Surface Heterogeneous Nucleation-Mediated Release of Beta-Carotene from Porous Silicon



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis of Porous Silicon
2.3. Relelase Studies of BCAR
2.4. Imaging Studies by Scanning Electron Microscopy (SEM)
3. Results
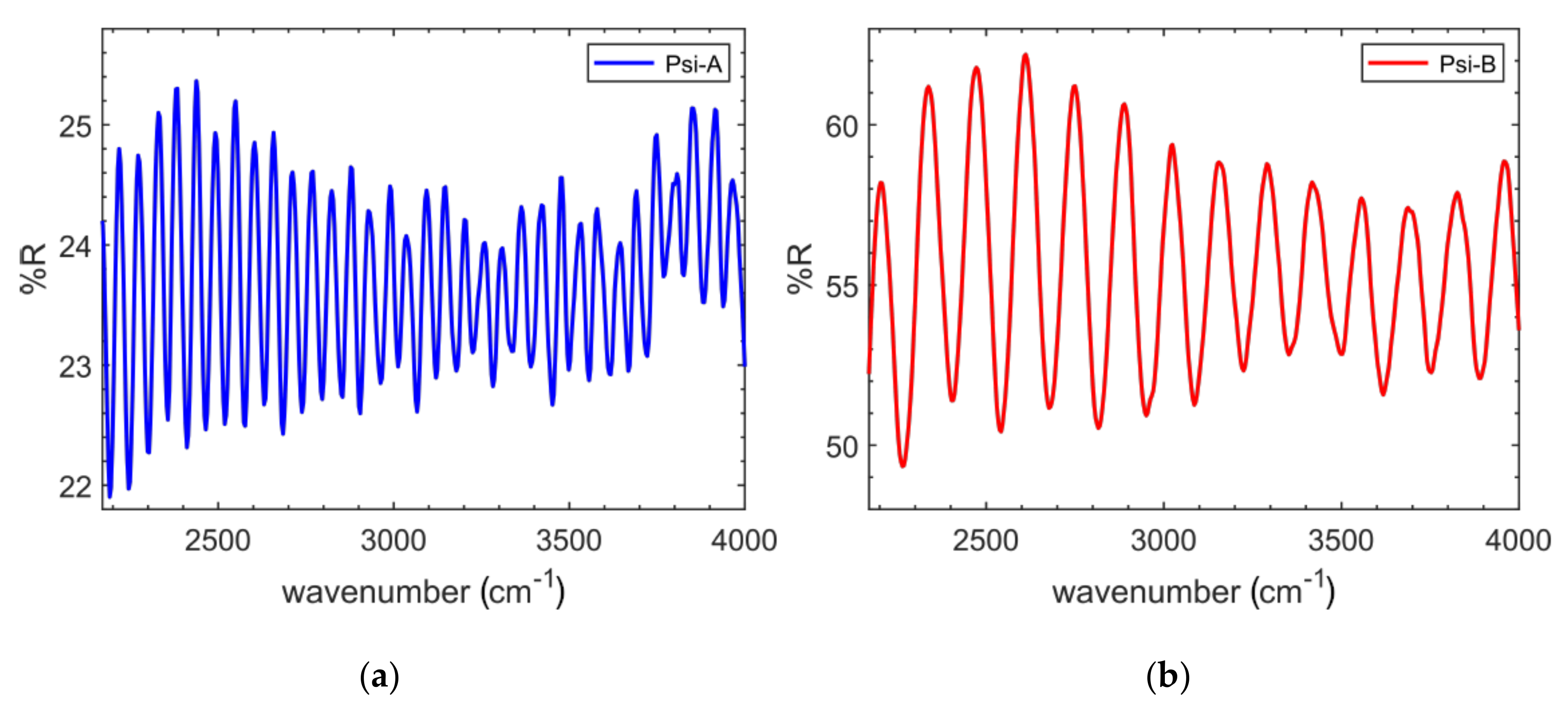
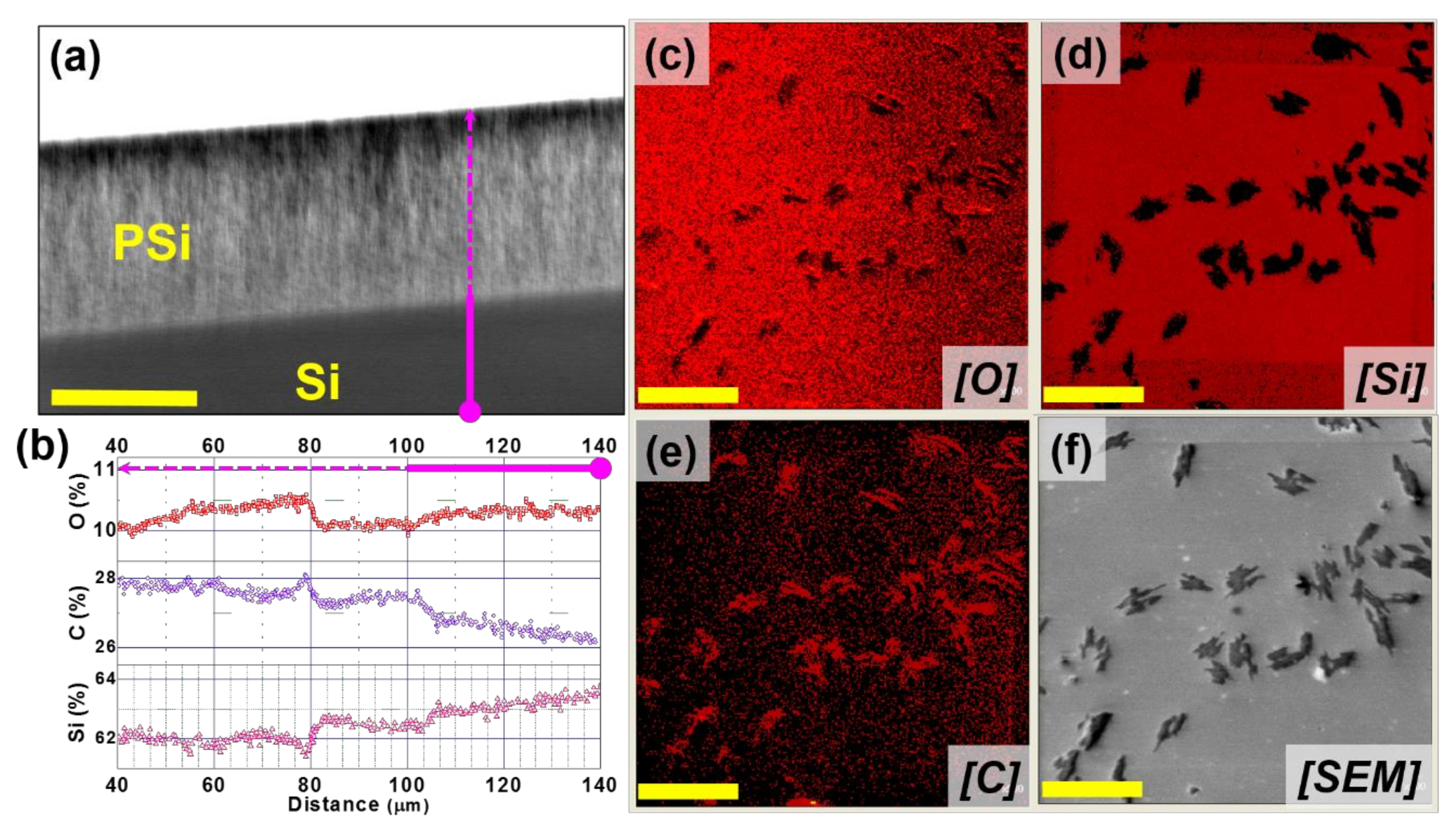
3.1. PSi Characterisation
3.2. Auger Electron Spectroscopy Analysis
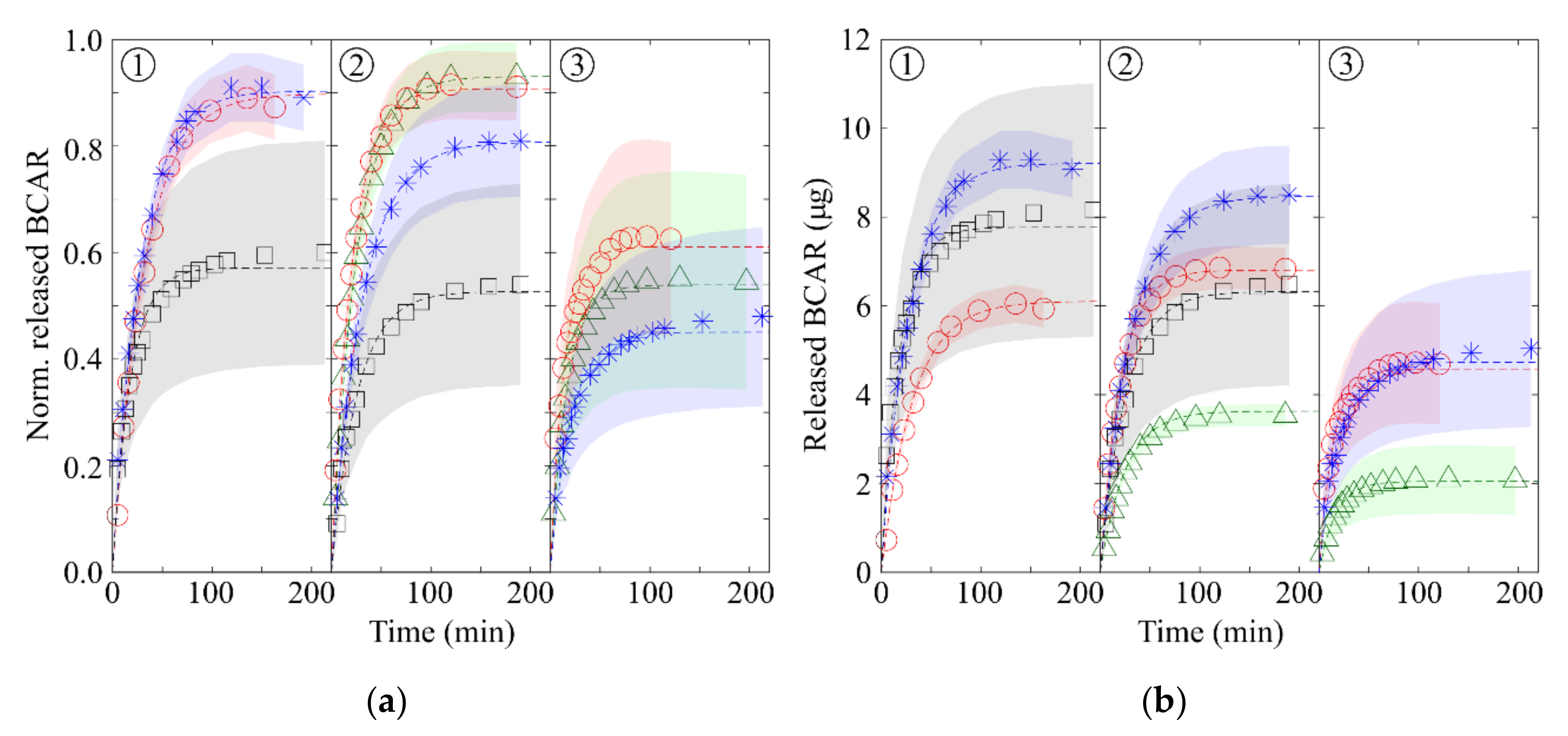
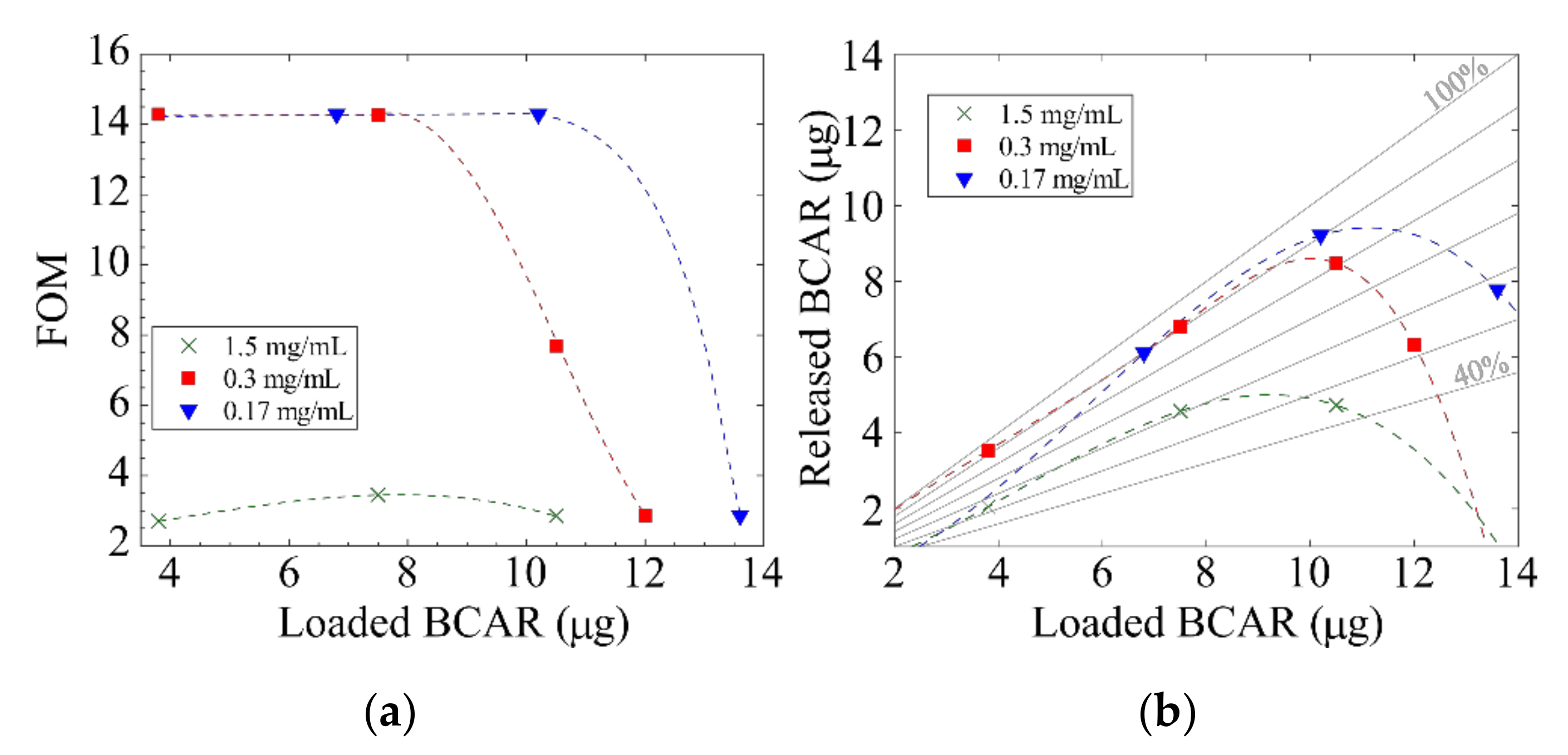
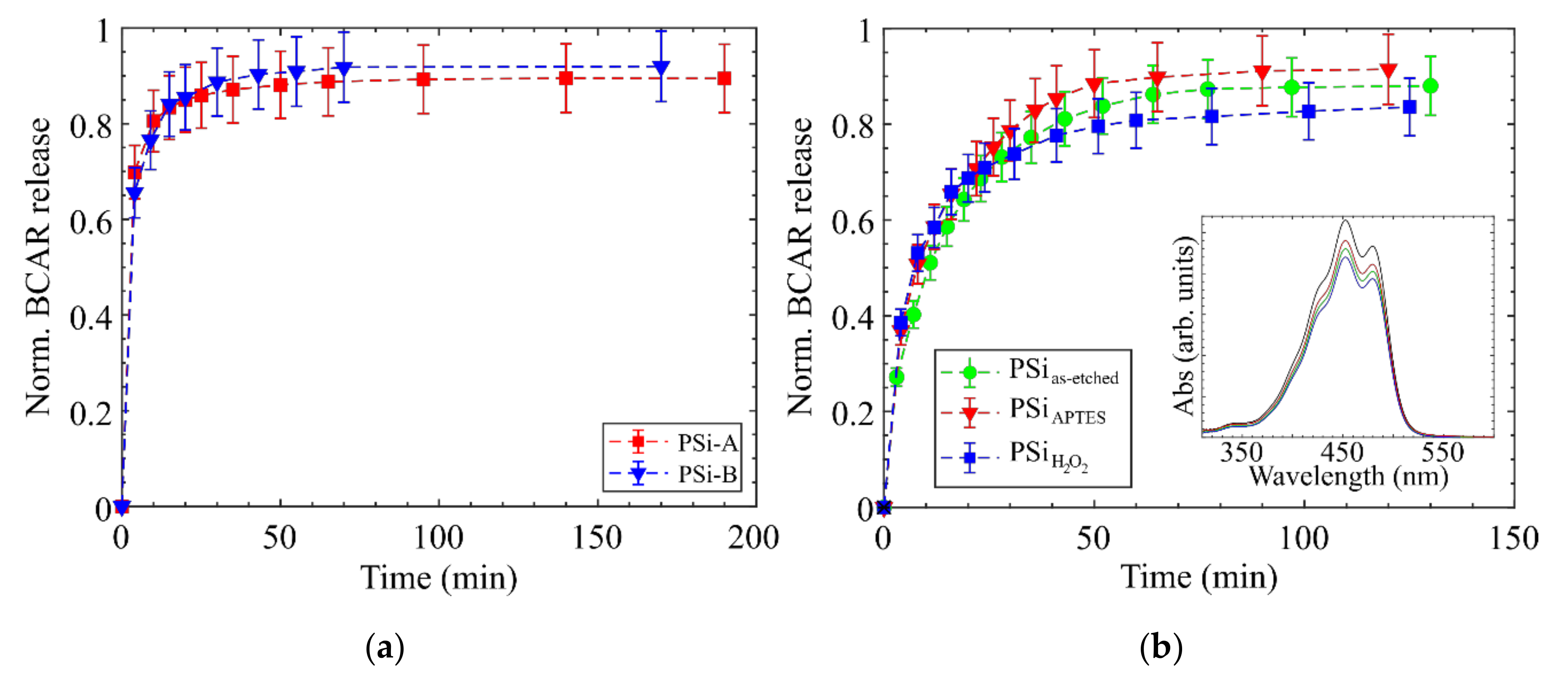
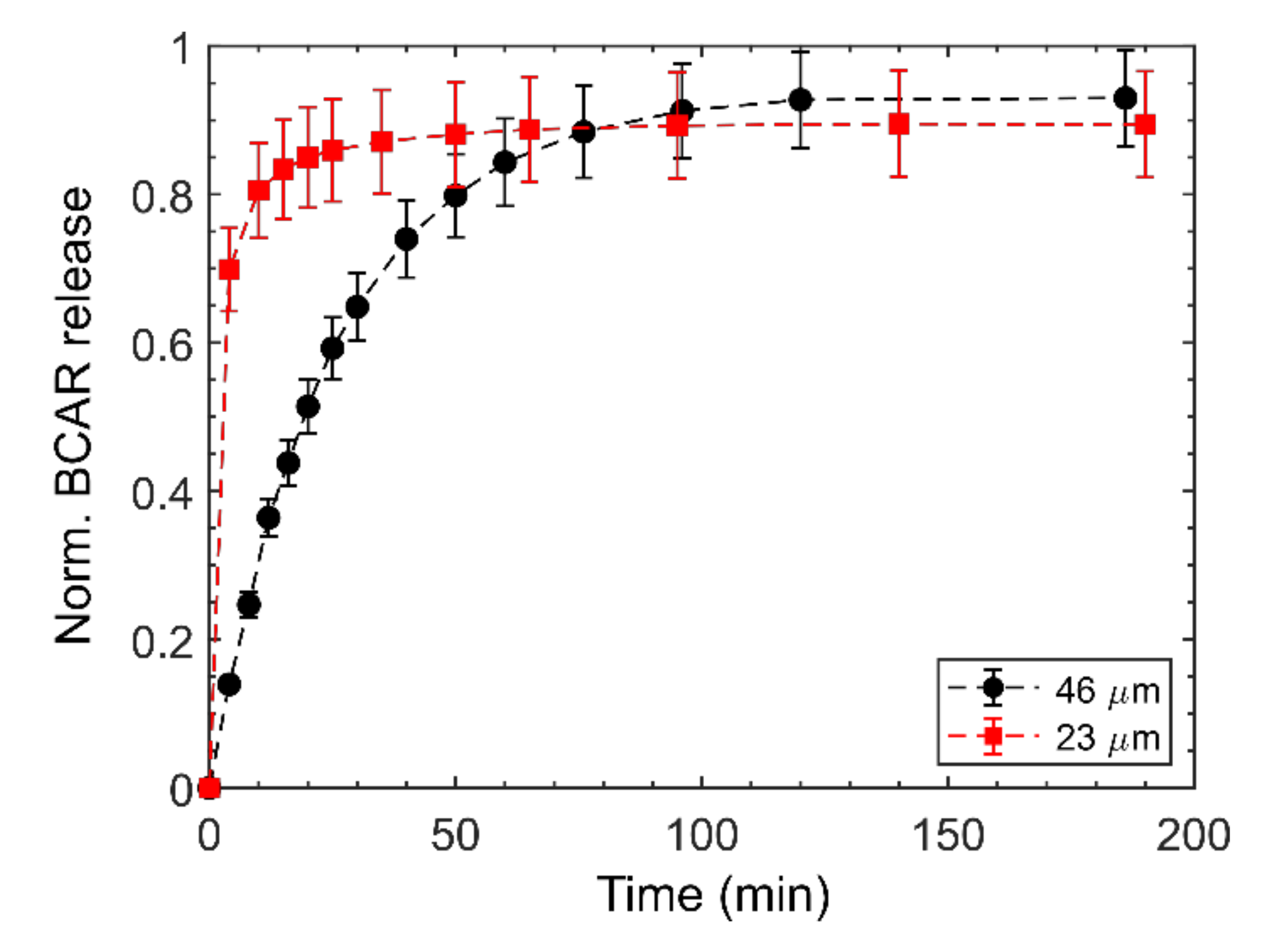
3.3. Release Experiments
4. Discussion
- release curves measure a global system response, averaged over the entire surface of the porous sample, rather than the local analysis given by the ITM whose precision depends on the spatial resolution of the technique used;
- the sensitivity achieved is much greater. For example, in our case agglomerates form and remain attached to the PSi inner surface, thus they will be invisible in standard ITM characterization based on optical scattering.
About of the Choice of the Model to Fit the Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, K.K.; Anan, A.; Buckley, R.P.; Ouellette, W.; Asefa, T. Toward Efficient Nanoporous Catalysts: Controlling Site-Isolation and Concentration of Grafted Catalytic Sites on Nanoporous Materials with Solvents and Colorimetric Elucidation of Their Site-Isolation. J. Am. Chem. Soc. 2008, 130, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, G.; Greenaway, A.G.; Yiu, H.H.P. The Potential Applications of Nanoporous Materials for the Adsorption, Separation, and Catalytic Conversion of Carbon Dioxide. Adv. Energy Mater. 2014, 4, 1301873. [Google Scholar] [CrossRef] [Green Version]
- Fechete, I.; Védrine, J.C. Nanoporous Materials as New Engineered Catalysts for the Synthesis of Green Fuels. Molecules 2015, 20, 5638–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, S.; Potter, M.E.; Raja, R. The Molecular Design of Active Sites in Nanoporous Materials for Sustainable Catalysis. Molecules 2017, 22, 2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churaman, W.; Currano, L.J.; Becker, C. Initiation and reaction tuning of nanoporous energetic silicon. J. Phys. Chem. Solids 2010, 71, 69–74. [Google Scholar] [CrossRef]
- Van Riet, R.; Amayuelas, E.; Lodewyckx, P.; Lefebvre, M.H.; Ania, C.O. Novel opportunities for nanoporous carbons as energetic materials. Carbon 2020, 164, 129–132. [Google Scholar] [CrossRef]
- Song, C.; Zheng, Z.; Zhang, W.; Yang, Y.; Zeng, G.; Yu, C.; Wang, J.; Chen, Y.; Ma, K.; Cheng, Z. Thermal-induced hydrosilylation to endow nanoporous silicon energetic films with long-term chemical stability. Chem. Eng. J. 2020, 401, 126074. [Google Scholar] [CrossRef]
- Lepoitevin, M.; Ma, T.; Bechelany, M.; Janot, J.M.; Balme, S. Functionalization of single solid state nanopores to mimic biological ion channels: A review. Adv. Colloid Interface Sci. 2017, 250, 195–213. [Google Scholar] [CrossRef]
- Cao, C.; Long, Y.-T. Biological Nanopores: Confined Spaces for Electrochemical Single-Molecule Analysis. Acc. Chem. Res. 2018, 51, 331–341. [Google Scholar] [CrossRef]
- Jackman, J.A.; Cho, N.-J.; Nishikawa, M.; Yoshikawa, G.; Mori, T.; Shrestha, L.K.; Ariga, K. Materials Nanoarchitectonics for Mechanical Tools in Chemical and Biological Sensing. Chem. Asian J. 2018, 13, 3366–3377. [Google Scholar] [CrossRef]
- Müller, K.; Helfferich, J.; Zhao, F.; Verma, R.; Kanj, A.B.; Meded, V.; Bléger, D.; Wenzel, W.; Heinke, L. Switching the Proton Conduction in Nanoporous, Crystalline Materials by Light. Adv. Mater. 2018, 30, 1706551. [Google Scholar] [CrossRef] [PubMed]
- Meloni, S.; Giacomello, A.; Casciola, C.M. Focus Article: Theoretical aspects of vapor/gas nucleation at structured surfaces. J. Chem. Phys. 2016, 145, 211802. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, H.; Durães, L.; García-González, C.A.; Del Gaudio, P.; Portugal, A.T.; Mahmoudi, M. Synthesis and biomedical applications of aerogels: Possibilities and challenges. Adv. Colloid Interface Sci. 2016, 236, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.A.; Mäkilä, E.; Airaksinen, A.J.; Bimbo, L.M.; Hirvonen, J. Porous silicon nanoparticles for nanomedicine: preparation and biomedical applications. Nanomedicine 2014, 9, 535–554. [Google Scholar] [CrossRef]
- Beg, S.; Rahman, M.; Jain, A.; Saini, S.; Midoux, P.; Pichon, C.; Ahmad, F.J.; Akhter, S. Nanoporous metal organic frameworks as hybrid polymer–metal composites for drug delivery and biomedical applications. Drug Discov. Today 2017, 22, 625–637. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [Green Version]
- Baeza, A.; Ruiz-Molina, D.; Vallet-Regí, M. Recent advances in porous nanoparticles for drug delivery in antitumoral applications: inorganic nanoparticles and nanoscale metal-organic frameworks. Expert Opin. Drug Deliv. 2016, 14, 1–14. [Google Scholar] [CrossRef]
- Jafari, S.; Derakhshankhah, H.; Alaei, L.; Fattahi, A.; Varnamkhasti, B.S.; Saboury, A.A. Mesoporous silica nanoparticles for therapeutic/diagnostic applications. Biomed. Pharmacother. 2019, 109, 1100–1111. [Google Scholar] [CrossRef]
- Zhou, M.; Shen, L.; Lin, X.; Hong, Y.; Feng, Y. Design and pharmaceutical applications of porous particles. RSC Adv. 2017, 7, 39490–39501. [Google Scholar] [CrossRef] [Green Version]
- Kumeria, T.; McInnes, S.; Maher, S.; Santos, A. Porous silicon for drug delivery applications and theranostics: recent advances, critical review and perspectives. Expert Opin. Drug Deliv. 2017, 14, 1407–1422. [Google Scholar] [CrossRef] [PubMed]
- Daldosso, N.; Ghafarinazari, A.; Cortelletti, P.; Marongiu, L.; Donini, M.; Paterlini, V.; Bettotti, P.; Guider, R.; Froner, E.; Dusi, S.; et al. Orange and blue luminescence emission to track functionalized porous silicon microparticles inside the cells of the human immune system. J. Mater. Chem. B 2014, 2, 6345–6353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kumeria, T.; Bezem, M.T.; Wang, J.; Sailor, M.J. Self-Reporting Photoluminescent Porous Silicon Microparticles for Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 3200–3209. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef]
- Fry, N.L.; Boss, G.R.; Sailor, M.J. Oxidation-Induced Trapping of Drugs in Porous Silicon Microparticles. Chem. Mater. 2014, 26, 2758–2764. [Google Scholar] [CrossRef]
- Li, Y.; Calder, S.; Yaffe, O.; Cahen, D.; Haick, H.; Kronik, L.; Zuilhof, H. Hybrids of Organic Molecules and Flat, Oxide-Free Silicon: High-Density Monolayers, Electronic Properties, and Functionalization. Langmuir 2012, 28, 9920–9929. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Yoshikawa, C.; Welch, N.G.; Pasic, P.; Thissen, H.; Voelcker, N.H. Spatially Controlled Surface Modification of Porous Silicon for Sustained Drug Delivery Applications. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.; Robins, E.; Geders, T.; Allen, M.J.; Choi, H.C.; Buriak, J. Three Methods for Stabilization and Functionalization of Porous Silicon Surfaces via Hydrosilylation and Electrografting Reactions. Phys. Status Solidi 2000, 182, 109–115. [Google Scholar] [CrossRef]
- Salonen, J.; Mäkilä, E. Thermally Carbonized Porous Silicon and Its Recent Applications. Adv. Mater. 2018, 30, e1703819. [Google Scholar] [CrossRef] [Green Version]
- Salonen, J.; Laine, E.; Niinistö, L. Thermal carbonization of porous silicon surface by acetylene. J. Appl. Phys. 2002, 91, 456. [Google Scholar] [CrossRef]
- Mäkilä, E.; Kivelä, H.; Shrestha, N.; Correia, A.; Kaasalainen, M.; Kukk, E.; Hirvonen, J.; Santos, H.A.; Salonen, J. Influence of Surface Chemistry on Ibuprofen Adsorption and Confinement in Mesoporous Silicon Microparticles. Langmuir 2016, 32, 13020–13029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, W.; Liu, D.; Ge, Y.; Zhao, M.; Zhu, X.; Li, W.; Wang, L.; Zheng, T.; Li, J. Oral Curcumin via Hydrophobic Porous Silicon Carrier: Preparation, Characterization, and Toxicological Evaluation In Vivo. ACS Appl. Mater. Interfaces 2019, 11, 31661–31670. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, J.; Dong, Q.; Wang, S.; Jian, H.; Wang, M.; Ren, P.; Baid, S. Preparation of Microgels with Ultrahigh Payload of Various Hydrophilic and Hydrophobic Inorganic Nanoparticle Composites up to 92 wt.%. ACS Appl. Mater. Interfaces 2019, 11, 4408–4415. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.-A.; Almeida, P.V.; Correia, A.; Herranz-Blanco, B.; Shrestha, N.; Mäkilä, E.; Salonen, J.; Hirvonen, J.; Santos, H.A. Intracellular responsive dual delivery by endosomolytic polyplexes carrying DNA anchored porous silicon nanoparticles. J. Control. Release 2017, 249, 111–122. [Google Scholar] [CrossRef]
- Wang, M.; Hartman, P.S.; Loni, A.; Canham, L.T.; Bodiford, N.; Coffer, J.L. Influence of Surface Chemistry on the Release of an Antibacterial Drug from Nanostructured Porous Silicon. Langmuir 2015, 31, 6179–6185. [Google Scholar] [CrossRef]
- Aw, M.S.; Bariana, M.; Yu, Y.; Addai-Mensah, J.; Losic, D. Surface-functionalized diatom microcapsules for drug delivery of water-insoluble drugs. J. Biomater. Appl. 2012, 28, 163–174. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Saffari, M.; Langrish, T.; Ghadi, A.E. Improving the dissolution rate of hydrophobic drugs through encapsulation in porous lactose as a new biocompatible porous carrier. Int. J. Pharm. 2017, 521, 204–213. [Google Scholar] [CrossRef]
- Liu, X.Y. Heterogeneous nucleation or homogeneous nucleation? J. Chem. Phys. 2000, 112, 9949–9955. [Google Scholar] [CrossRef]
- Sear, R.P. Nucleation: theory and applications to protein solutions and colloidal suspensions. J. Phys. Condens. Matter 2007, 19, 033101. [Google Scholar] [CrossRef]
- Liu, X.Y. A new kinetic model for three-dimensional heterogeneous nucleation. J. Chem. Phys. 1999, 111, 1628–1635. [Google Scholar] [CrossRef] [Green Version]
- Singha, S.; Das, P.K.; Maiti, B. Inclusion of line tension effect in classical nucleation theory for heterogeneous nucleation: A rigorous thermodynamic formulation and some unique conclusions. J. Chem. Phys. 2015, 142, 104706. [Google Scholar] [CrossRef] [PubMed]
- Navascués, G.; Tarazona, P. Line tension effects in heterogeneous nucleation theory. J. Chem. Phys. 1981, 75, 2441–2446. [Google Scholar] [CrossRef]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Wi, H.S.; Jo, W.; Cho, Y.C.; Lee, H.H.; Jeong, S.-Y.; Kim, Y.-I.; Lee, G. Multiple pathways of crystal nucleation in an extremely supersaturated aqueous potassium dihydrogen phosphate (KDP) solution droplet. Proc. Natl. Acad. Sci. USA 2016, 113, 13618–13623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolde, P.R.T.; Frenkel, D. Enhancement of Protein Crystal Nucleation by Critical Density Fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Kovács, T.; Meldrum, F.C.; Christenson, H.K. Crystal Nucleation without Supersaturation. J. Phys. Chem. Lett. 2012, 3, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hu, C.; Ward, M.D. Stereochemical Control of Polymorph Transitions in Nanoscale Reactors. J. Am. Chem. Soc. 2013, 135, 2144–2147. [Google Scholar] [CrossRef]
- Blagden, N.; De Matas, M.; Gavan, P.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [Green Version]
- Stanford, V.L.; Vyazovkin, S. Crystallization of ammonium perchlorate from solution confined to native and organically modified silica nanopores. Thermochim. Acta 2019, 677, 109–116. [Google Scholar] [CrossRef]
- Cooper, S.J.; Nicholson, C.E.; Liu, J. A simple classical model for predicting onset crystallization temperatures on curved substrates and its implications for phase transitions in confined volumes. J. Chem. Phys. 2008, 129, 124715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanh, N.T.K.; MacLean, N.; Mahiddine, S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Eden, K.; Morris, R.J.; Gillam, J.; Macphee, C.E.; Allen, R.J. Competition between Primary Nucleation and Autocatalysis in Amyloid Fibril Self-Assembly. Biophys. J. 2015, 108, 632–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y. Interfacial Effect of Molecules on Nucleation Kinetics†. J. Phys. Chem. B 2001, 105, 11550–11558. [Google Scholar] [CrossRef]
- Ling, J.; Chadwick, K. Heterogeneous Crystallization Inside Microporous Polymer Particles as a Process Intensification Technology for the Manufacture of Drug Formulations. Org. Process Res. Dev. 2017, 21, 827–834. [Google Scholar] [CrossRef]
- Bannigan, P.; Verma, V.; Hudson, S.P. Overcoming the Common Ion Effect for Weakly Basic Drugs: Inhibiting the Crystallization of Clofazimine Hydrochloride in Simulated Gastrointestinal Media. Cryst. Growth Des. 2019, 19, 1599–1609. [Google Scholar] [CrossRef] [Green Version]
- Brandel, C.; Ter Horst, J.H. Measuring induction times and crystal nucleation rates. Faraday Discuss. 2015, 179, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.A.; Kadam, S.S.; Meekes, H.; Stankiewicz, A.I.; Ter Horst, J.H. Crystal Nucleation Kinetics from Induction Times and Metastable Zone Widths. Cryst. Growth Des. 2013, 13, 2435–2440. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, J. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef]
- Izadifar, M.; Haddadi, A.; Chen, X.; E Kelly, M. Rate-programming of nano-particulate delivery systems for smart bioactive scaffolds in tissue engineering. Nanotechnology 2014, 26, 12001. [Google Scholar] [CrossRef]
- Ping, I.L. Modeling of drug release from matrix systems involving moving boundaries: Approximate analytical solutions. Int. J. Pharm. 2011, 418, 18–27. [Google Scholar]
- Ziemys, A.; Kojic, M.; Milosevic, M.; Ferrari, M. Interfacial effects on nanoconfined diffusive mass transport regimes. Phys. Rev. Lett. 2012, 108, 236102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz, M.; Pawlik, A.; Szuwarzyński, M.; Jaskuła, M.; Sulka, G.D. Nanoporous anodic titanium dioxide layers as potential drug delivery systems: Drug release kinetics and mechanism. Colloids Surf. B Biointerfaces 2016, 143, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Coulson, C.A.; Crank, J. The Mathematics of Diffusion. Math. Gaz. 1958, 42, 165. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, W.I. Diffusional Models Useful in Biopharmaceutics. J. Pharm. Sci. 1967, 56, 315–324. [Google Scholar] [CrossRef]
- Washburn, E.W. The Dynamics of Capillary Flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BCAR Loading Sol. Conc. (mg/mL) | Loaded BCAR (g) | |||
|---|---|---|---|---|
| 1.5 | 3.8 | 7.5 | 10.5 | N.P. |
| 0.3 | 3.8 | 7.5 | 10.5 | 12.0 |
| 0.17 | N.P. | 6.8 | 20.2 | 13.6 |
| PSi Type | Etching Recipe | Etch Rate (nm/s) | Thickness (μm) | neff | P (%) |
|---|---|---|---|---|---|
| PSi-A | 16% v/v HF (48%) in ethanol | 52 | 46.5 | 2.2 | 45 |
| PSi-B | 6% v/v HF (48%) in water + 0.5%v Triton-100 | 22 | 26.5 | 2.0 | 55 |
| Analytical | 1st Order Kinetic | ||||
|---|---|---|---|---|---|
| PSi-A 46 μm | PSi-B 26 μm | PSi-A 46 μm | PSi-B 26 μm | ||
| M∞ | 0.96 | 0.89 | 0.96 | 0.87 | M∞ |
| L (μm) | 48 | 24 | 0.04 | 0.3 | k (s−1) |
| D (cm2/s) | 4 × 10−6 | 8 × 10−6 | - | - | - |
| R2 | 0.984 | 0.990 | 0.999 | 0.986 | R2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piotto, C.; Pujari, S.P.; Zuilhof, H.; Bettotti, P. Surface Heterogeneous Nucleation-Mediated Release of Beta-Carotene from Porous Silicon. Nanomaterials 2020, 10, 1659. https://doi.org/10.3390/nano10091659
Piotto C, Pujari SP, Zuilhof H, Bettotti P. Surface Heterogeneous Nucleation-Mediated Release of Beta-Carotene from Porous Silicon. Nanomaterials. 2020; 10(9):1659. https://doi.org/10.3390/nano10091659
Chicago/Turabian StylePiotto, Chiara, Sidharam P. Pujari, Han Zuilhof, and Paolo Bettotti. 2020. "Surface Heterogeneous Nucleation-Mediated Release of Beta-Carotene from Porous Silicon" Nanomaterials 10, no. 9: 1659. https://doi.org/10.3390/nano10091659
APA StylePiotto, C., Pujari, S. P., Zuilhof, H., & Bettotti, P. (2020). Surface Heterogeneous Nucleation-Mediated Release of Beta-Carotene from Porous Silicon. Nanomaterials, 10(9), 1659. https://doi.org/10.3390/nano10091659