
Silica Microspheres-in-Pores Composite Monoliths with Fluorescence and Potential for Water Remediation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Preparation of Polyacrylamide (PAM) and Ag/PAM Composite Monoliths
2.3. Ship-in-a-Bottle Synthesis of Silica Microspheres in Macroporous PAM Scaffolds
2.4. Fluorescent MPTMS Silica Microspheres and Composite Monoliths
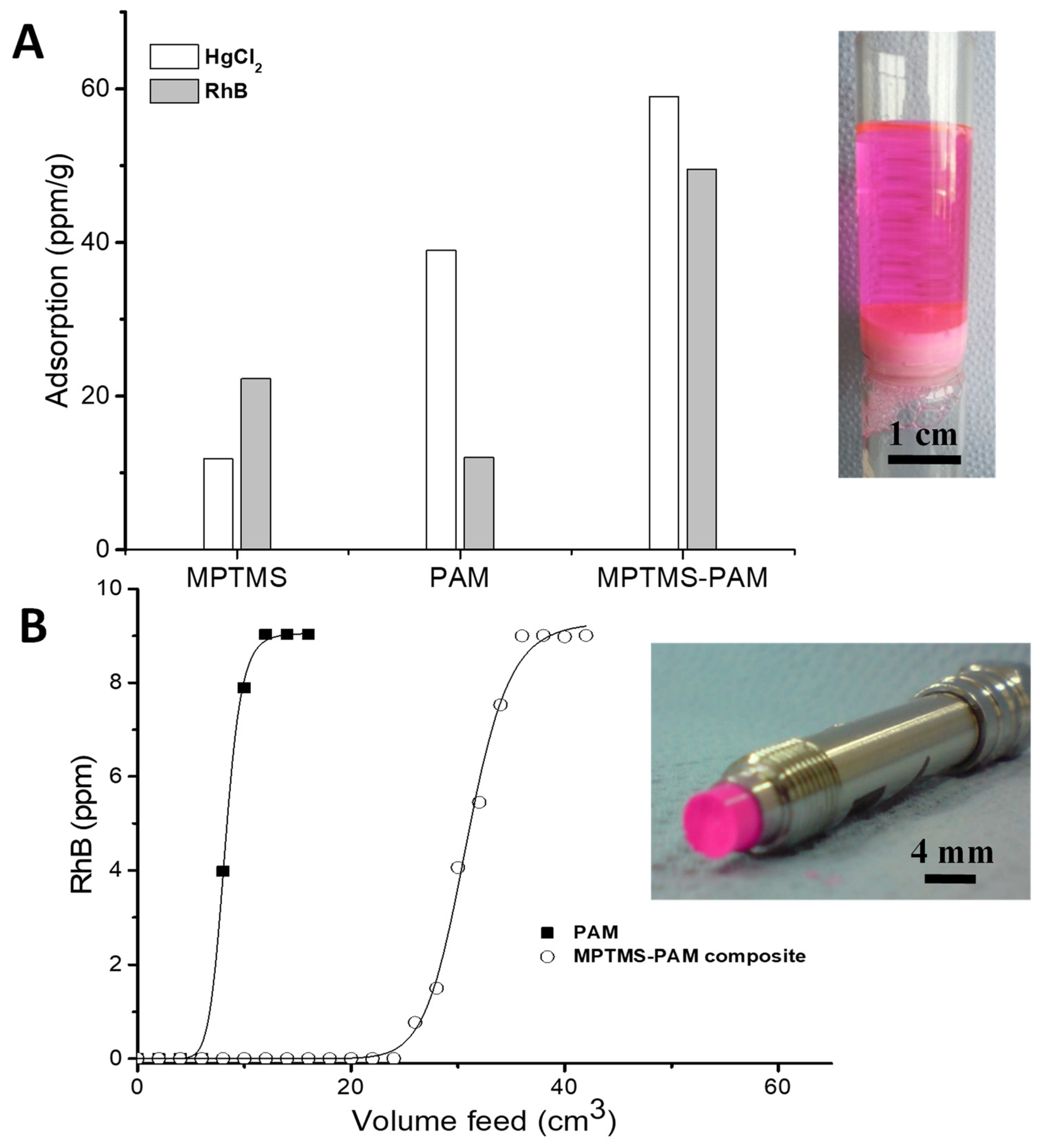
2.5. Removal of Hg2+ and Rhodamine B from Water
2.6. Characterisation
3. Results and Discussion
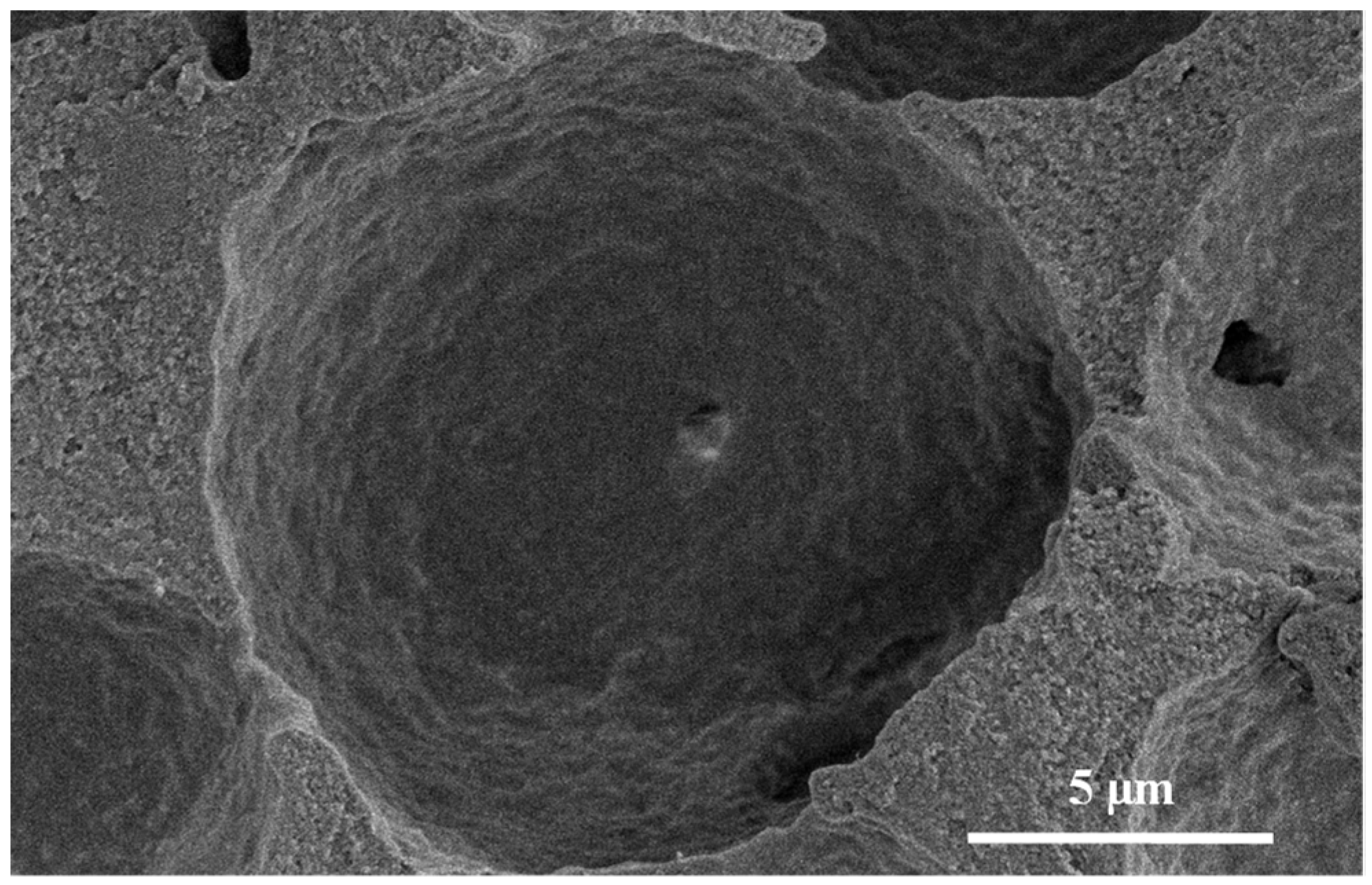
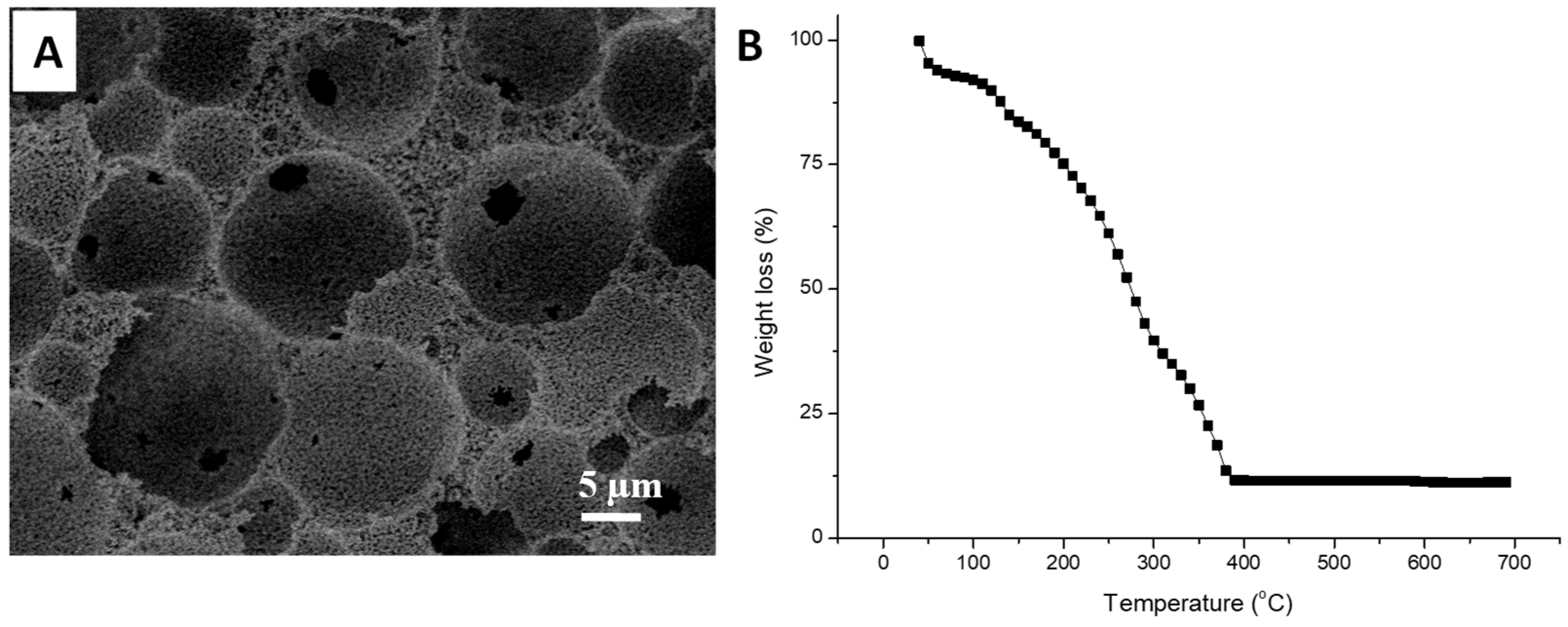
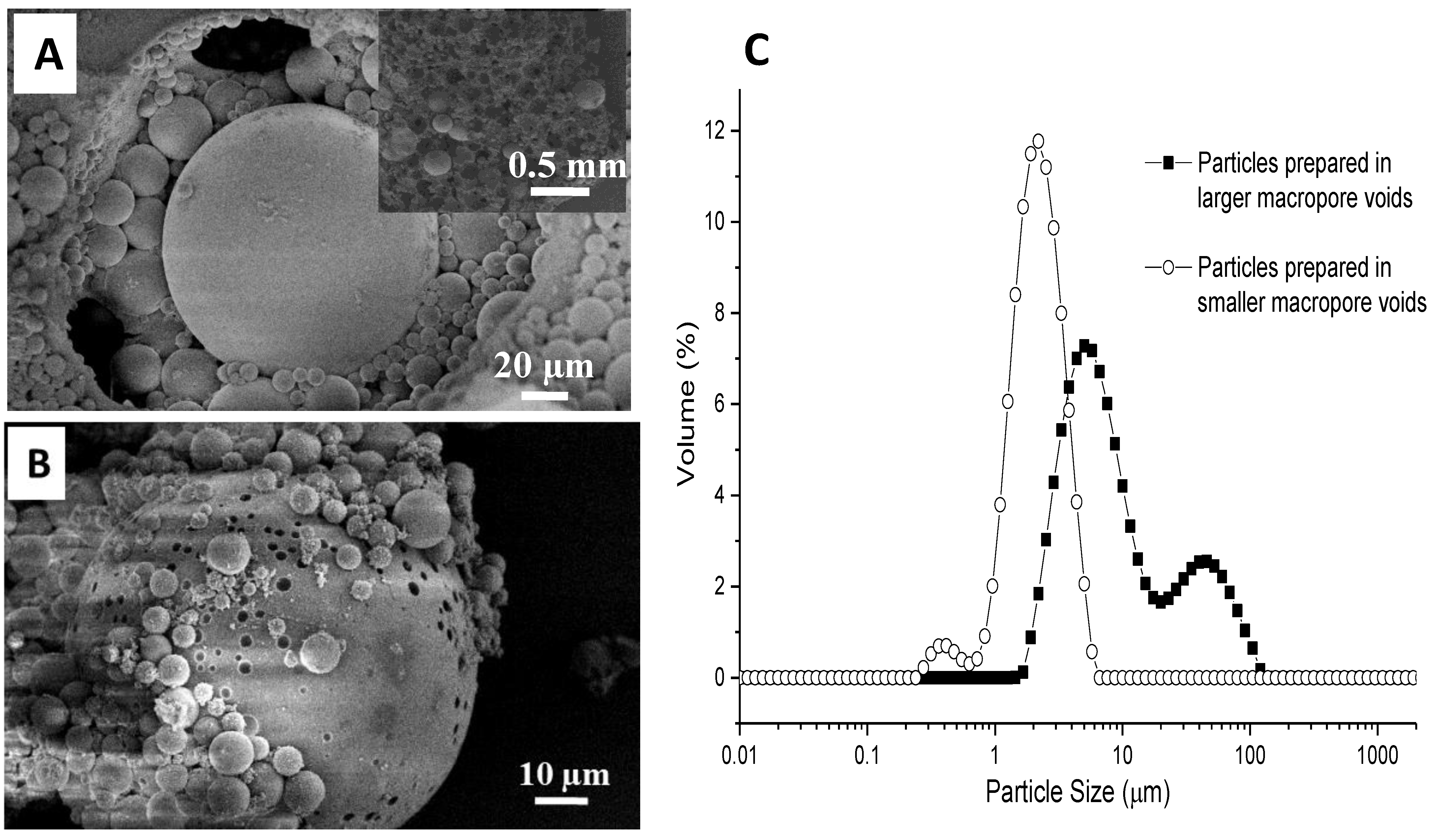
3.1. Silica Microspheres in PAM Beads
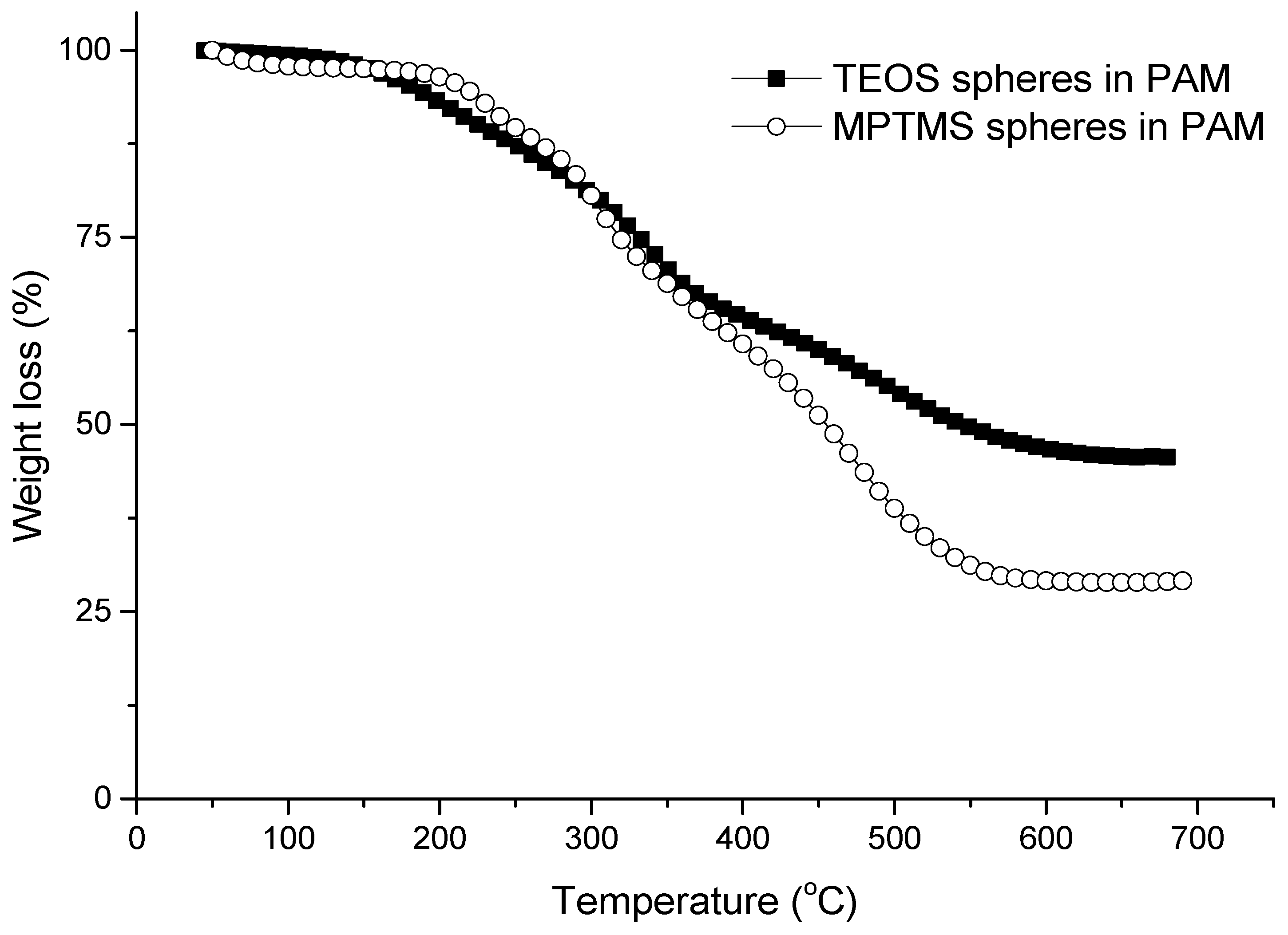
3.2. Synthesis of Organosilica Spheres in Emulsion-Templated PAM Monoliths
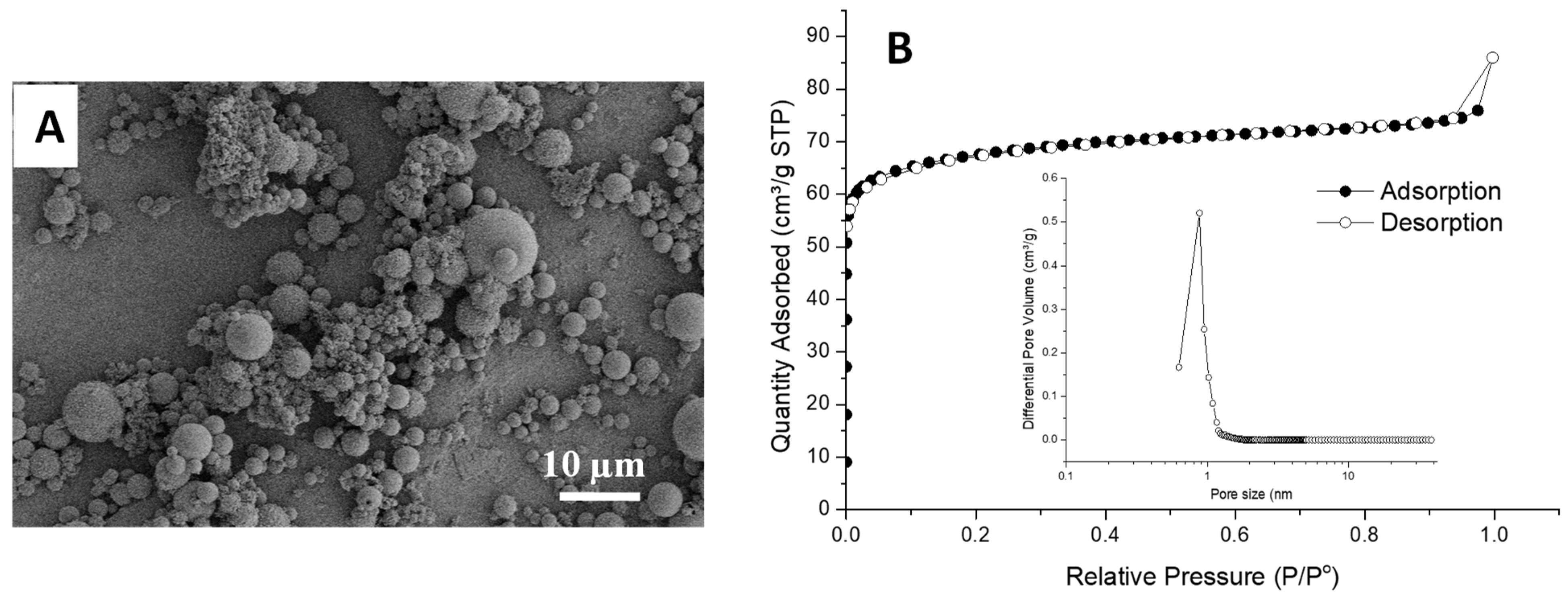
3.3. Synthesis of Fluorescent Spheres in Emulsion-Templated Scaffold
3.4. Monolithic Filtration System for Adsorption of Metal Ions and Organic Dye
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Nicole, L.; Laberty-Robert, C.; Rozes, L.; Sanchez, C. Hybrid materials science: A promised land for the integrative design of multifunctional materials. Nanoscale 2014, 6, 6267–6292. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Chen, L.-H.; Li, Y.; Rooke, J.C.; Sanchez, C.; Su, B.-L. Hierarchically porous materials: Synthesis strategies and structure design. Chem. Soc. Rev. 2017, 46, 481–558. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.-M.; Zhang, M.; Wu, Z.-F. Toward delivery of multiple growth factors in tissue engineering. Biomaterials 2010, 31, 6279–6308. [Google Scholar] [CrossRef] [PubMed]
- Kickelbick, G. (Ed.) Introduction to Hybrid Materials. In Hybrid Materials: Synthesis, Characterization, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 1; pp. 1–48. [Google Scholar]
- Habraken, W.J.E.M.; de Jonge, L.T.; Wolke, J.G.C.; Yubao, L.; Mikos, A.G.; Jansen, J.A. Introduction of gelatin microspheres into an injectable calcium phosphate cement. J. Biomed. Mater. Res. 2008, 87, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Maligal-Ganesh, R.V.; Xiao, C.; Goh, T.W.; Wang, L.-L.; Gustafson, J.; Pei, Y.; Qi, Z.; Johnson, D.D.; Zhang, S.; Tao, F.; et al. A Ship-in-a-Bottle Strategy to Synthesize Encapsulated Intermetallic Nanoparticle Catalysts: Exemplified for Furfural Hydrogenation. ACS Catal. 2016, 6, 1754–1763. [Google Scholar] [CrossRef]
- Amooghin, A.E.; Sanaeepur, H.; Omidkhah, M.; Kargari, A. “Ship-in-a-bottle”, a new synthesis strategy for preparing novel hybrid host—guest nanocomposites for highly selective membrane gas separation. J. Mater. Chem. A 2018, 6, 1751–1771. [Google Scholar] [CrossRef]
- Wang, T.; Gao, L.; Hou, J.; Herou, S.J.A.; Griffiths, J.T.; Li, W.; Dong, J.; Gao, S.; Titirici, M.-M.; Kumar, R.V.; et al. Rational approach to guest confinement inside MOF cavities for low-temperature catalysis. Nat. Commun. 2019, 10, 1340. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.-A.; Huo, Q.; Chi, M.; Veith, G.M.; Binder, A.J.; Dai, S. A “Ship-In-A-Bottle” Approach to Synthesis of Polymer Dots@Silica or Polymer Dots@Carbon Core-Shell Nanospheres. Adv. Mater. 2012, 24, 6017–6021. [Google Scholar] [CrossRef]
- Dong, L.; Hu, D.; Wang, Y.; Sheng, Z.; Hong, M.; Yang, S. A zeolite-based ship-in-a-bottle route to ultrasmall carbon dots for live cell labeling and bioimaging. Nanoscale Adv. 2020, 2, 5803–5809. [Google Scholar] [CrossRef]
- Petkovich, N.D.; Stein, A. Controlling macro- and mesostructures with hierarchical porosity through combined hard and soft templating. Chem. Soc. Rev. 2013, 42, 3721–3739. [Google Scholar] [CrossRef]
- Zhang, H. Ice Templating and Freeze-Drying for Porous Materials and Their Applications; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Zhang, T.; Sanguramath, R.A.; Israel, S.; Silverstein, M.S. Emulsion templating: Porous polymers and beyond. Macromolecules 2019, 52, 5445–5479. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Yan, P.; Zhang, L.; Zhou, H.-C.; Jiang, H.-L. Metal–organic framework-based hierarchically porous materials: Synthesis and applications. Chem. Rev. 2021, 121. [Google Scholar]
- Cai, Y.; Chen, Y.; Hong, X.; Liu, Z.; Yuan, W. Porous microsphere and its applications. Int. J. Nanomed. 2013, 8, 1111–1120. [Google Scholar]
- Kuppusamy, S.; Venkateswarlu, K.; Megharaj, M.; Mayilswami, S.; Lee, Y.B. Risk-based remediation of polluted sites: A critical perspective. Chemosphere 2017, 186, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, A.K.; Zeng, X.; Li, J. Environmental pollution of electronic waste recycling in India: A critical review. Environ. Pollut. 2016, 211, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Khin, M.M.; Nair, A.S.; Babu, V.J.; Murugan, R.; Ramakrishna, S. A review on nanomaterials for environmental remediation. Energy Environ. Sci. 2012, 5, 8075–8109. [Google Scholar] [CrossRef]
- Samanta, P.; Desai, A.V.; Let, S.; Ghosh, S.K. Advanced porous materials for sensing, capture and detoxification of organic pollutants toward water remediation. ACS Sustain. Chem. Eng. 2019, 7, 7456–7478. [Google Scholar] [CrossRef]
- Zhang, H.; Cooper, A.I. Synthesis of monodisperse emulsion-templated polymer beads by oil-in-water-in-oil (O/W/O) sedimentation polymerization. Chem. Mater. 2002, 14, 4017–4020. [Google Scholar] [CrossRef]
- Qian, L.; Ahmed, A.; Foster, A.; Rannard, S.P.; Cooper, A.I.; Zhang, H. Systematic tuning of pore morphologies and pore volumes in macroporous materials by freezing. J. Mater. Chem. 2009, 19, 5212–5219. [Google Scholar] [CrossRef]
- Herz, E.; Ow, H.; Bonner, D.; Burns, A.; Wiesner, U. Dye structure–optical property correlations in near-infrared fluorescent core-shell silica nanoparticles. J. Mater. Chem. 2009, 19, 6341–6347. [Google Scholar] [CrossRef]
- Singh, P.; Srivastava, S.; Singh, S.K. Nanosilica: Recent progress in synthesis, functionalization, biocompatibility, and biomedical applications. ACS Biomater. Sci. Eng. 2019, 5, 4882–4898. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, F.; Roointan, A.; Mohammadi-Samani, S.; Hosseini, M. Mesoporous silica nanoparticles: Synthesis, pharmaceutical applications, biodistribution, and biosafety assessment. Chem. Eng. J. 2019, 359, 684–705. [Google Scholar] [CrossRef]
- Hayes, R.; Ahmed, A.; Edge, T.; Zhang, H. Core–shell particles: Preparation, fundamentals and applications in high performance liquid chromatography. J. Chromatogr. A 2014, 1357, 36–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Clowes, R.; Willneff, E.; Ritchie, H.; Myers, P.; Zhang, H. Synthesis of uniform porous silica microspheres with hydrophilic polymer as stabilizing agent. Ind. Eng. Chem. Res. 2010, 49, 602–608. [Google Scholar] [CrossRef]
- Diagboya, P.N.E.; Dikio, E.D. Silica-based mesoporous materials; emerging designer adsorbents for aqueous pollutants removal and water treatment. Micropor. Mesopor. Mater. 2018, 266, 252–267. [Google Scholar] [CrossRef]
- Soltani, R.; Marjani, A.; Shirazian, S. Facile one-pot synthesis of thiol-functionalized mesoporous silica submicrospheres for Tl(I) adsorption: Isotherm, kinetic and thermodynamic studies. J. Hazard. Mater. 2019, 371, 146–155. [Google Scholar] [CrossRef]
- Yan, X.; Meng, J.; Hu, X.; Feng, R.; Zhou, M. Synthesis of thiol-functionalized mesoporous silica nanoparticles for adsorption of Hg2+ from aqueous solution. J. Sol-Gel Sci. Technol. 2019, 89, 617–622. [Google Scholar] [CrossRef]
- Liang, R.; Fang, X.; Qiu, B.; Zou, H. One-step synthesis of golf ball-like thiol-functionalized silica particles. Soft Matter 2020, 16, 9113–9120. [Google Scholar] [CrossRef] [PubMed]
- Eeltink, S.; Wouters, S.; Dores-Sousa, J.L.; Svec, F. Advances in organic polymer-based monolithic column technology for high-resolution liquid chromatography-mass spectrometry profiling of antibodies, intact proteins, oligonucleotides, and peptides. J. Chromatogr. A 2017, 1498, 8–21. [Google Scholar] [CrossRef]
- Huh, S.; Wiench, J.W.; Yoo, J.-C.; Pruski, M.; Lin, V.S.Y. Organic functionalization and morphology control of mesoporous silicas via a co-condensation synthesis method. Chem. Mater. 2003, 15, 4247–4256. [Google Scholar] [CrossRef]
- Barczak, M.; Dobrowolski, R.; Borowski, P.; Giannakoudakis, D.A. Pyridine-, thiol- and amine-functionalized mesoporous silicas for adsorptive removal of pharmaceuticals. Micropor. Mesopor. Mater. 2020, 299, 110312. [Google Scholar] [CrossRef]
- Li, B.; Zou, X.; Zhao, Y.; Sun, L.; Li, S. Biofunctionalization of silica microspheres for protein separation. Mater. Sci. Eng. C 2013, 33, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.W.; Tan, W.; Hong, J.-I. Fluorescent dye-doped silica nanoparticles: New tools for bioapplications. Chem. Commun. 2012, 48, 2270–2282. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Li, N.; Chi, Y.; Geng, W.; Yan, W.; Zhao, Y.; Li, X.; Dong, B. Effect of large pore size of multifunctional mesoporous microsphere on removal of heavy metal ions. J. Hazard. Mater. 2013, 254–255, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sun, Q.; Zhao, Q.; Marin, L.; Cheng, X. Fluorescent porous silica microspheres for highly and selectively detecting Hg2+ and Pb2+ ions and imaging in living cells. ACS Omega 2019, 4, 18381–18391. [Google Scholar] [CrossRef]
- Karimi, M.; Shojaei, A.; Nematollahzadeh, A.; Abdekhodaie, M.J. Column study of Cr (VI) adsorption onto modified silica–polyacrylamide microspheres composite. Chem. Eng. J. 2012, 210, 280–288. [Google Scholar] [CrossRef]
- Ahmed, A.; Ritchie, H.; Myers, P.; Zhang, H. One-pot synthesis of spheres-on-sphere silica particles from a single precursor for fast HPLC with low back pressure. Adv. Mater. 2012, 24, 6042–6048. [Google Scholar] [CrossRef]
- Zhu, X.; Su, Q.; Feng, W.; Li, F. Anti-Stokes shift luminescent materials for bio-applications. Chem. Soc. Rev. 2017, 46, 1025–1039. [Google Scholar] [CrossRef]
- Aslan, K.; Wu, M.; Lakowicz, J.R.; Geddes, C.D. Fluorescent core-shell Ag@SiO2 nanocomposites for metal-enhanced fluorescence and single nanoparticle sensing platforms. J. Am. Chem. Soc. 2007, 129, 1524–1525. [Google Scholar] [CrossRef]
- Viger, M.L.; Live, L.S.; Therrien, O.D.; Boudreau, D. Reduction of self-quenching in fluorescent silica-coated silver nanoparticles. Plasmonics 2008, 3, 33–40. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, Y.; Li, H.; Fang, C.; Li, H.; Wang, K. Synthesis of stable Ag NPs solution via anionic polyacrylamide template method as sensitive fluorescence sensor for detecting heavy metal ions. Chem. Lett. 2019, 48, 1448–1451. [Google Scholar] [CrossRef]
- Ptaszkowska-Konizar, M.; Goscianska, J.; Pietrzak, R. Removal of rhodamine B from water by modified carbon xerogels. Colloids Surf. A 2018, 543, 109–117. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | MPTMS (cm3) a | Mass Gained (%) | Surface Area (m2/g) b | Intrusion Volume (cm3/g) c | Bulk Density (g/cm3) d | Macropores (µm) e |
|---|---|---|---|---|---|---|
| PAM | --- | --- | 6.13 | 2.878 | 0.109 | 1.08 |
| S1 | 0.5 | 112 | 9.36 | 1.503 | 0.463 | 1.08 |
| S2 | 2.5 | 114 | 7.64 | 1.194 | 0.563 | 1.08 |
| S3 | 4.0 | 118 | 8.69 | 0.945 | 0.595 | 0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, A.; Myers, P.; Zhang, H. Silica Microspheres-in-Pores Composite Monoliths with Fluorescence and Potential for Water Remediation. Nanomaterials 2021, 11, 2681. https://doi.org/10.3390/nano11102681
Ahmed A, Myers P, Zhang H. Silica Microspheres-in-Pores Composite Monoliths with Fluorescence and Potential for Water Remediation. Nanomaterials. 2021; 11(10):2681. https://doi.org/10.3390/nano11102681
Chicago/Turabian StyleAhmed, Adham, Peter Myers, and Haifei Zhang. 2021. "Silica Microspheres-in-Pores Composite Monoliths with Fluorescence and Potential for Water Remediation" Nanomaterials 11, no. 10: 2681. https://doi.org/10.3390/nano11102681
APA StyleAhmed, A., Myers, P., & Zhang, H. (2021). Silica Microspheres-in-Pores Composite Monoliths with Fluorescence and Potential for Water Remediation. Nanomaterials, 11(10), 2681. https://doi.org/10.3390/nano11102681