
Ordered Porous TiO2@C Layer as an Electrocatalyst Support for Improved Stability in PEMFCs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
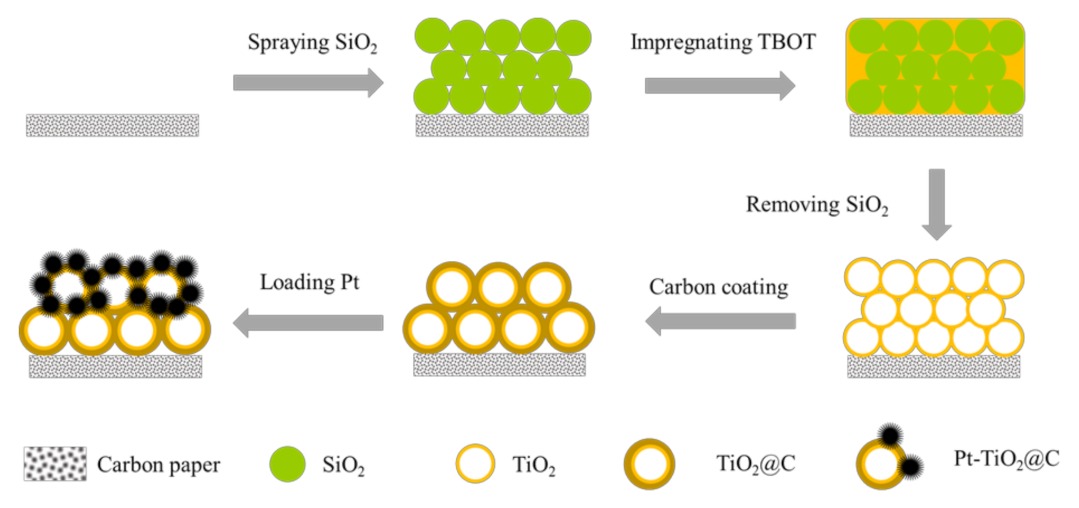
2.2. Preparation Ordered Porous Pt-TiO2@C Array
2.2.1. Preparation of SiO2 Sphere Array
2.2.2. Preparation of Porous TiO2/CP Array
2.2.3. Preparation of Porous TiO2@C/CP Array
2.2.4. Preparation of Pt nanoparticle on Porous TiO2@C/CP Array
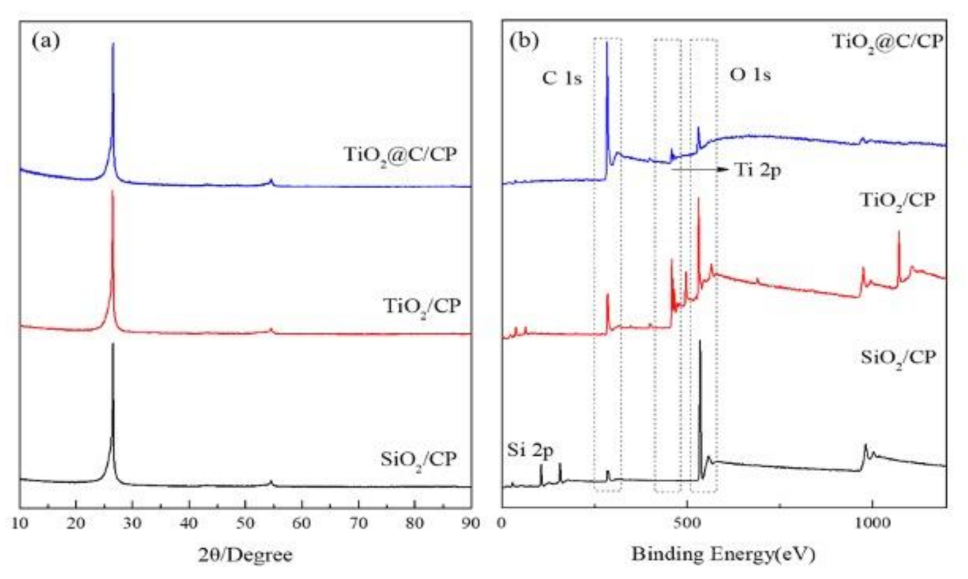
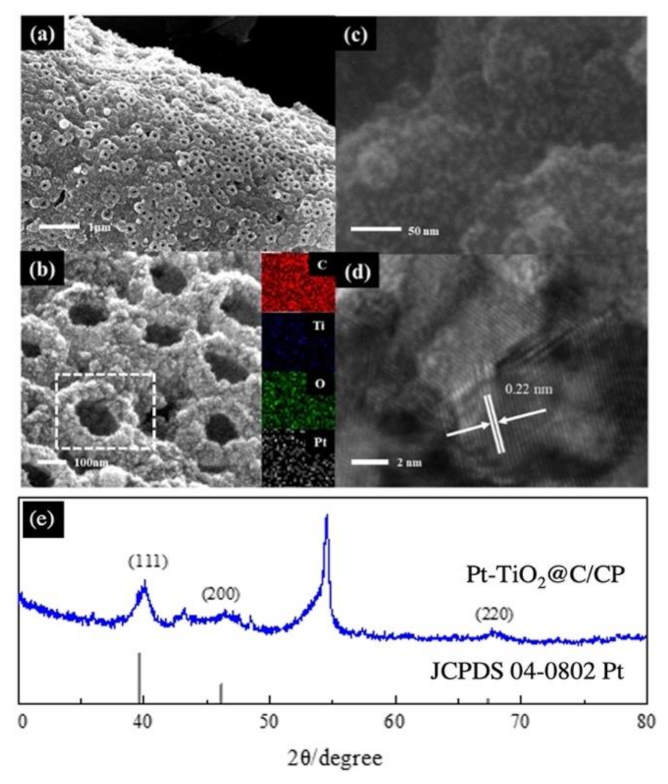
2.3. Structural Characterizations
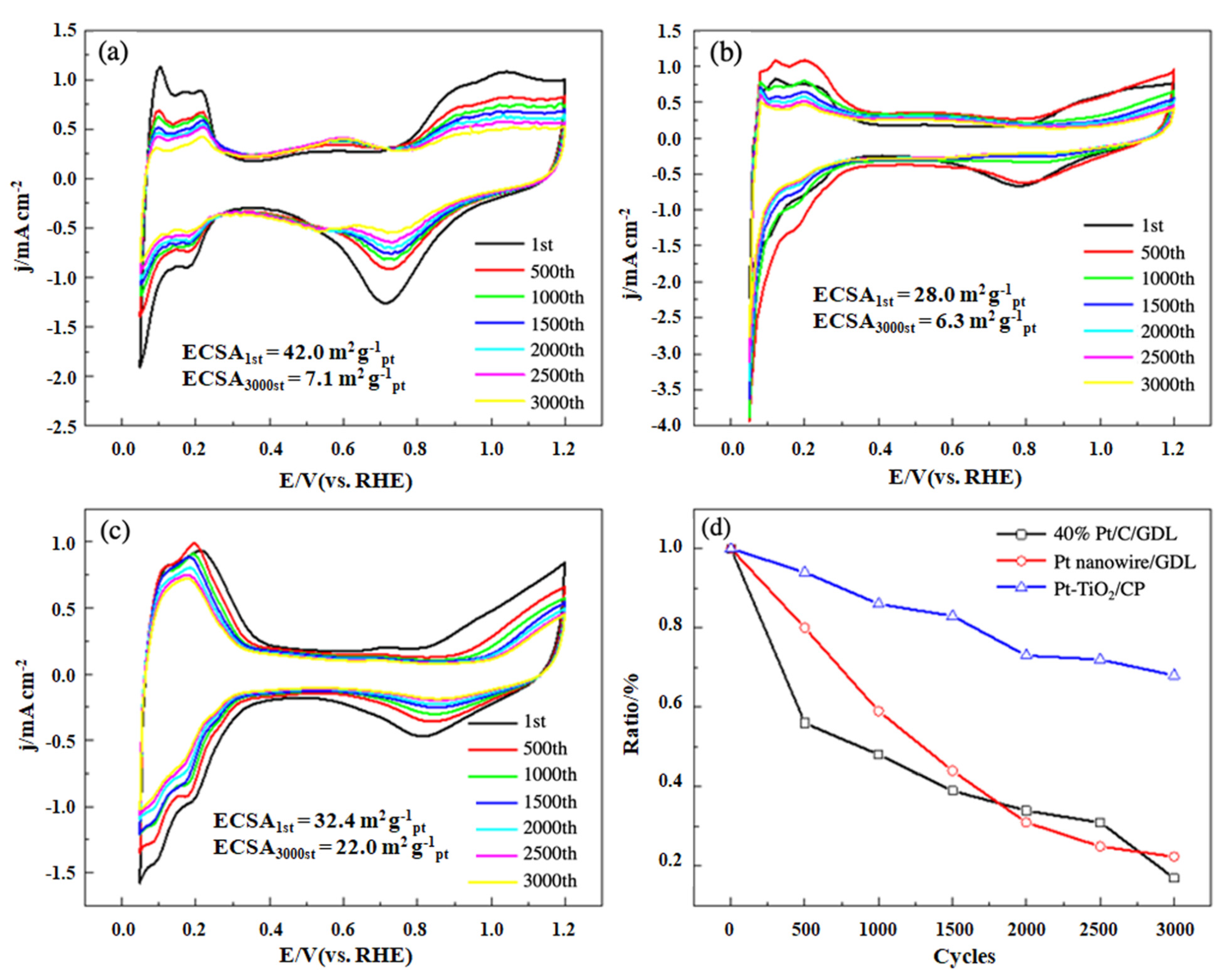
2.4. Electrochemical Tests
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mobarakeh, M.D. Synthesis of electrospun carbon nanofiber/WO3 supported Pt, Pt/Pd and Pt/Ru catalysts for fuel cells. Fiber. Polym. 2017, 18, 95–101. [Google Scholar] [CrossRef]
- Fang, B.; Chaudhari, N.; Kim, M.; Kim, J.; Yu, J. Homogeneous deposition of platinum nanoparticles on carbon black for proton exchange membrane fuel cell. J. Am. Chem. Soc. 2009, 131, 15330–15338. [Google Scholar] [CrossRef] [PubMed]
- Cleve, T.V.; Moniri, S.; Belok, G.; More, K.L.; Linic, S. Nanoscale engineering of efficient oxygen reduction electrocatalysts by tailoring the local chemical environment of Pt surface sites. ACS Catal. 2017, 7, 17–24. [Google Scholar] [CrossRef]
- Chen, Q.; Jia, Y.; Xie, S.; Xie, Z. Well-faceted noble-metal nanocrystals with nonconvex polyhedral shapes. Chem. Soc. Rev. 2016, 45, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.S.; Popov, B.N. Hybrid cathode catalyst with synergistic effect between carbon composite catalyst and Pt for ultra-low Pt loading in PEMFCs. Catal. Today 2017, 295, 65–74. [Google Scholar] [CrossRef]
- Lamy, C.; Jones, D.J.; Coutanceau, C.; Brault, P.; Martemianov, S.; Bultel, Y. Do not forget the electrochemical characteristics of the membrane electrode assembly when designing a proton exchange membrane fuel cell stack. Electrochim. Acta 2011, 56, 10406–10423. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Wang, S.; Li, Y.; Jiang, L.; Sun, H.; Zhu, S.; Su, D.; Sun, G. Vertically oriented polypyrrole nanowire arrays on Pd-plated Nafion® membrane and its application in direct methanol fuel cells. J. Mater. Chem. A 2013, 1, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Xia, Z.; Shang, L.; Fu, X.; Li, H.; Wang, S.; Sun, G. Hierarchically ordered arrays with platinum coated PANI nanowires for highly efficient fuel cell electrodes. J. Mater. Chem. A 2017, 5, 15260–15265. [Google Scholar] [CrossRef]
- Xia, Z.; Wang, S.; Jiang, L.; Sun, H.; Liu, S.; Fu, X.; Zhang, B.; Su, D.S.; Wang, J.; Sun, G. Bio-inspired construction of advanced fuel cell cathode with Pt anchored in ordered hybrid polymer matrix. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Wilckens, R.S.; Du, S. Evolution of gas diffusion layer structures for aligned Pt nanowire electrodes in PEMFC applications. Electrochim. Acta 2018, 279, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, M.; Yang, Z.; Wang, X. High performance and durability of order-structured cathode catalyst layer based on TiO2@PANI core-shell nanowire arrays. Appl. Surf. Sci. 2017, 406, 69–76. [Google Scholar] [CrossRef]
- Salernitano, E.; Giorgi, L.; Makris, T.D. Direct growth of carbon nanofibers on carbon- based substrates as integrated gas diffusion and catalyst layer for polymer electrolyte fuel cells. Int. J. Hydrog. Energ. 2014, 39, 15005–15016. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Jiang, S.; Yi, B.; Cao, L.; Song, W.; Zhao, Q.; Yu, H.; Shao, Z. Development of advanced catalytic layer based on vertically aligned conductive polymer arrays for thin-film fuel cell electrodes. J. Power Sources 2016, 329, 347–354. [Google Scholar] [CrossRef]
- Debe, M.K.; Hendricks, S.M.; Vernstrom, G.D.; Meyers, M.; Brostrom, M.; Stephens, M.; Chan, Q.; Willey, J.; Hamden, M.; Mittelsteadt, C.K.; et al. Initial performance and durability of ultra-low loaded NSTF electrodes for PEM electrolyzers. J. Electrochem. Soc. 2012, 159, K165–K176. [Google Scholar] [CrossRef]
- Hussain, S.; Erikson, H.; Kongi, N.; Merisalu, M.; Ritslaid, P.; Sammelselg, V.; Tammeveski, K. Heat-treatment effects on the ORR activity of Pt nanoparticles deposited on multi-walled carbon nanotubes using magnetron sputtering technique. Int. J. Hydrogen Energ. 2017, 42, 5958–5970. [Google Scholar] [CrossRef]
- Sun, S.; Jaouen, F.; Dodelet, J. Controlled growth of Pt nanowires on carbon nanospheres and their enhanced performance as electrocatalysts in PEM fuel cells. Adv. Mater. 2008, 20, 3900–3904. [Google Scholar] [CrossRef]
- Saha, M.S.; Banis, M.N.; Zhang, Y.; Li, R.; Sun, X.; Cai, M.; Wagner, F.T. Tungsten oxide nanowires grown on carbon paper as Pt electrocatalyst support for high performance proton exchange membrane fuel cells. J. Power Sources 2009, 192, 330–335. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, H.; Li, Y.; Song, W.; Yi, B.; Shao, Z. Preparation of Pt catalysts decorated TiO2 nanotube arrays by redox replacement of Ni precursors for proton exchange membrane fuel cells. Electrochim. Acta 2012, 80, 1–6. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fang, B.; Li, H.; Bi, X.T.; Wang, H. Progress in modified carbon support materials for Pt and Pt-alloy cathode catalysts in polymer electrolyte membrane fuel cells. Prog. Mater. Sci. 2016, 82, 445–498. [Google Scholar] [CrossRef]
- Chung, D.Y.; Yoo, J.M.; Sung, Y.E. Highly durable and active Pt-based nanoscale design for fuel-cell oxygen-reduction electrocatalysts. Adv. Mater. 2018, 30, 1704123–1704143. [Google Scholar] [CrossRef]
- Park, Y.C.; Kakinuma, K.; Uchida, M.; Tryk, D.A.; Kamino, T.; Uchida, H.; Watanabe, M. Investigation of the corrosion of carbon supports in polymer electrolyte fuel cells using simulated start-up/shutdown cycling. Electrochim. Acta 2013, 91, 195–207. [Google Scholar] [CrossRef]
- Spernjak, D.; Fairweather, J.; Mukundan, R.; Rockward, T.; Borup, R.L. Influence of the microporous layer on carbon corrosion in the catalyst layer of a polymer electrolyte membrane fuel cell. J. Power Sources 2014, 214, 386–398. [Google Scholar] [CrossRef]
- Wang, X.; Lee, J.S.; Zhu, Q.; Liu, J.; Wang, Y.; Dai, S. Ammonia-treated ordered mesoporous carbons as catalytic materials for oxygen reduction reaction. Chem. Mater. 2010, 22, 2178–2180. [Google Scholar] [CrossRef]
- Abdullah, A.M.; Al-Thani, N.J.; Tawbi, K.; Al-Kandari, H. Carbon/nitrogen-doped TiO2: New synthesis route, characterization and application for phenol degradation. Arab. J. Chem. 2016, 9, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Fang, B.; Bonakdarpour, A.; Reilly, K.; Xing, Y.; Taghipour, F.; Wilkinson, D.P. Large-scale synthesis of TiO2 microspheres with hierarchical nanostructure for highly efficient photodriven reduction of CO2 to CH4. ACS Appl. Mater. Interfaces 2014, 6, 15488–15498. [Google Scholar] [CrossRef]
- Jin, J.; Ren, X.N.; Lu, Y.; Zheng, X.F.; Wang, H.E.; Chen, L.H.; Yang, X.Y.; Li, Y.; Su, B.L. High lithium ion battery performance enhancement by controlled carbon coating of TiO2 hierarchically porous hollow spheres. RSC Adv. 2016, 6, 70485–70492. [Google Scholar] [CrossRef]
- Meng, H.; Xie, F.; Chen, J.; Sun, S.; Shen, P. Morphology controllable growth of Pt nanoparticles/nanowires on carbon powders and its application as novel electro-catalyst for methanol oxidation. Nanoscale 2011, 3, 5041–5048. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Daniel, L.; Bonakdarpour, A.; Govindarajan, R.; Sharman, J.; Wilkinson, D.P. Dense Pt nanowire electrocatalyst for improved fuel cell performance using a graphitic carbon nitride-decorated hierarchical nanocarbon support. Small 2021, 17, 2102288. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xu, J.; Wang, Y.; Jiang, J.; Wang, X. A novel catalyst coated membrane embedded with Cs-substituted phosphotungstates for proton exchange membrane water electrolysis. Int. J. Hydrog. Energ. 2014, 39, 14531–14539. [Google Scholar] [CrossRef]
- Fang, B.; Pinaud, B.A.; Wilkinson, D.P. Carbon-supported Pt hollow nanospheres as a highly efficient electrocatalyst for the oxygen reduction reaction. Electrocatalysis 2016, 7, 336–344. [Google Scholar] [CrossRef]
- Chen, S.; Xin, Y.; Zhou, Y.; Zhang, F.; Ma, Y.; Zhou, H.; Qi, L. Branched CNT@SnO2 nanorods@ carbon hierarchical heterostructures for lithium ion batteries with high reversibility and rate capability. J. Mater. Chem. A 2014, 2, 15582–15589. [Google Scholar] [CrossRef]
- Wei, Z.; Su, K.; Sui, S.; He, A.; Du, S. High performance polymer electrolyte membrane fuel cells (PEMFCs) with gradient Pt nanowire cathodes prepared by decal transfer method. Int. J. Hydrogen Energ. 2015, 40, 3068–3074. [Google Scholar] [CrossRef]
- Narayanamoorthy, B.; Linkov, V.; Sita, C.; Pasupathi, S. Pt3M(M: Co, Ni and Fe) bimetallic alloy nanoclusters as support-free electrocatalysts with improved activity and durability for dioxygen reduction in PEM fuel cells. Electrocatalysis 2016, 7, 400–410. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, M.; Fang, B.; Liu, G. Ordered SnO2@C flake array as catalyst support for improved electrocatalytic activity and cathode durability in PEMFCs. Nanomaterials 2020, 10, 2412–2427. [Google Scholar] [CrossRef]
- Li, B.; Yan, Z.; Higgins, D.C.; Yang, D.; Chen, Z.; Ma, J. Carbon-supported Pt nanowire as novel cathode catalysts for proton exchange membrane fuel cells. J. Power Sources 2014, 262, 488–493. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, S.; Liu, Q.; Gu, J.; Guo, Z.; Chen, Z.; Feng, C.; Zhang, D.; Moon, W.J. Carbon-coated SnO2@C with hierarchically porous structures and graphite layers inside for a high-performance lithium-ion battery. J. Mater. Chem. 2012, 22, 2766–2773. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, S.; Li, Z.; Ma, D.; Xu, G.; Fang, B. Mesoporous g-C3N4 nanosheets with improved photocatalytic performance for hydrogen evolution. Mater. Charact. 2021, 174, 111031. [Google Scholar] [CrossRef]
- Yu, S.; Song, S.; Li, R.; Fang, B. The lightest solid meets the lightest gas: An overview of carbon aerogels and their composites for hydrogen related applications. Nanoscale 2020, 12, 19536–19556. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, X.; Wang, Y.J.; Song, S.; Cui, L.; Fan, H.; Qiao, X.; Fang, B. A new perspective of the lanthanide metal–organic frameworks: Tailoring Dy-BTC nanospheres for rechargeable Li-O2 batteries. Nanoscale 2020, 12, 9524–9532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Luo, F.; Tan, Y.; Zeng, L.; Fang, B.; Liu, A. Rock salt type NiCo2O3 supported on ordered mesoporous carbon as a highly efficient electrocatalyst for oxygen evolution reaction. Appl. Catal. B Environ. 2019, 256, 117852. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, G.; Ma, D.; Li, Z.; Yan, Z.; Xu, A.; Zhong, W.; Fang, B. Synergistic effects of g-C3N4 three-dimensional inverse opals and Ag modification on high-efficiency photocatalytic H2 evolution. J. Clean. Prod. 2021, 328, 129745. [Google Scholar] [CrossRef]
- Fang, B.; Wei, Y.Z.; Suzuki, K.; Kumagai, M. Surface modification of carbonaceous materials for EDLCs application. Electrochim. Acta 2005, 50, 3616–3621. [Google Scholar] [CrossRef]
- Xing, Y.; Fang, B.; Bonakdarpour, A.; Zhang, S.; Wilkinson, D.P. Facile fabrication of mesoporous carbon nanofibers with unique hierarchical nanoarchitecture for electrochemical hydrogen storage. Int. J. Hydrog. Energy 2014, 39, 7859–7867. [Google Scholar] [CrossRef]
- Xing, Y.; Wang, S.; Fang, B.; Feng, Y.; Zhang, S. Three-dimensional nanoporous Cu6Sn5/Cu composite from dealloying as anode for Lithium ion batteries. Microporous Mesoporous Mater. 2018, 261, 237–243. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Yang, Z.; Wang, X.; Fang, B. Ordered Porous TiO2@C Layer as an Electrocatalyst Support for Improved Stability in PEMFCs. Nanomaterials 2021, 11, 3462. https://doi.org/10.3390/nano11123462
Liu G, Yang Z, Wang X, Fang B. Ordered Porous TiO2@C Layer as an Electrocatalyst Support for Improved Stability in PEMFCs. Nanomaterials. 2021; 11(12):3462. https://doi.org/10.3390/nano11123462
Chicago/Turabian StyleLiu, Gaoyang, Zhaoyi Yang, Xindong Wang, and Baizeng Fang. 2021. "Ordered Porous TiO2@C Layer as an Electrocatalyst Support for Improved Stability in PEMFCs" Nanomaterials 11, no. 12: 3462. https://doi.org/10.3390/nano11123462
APA StyleLiu, G., Yang, Z., Wang, X., & Fang, B. (2021). Ordered Porous TiO2@C Layer as an Electrocatalyst Support for Improved Stability in PEMFCs. Nanomaterials, 11(12), 3462. https://doi.org/10.3390/nano11123462