5750. [Google Scholar] [CrossRef]
- Sapsford, K.E.; Algar, W.S.; Berti, L.; Gemmill, K.B.; Casey, B.J.; Oh, E.; Stewart, M.H.; Medintz, I.L. Funtionalizing nanoparticles with biological molecules: Developing chemistries that facilitate nanotechnology. Chem. Rev. 2013, 113, 1904–2074. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Naik, R.R.; Stringer, S.J.; Agarwal, G.; Jones, S.E.; Stone, M.O. Biomimetic synthesis and patterning of silver nanoparticles. Nat. Mater. 2002, 1, 169–172. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Isozaki, K.; Nakamura, M.; Takaya, H.; Watanabe, T. Discovery of 12-mer peptides that bind to wood lignin. Sci. Rep. 2016, 6, 21833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, A.M.; Westcott, Z.; Hughes, Z.E.; Palafox-Hernandez, J.P.; Giesa, T.; Puddu, V.; Buehler, M.J.; Perry, C.C.; Walsh, T.R. Aqueous Peptide–TiO2 Interfaces: Isoenergetic Binding via Either Entropically or Enthalpically Driven Mechanisms. ACS Appl. Mater. Interfaces 2016, 8, 18620–18630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, J.W.; Raorane, D.; Huh, J.H.; Majumdar, A.; Lee, S.-W. Evolutionary Screening of Biomimetic Coatings for Selective Detection of Explosives. Langmuir 2008, 24, 4938–4943. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Okeya, Y.; Hashizume, M.; Serizawa, T. Screening of peptides recognizing simple polycyclic aromatic hydrocarbons. Chem. Commun. 2013, 49, 5088. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shindo, H. Binding sites and structure of peptides bound to SiO2 nanoparticles studied by solution NMR spectroscopy. Polym. J. 2018, 50, 989. [Google Scholar] [CrossRef]
- Gwak, Y.; Park, J.-I.; Kim, M.; Kim, H.S.; Kwon, M.J.; Oh, S.J.; Kim, Y.-P.; Jin, E. Creating Anti-icing Surfaces via the Direct Immobilization of Antifreeze Proteins on Aluminum. Sci. Rep. 2015, 5, 12019. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Becker, M.L.; Carri, G.A. The Influence of Amino Acid Sequence and Functionality on the Binding Process of Peptides onto Gold Surfaces. Langmuir 2011, 28, 1408–1417. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Zhou, B.; Lai, L.; Pei, J. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinform. 2017, 18, 277. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.R.; Knecht, M.R. Biointerface Structural Effects on the Properties and Applications of Bioinspired Peptide-Based Nanomaterials. Chem. Rev. 2017, 117, 12641–12704. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Kim, S.N.; Crookes-Goodson, W.J.; Farmer, B.L.; Naik, R.R. Biomimetic chemosensor: Designing peptide recognition elements for surface functionalisation of carbon nanotube field effect transistors. ACS Nano 2010, 1, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Seker, U.O.S.; Wilson, B.; Kulp, J.; Evans, J.S.; Tamerler, C.; Sarikaya, M. Thermodynamics of Engineered Gold Binding Peptides: Establishing the Structure–Activity Relationships. Biomacromolecules 2014, 15, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.-H.; Cheong, T.-C.; Min, J.H.; Wu, J.H.; Lee, S.J.; Kim, D.; Yang, J.-S.; Kim, S.; Kim, Y.K.; Seong, S.-Y. A multifunctional core–shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, J.; Zhang, R.; Liu, Y.; Liu, D.; Goldys, E.M.; Yang, X.; Xi, P.; Sunna, A.; Lu, J.; et al. Tunable lifetime multiplexing using luminescent nanocrystals. Nat. Photonics 2013, 8, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Care, A.; Zhang, R.; Lu, Y.; Packer, N.H.; Sunna, A.; Qian, Y.; Zvyagin, A.V. Facile Assembly of Functional Upconversion Nanoparticles for Targeted Cancer Imaging and Photodynamic Therapy. ACS Appl. Mater. Interfaces 2016, 8, 11945–11953. [Google Scholar] [CrossRef]
- Hall, V.; Sklepari, M.; Rodger, A. Protein Secondary Structure Prediction from Circular Dichroism Spectra Using a Self-Organizing Map with Concentration Correction. Chirality 2014, 26, 471–482. [Google Scholar] [CrossRef]
- Sklepari, M.; Rodger, A.; Reason, A.; Jamshidi, S.; Prokes, I.; Blindauer, C.A. Biophysical characterization of a protein for structure comparison: Methods for identifying insulin structural changes. Anal. Methods 2016, 8, 7460–7471. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.G.; Miles, A.J.; Wien, F.; Wallace, B. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 2006, 22, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, M.A.; Marrington, R.; Rodger, P.M.; Rodger, A.; Robinson, C. An Escherichia coli twin-arginine signal peptide switches between helical and unstructured conformations depending on the hydrophobicity of the environment. JBIC J. Boil. Inorg. Chem. 2003, 270, 3345–3352. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Elgundi, Z.; Care, A.; Goodchild, S.C.; Lord, M.; Rodger, A.; Sunna, A. Elucidating the Binding Mechanism of a Novel Silica-Binding Peptide. Biomolecules 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seker, U.O.S.; Wilson, B.; Sahin, D.; Tamerler, C.; Sarikaya, M. Quantitative Affinity of Genetically Engineered Repeating Polypeptides to Inorganic Surfaces. Biomacromolecules 2009, 10, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.; Whitelock, J.; Simmons, A.; Williams, R.L.; Milthorpe, B.K. Fibrinogen adsorption and platelet adhesion to silica surfaces with stochastic nanotopography. Biointerphases 2014, 9, 41002. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Kim, J.-T. Application of Kevin–Voigt Model in Quantifying Whey Protein Adsorption on Polyethersulfone Using QCM-D. J. Lab. Autom. 2009, 14, 213–220. [Google Scholar] [CrossRef]
- Hovgaard, M.B.; Dong, M.; Otzen, D.E.; Besenbacher, F. Quartz Crystal Microbalance Studies of Multilayer Glucagon Fibrillation at the Solid-Liquid Interface. Biophys. J. 2007, 93, 2162–2169. [Google Scholar] [CrossRef] [Green Version]
- Goward, C.R.; Irons, L.I.; Murphy, J.P.; Atkinson, T. The secondary structure of protein G′, a robust molecule. Biochem. J. 1991, 274, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Provencher, S.W.; Gloeckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Lian, L.-Y.; Derrick, J.P.; Sutcliffe, M.; Yang, J.; Roberts, G. Determination of the solution structures of domains II and III of protein G from Streptococcus by 1H nuclear magnetic resonance. J. Mol. Boil. 1992, 228, 1219–1234. [Google Scholar] [CrossRef]
- Brown, S. Metal-recognition by repeating polypeptides. Nat. Biotechnol. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Taniguchi, K.; Nomura, K.; Hata, Y.; Nishimura, T.; Asami, Y.; Kuroda, A. The Si-tag for immobilizing proteins on a silica surface. Biotechnol. Bioeng. 2007, 96, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarikaya, M.; Tamerler, C.; Jen, A.-Y.; Schulten, K.; Baneyx, F. Molecular biomimetics: Nanotechnology through biology. Nat. Mater. 2003, 2, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, J.E.; Millett, I.S.; Jacob, J.; Zagrovic, B.; Dillon, T.M.; Cingel, N.; Dothager, R.S.; Seifert, S.; Thiyagarajan, P.; Sosnick, T.R.; et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Kuroda, A. Why does the silica-binding protein “Si-tag” bind strongly to silica surfaces? Implications of conformational adaptation of the intrinsically disordered polypeptide to solid surfaces. Colloids Surf. B Biointerfaces 2011, 86, 359–363. [Google Scholar] [CrossRef]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [Green Version]
- Gillams, R.J.; Jia, T.Z. Mineral Surface-Templated Self-Assembling Systems: Case Studies from Nanoscience and Surface Science towards Origins of Life Research. Life 2018, 8, 10. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-Helix | β-Sheet | Bends | Turns | Other | NRMSD | |
|---|---|---|---|---|---|---|
| PG | 0.38 | 0.16 | 0.16 | 0.10 | 0.20 | 0.01 |
| 1 × LPG | 0.29 | 0.21 | 0.16 | 0.10 | 0.25 | 0.05 |
| 2 × LPG | 0.23 | 0.28 | 0.13 | 0.10 | 0.25 | 0.06 |
| 3 × LPG | 0.21 | 0.31 | 0.13 | 0.10 | 0.25 | 0.07 |
| 4 × LPG | 0.17 | 0.36 | 0.14 | 0.10 | 0.23 | 0.06 |
| Derandomized proteins | ||||||
| 1 × LPG–0.22RC | 0.39 | 0.15 | 0.17 | 0.10 | 0.20 | 0.03 |
| 2 × LPG–0.31RC | 0.36 | 0.14 | 0.19 | 0.09 | 0.23 | 0.03 |
| 3 × LPG–0.36RC | 0.36 | 0.14 | 0.19 | 0.09 | 0.23 | 0.03 |
| 4 × LPG–0.40 | 0.34 | 0.17 | 0.14 | 0.12 | 0.23 | 0.04 |
| Reconstructed proteins | ||||||
| 1 × LPG | 0.38 | 0.16 | 0.16 | 0.10 | 0.34 | 0.38 |
| 2 × LPG | 0.30 | 0.12 | 0.13 | 0.08 | 0.38 | 0.30 |
| 3 × LPG | 0.25 | 0.10 | 0.13 | 0.06 | 0.47 | 0.25 |
| 4 × LPG | 0.23 | 0.09 | 0.12 | 0.06 | 0.51 | 0.23 |
| α-Helix | β-Sheet | Bends | Turns | Other | Total | |
|---|---|---|---|---|---|---|
| PG | 75 | 31 | 31 | 20 | 39 | 196 |
| Derandomized proteins | ||||||
| 1 × LPG–0.22RC | 65 | 25 | 28 | 17 | 33 | 168 |
| 2 × LPG–0.31RC | 59 | 23 | 30 | 14 | 37 | 163 |
| 3 × LPG–0.36RC | 59 | 23 | 31 | 14 | 38 | 164 |
| 4 × LPG–0.40 | 56 | 28 | 24 | 20 | 39 | 167 |
| Reconstructed proteins | ||||||
| 1 × LPG | 65 | 25 | 28 | 17 | 81 | 215 |
| 2 × LPG | 59 | 23 | 30 | 14 | 110 | 236 |
| 3 × LPG | 59 | 23 | 31 | 14 | 130 | 257 |
| 4 × LPG | 56 | 28 | 24 | 20 | 150 | 278 |
| Thickness (@Req) a (nm) | Thickness (@kd) b (nm) | Mass Deposited (@Req) (×108 ng/cm2) | Mass Deposited (@kd) (×108 ng/cm2) | Viscosity (@Req) × 10−4 (kg/ms) | Viscosity (@Req) × 10−4 (kg/ms) | |
|---|---|---|---|---|---|---|
| 3 × LPG/SiO2 | 5.76 ± 0.15 | 4.36 ± 0.05 | 680 ± 34 | 589 ± 24 | 19.7 ± 0.4 | 17.56 ± 0.15 |
| Immobilized Truncated Derivative | Thickness of Bound Protein (nm) | Mass of Protein Deposited (ng/cm2) | Thickness of Bound Trastuzumab (nm) | Mass Deposited for Bound Trastuzumab (ng/cm2) | Thickness of Bound HER2 (nm) | Mass deposited for Bound HER2 (ng/cm2) |
|---|---|---|---|---|---|---|
| 3 × LPG | 5.76 ± 0.22 | 681 ± 32 | 15.0 ± 0.20 | 1019 ± 48 | 20.0 ± 0.4 | 1251 ± 51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bansal, R.; Elgundi, Z.; Goodchild, S.C.; Care, A.; Lord, M.S.; Rodger, A.; Sunna, A. The Effect of Oligomerization on A Solid-Binding Peptide Binding to Silica-Based Materials. Nanomaterials 2020, 10, 1070. https://doi.org/10.3390/nano10061070
Bansal R, Elgundi Z, Goodchild SC, Care A, Lord MS, Rodger A, Sunna A. The Effect of Oligomerization on A Solid-Binding Peptide Binding to Silica-Based Materials. Nanomaterials. 2020; 10(6):1070. https://doi.org/10.3390/nano10061070
Chicago/Turabian StyleBansal, Rachit, Zehra Elgundi, Sophia C. Goodchild, Andrew Care, Megan S. Lord, Alison Rodger, and Anwar Sunna. 2020. "The Effect of Oligomerization on A Solid-Binding Peptide Binding to Silica-Based Materials" Nanomaterials 10, no. 6: 1070. https://doi.org/10.3390/nano10061070
APA StyleBansal, R., Elgundi, Z., Goodchild, S. C., Care, A., Lord, M. S., Rodger, A., & Sunna, A. (2020). The Effect of Oligomerization on A Solid-Binding Peptide Binding to Silica-Based Materials. Nanomaterials, 10(6), 1070. https://doi.org/10.3390/nano10061070