
Graphene Oxide Chemistry Management via the Use of KMnO4/K2Cr2O7 Oxidizing Agents

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. GO Synthesis
2.2. Characterization
3. Results and Discussion
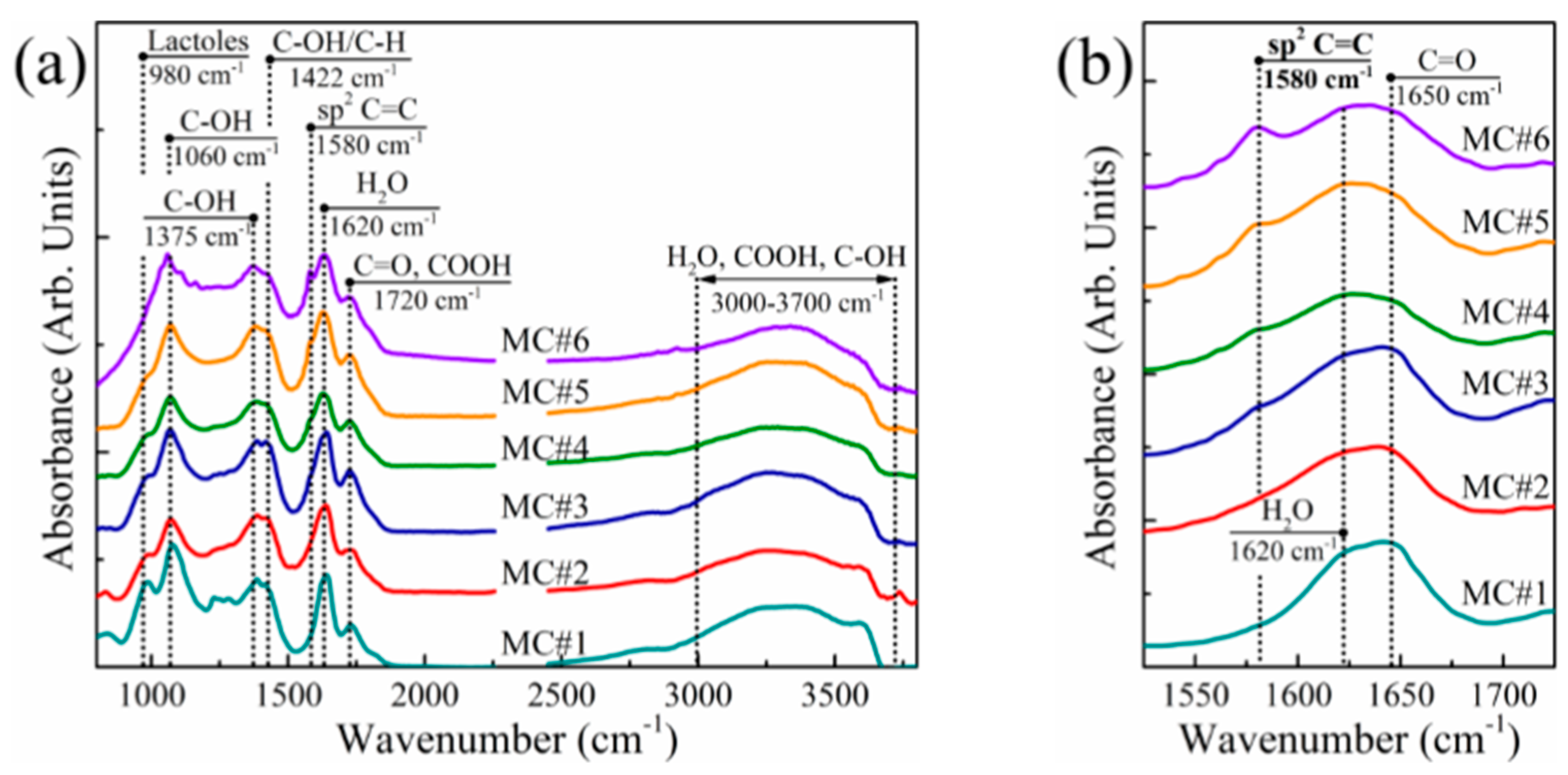
3.1. FTIR Analysis
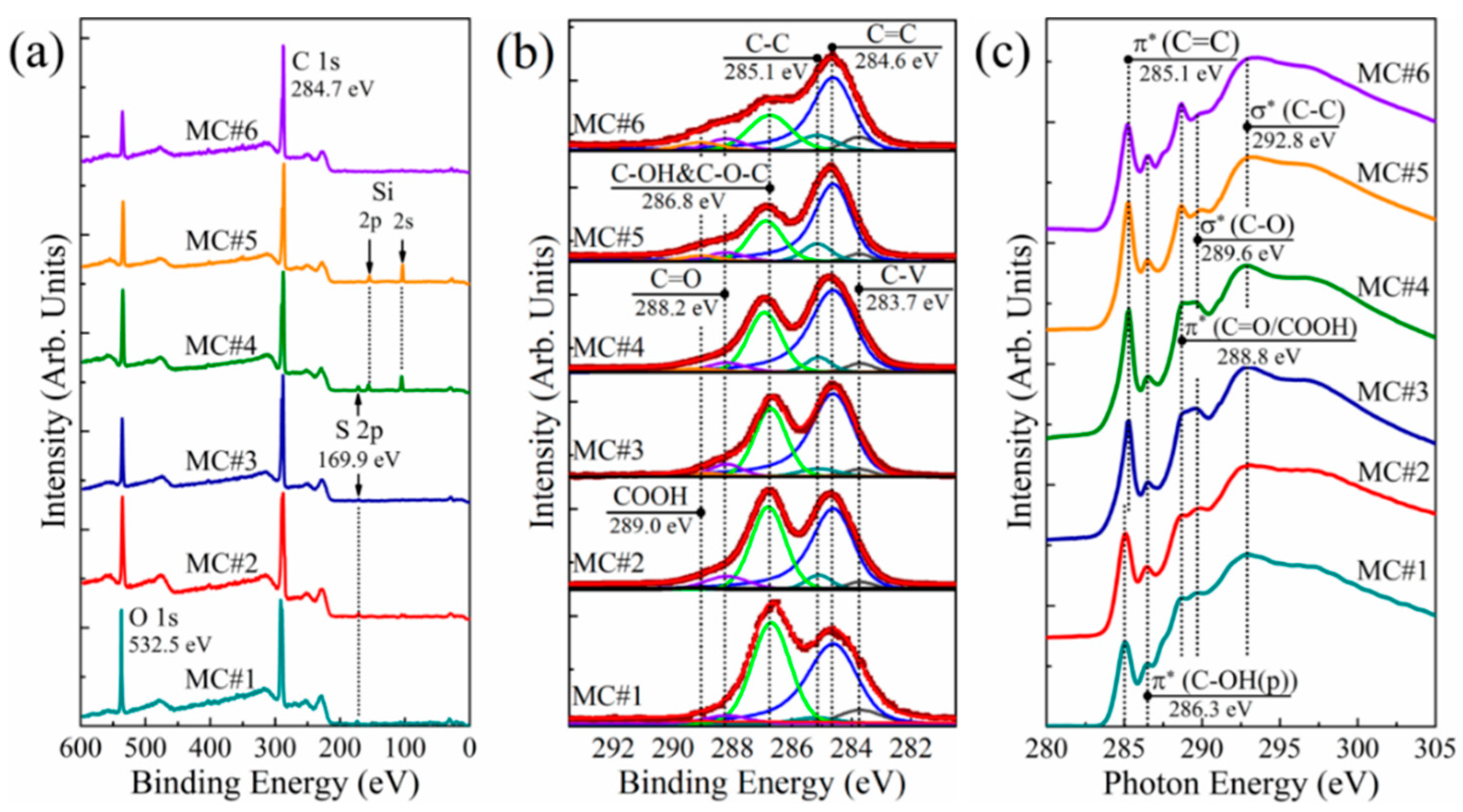
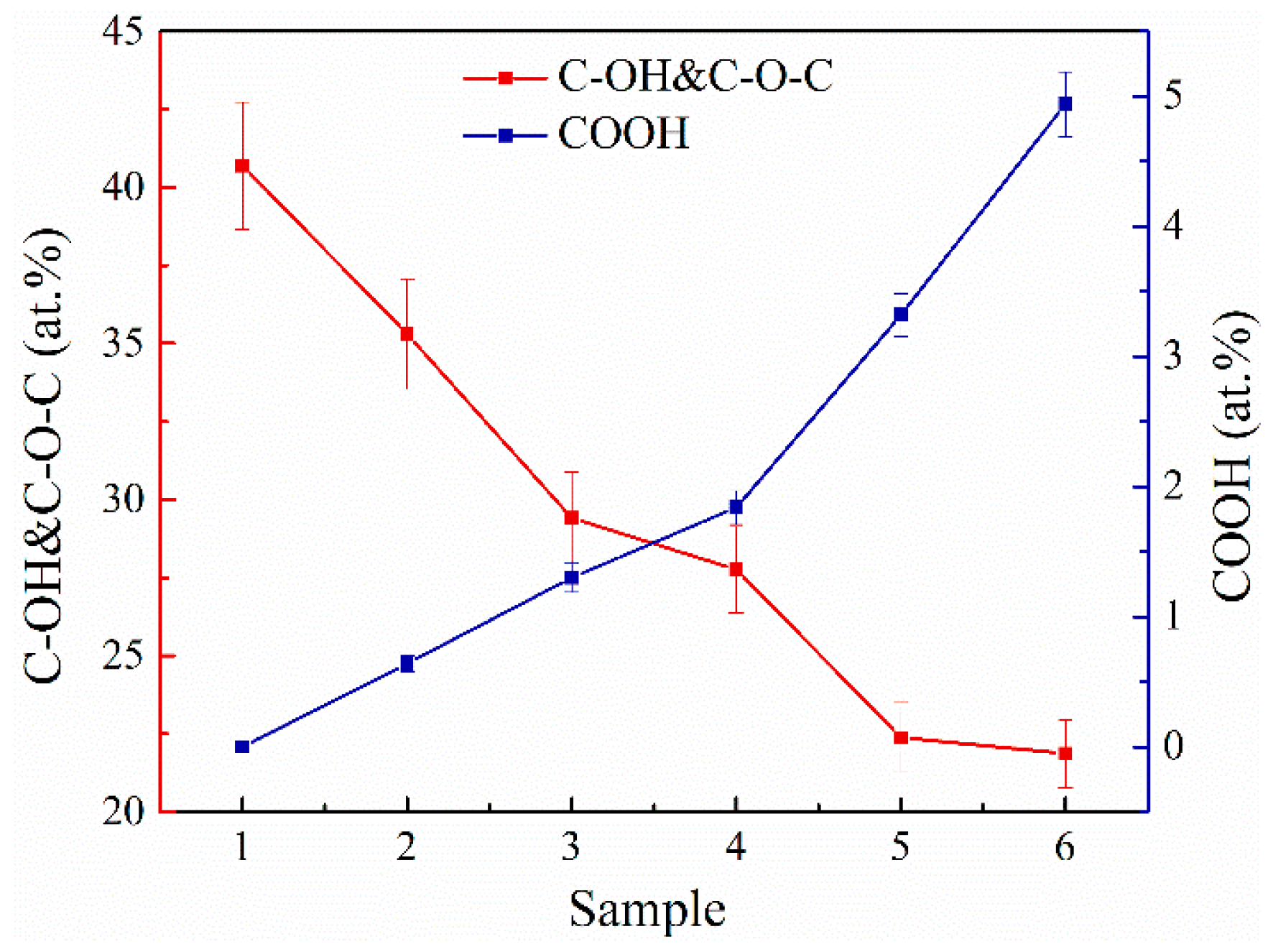
3.2. XPS and XAS Analysis
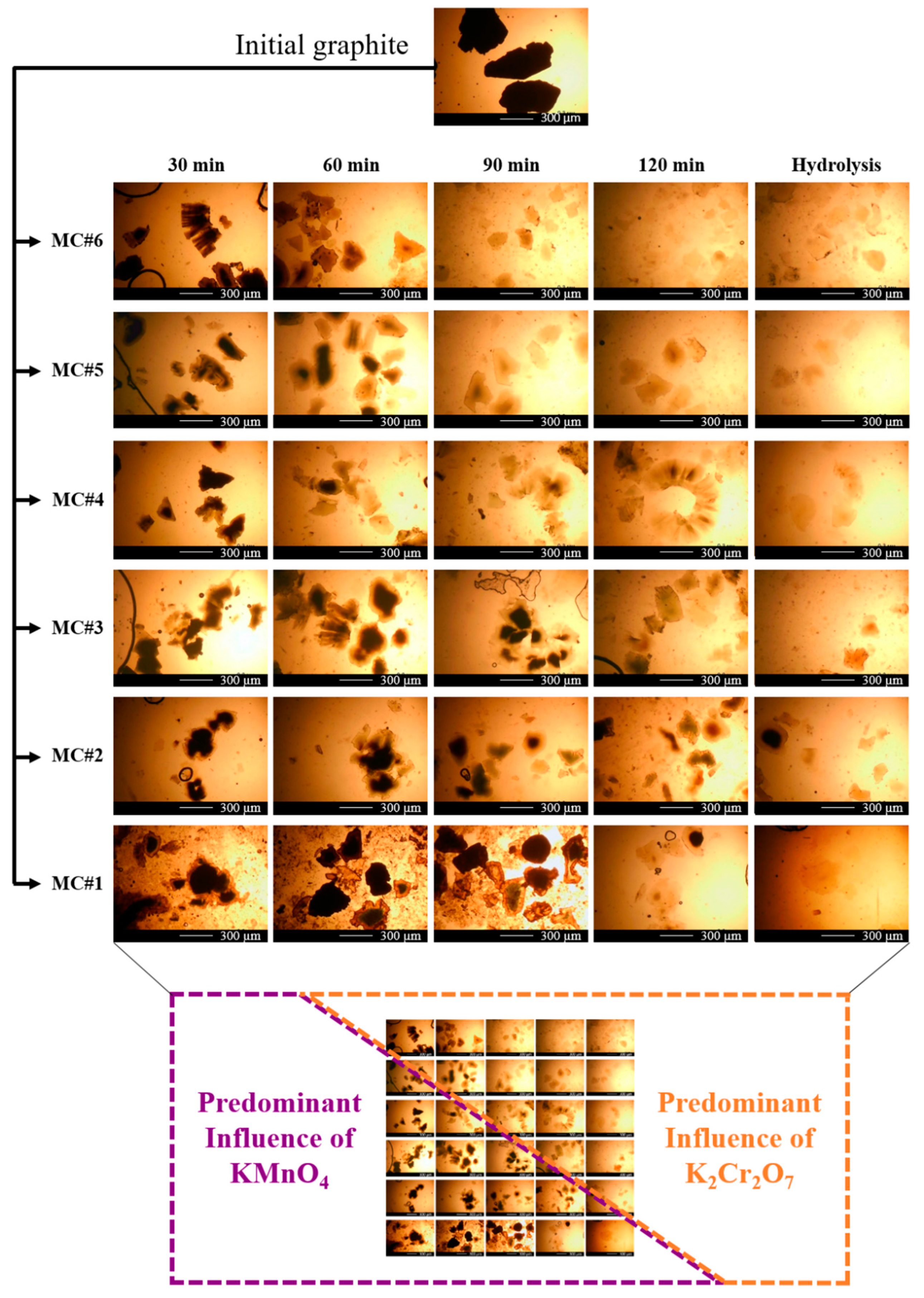
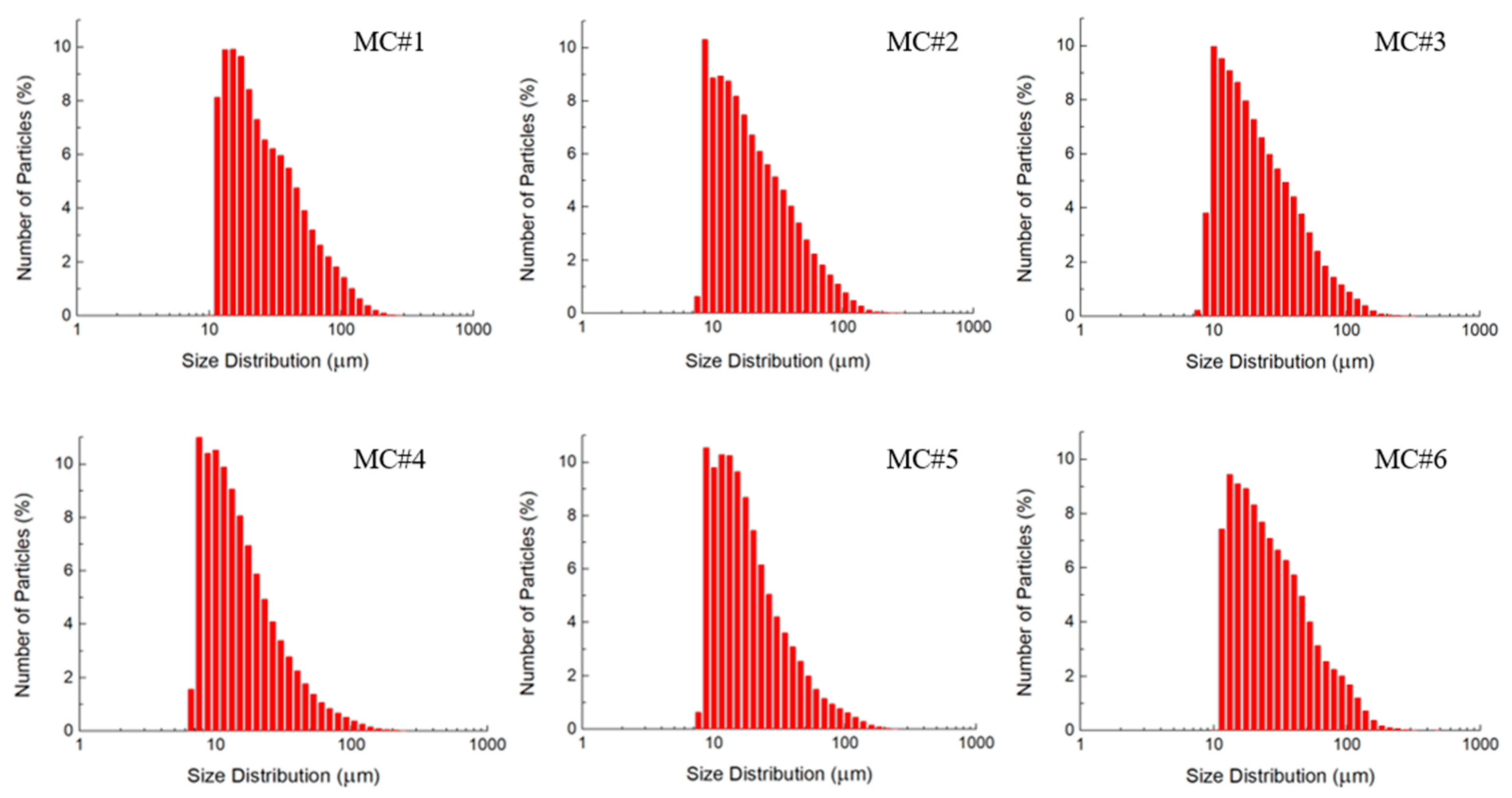
3.3. Optical Microscopy and Laser Diffraction Analysis
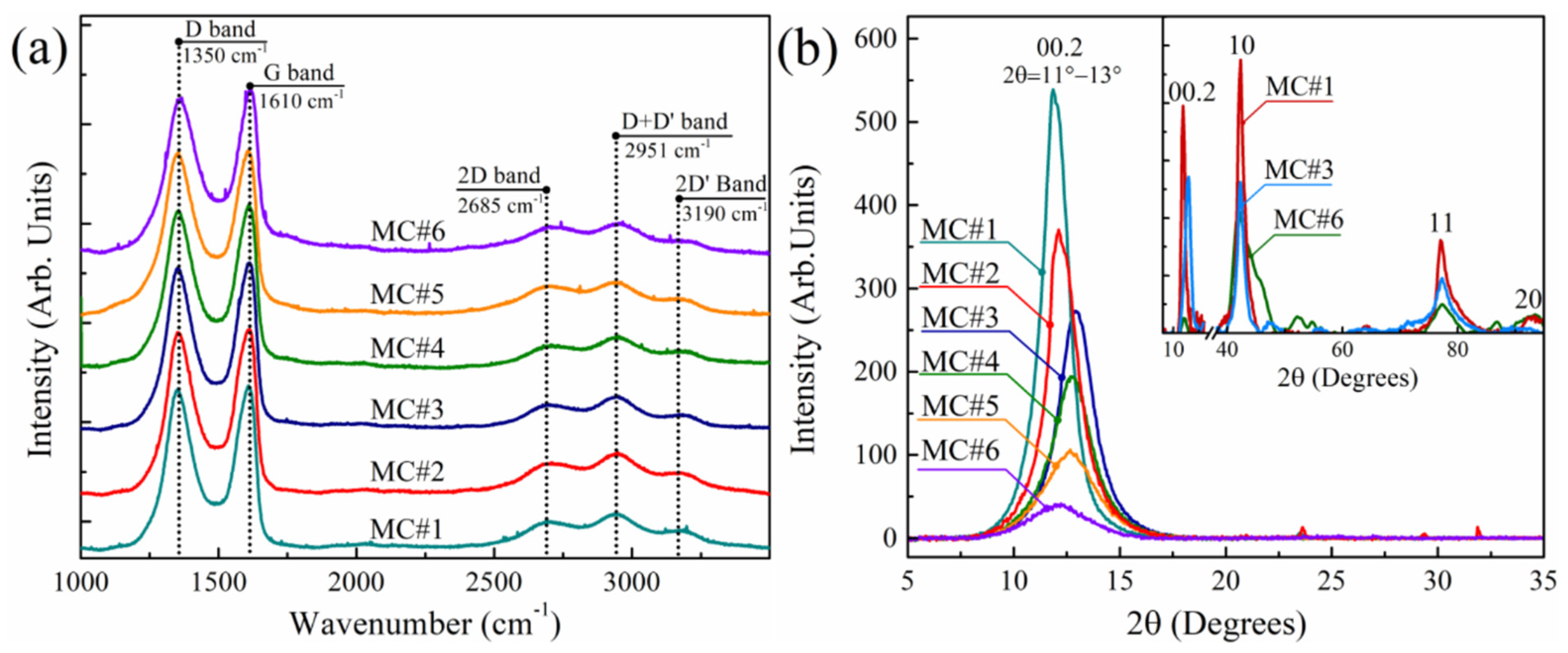
3.4. Raman Spectra and XRD Patterns
3.5. Morphology: TEM Images and SAED Patterns
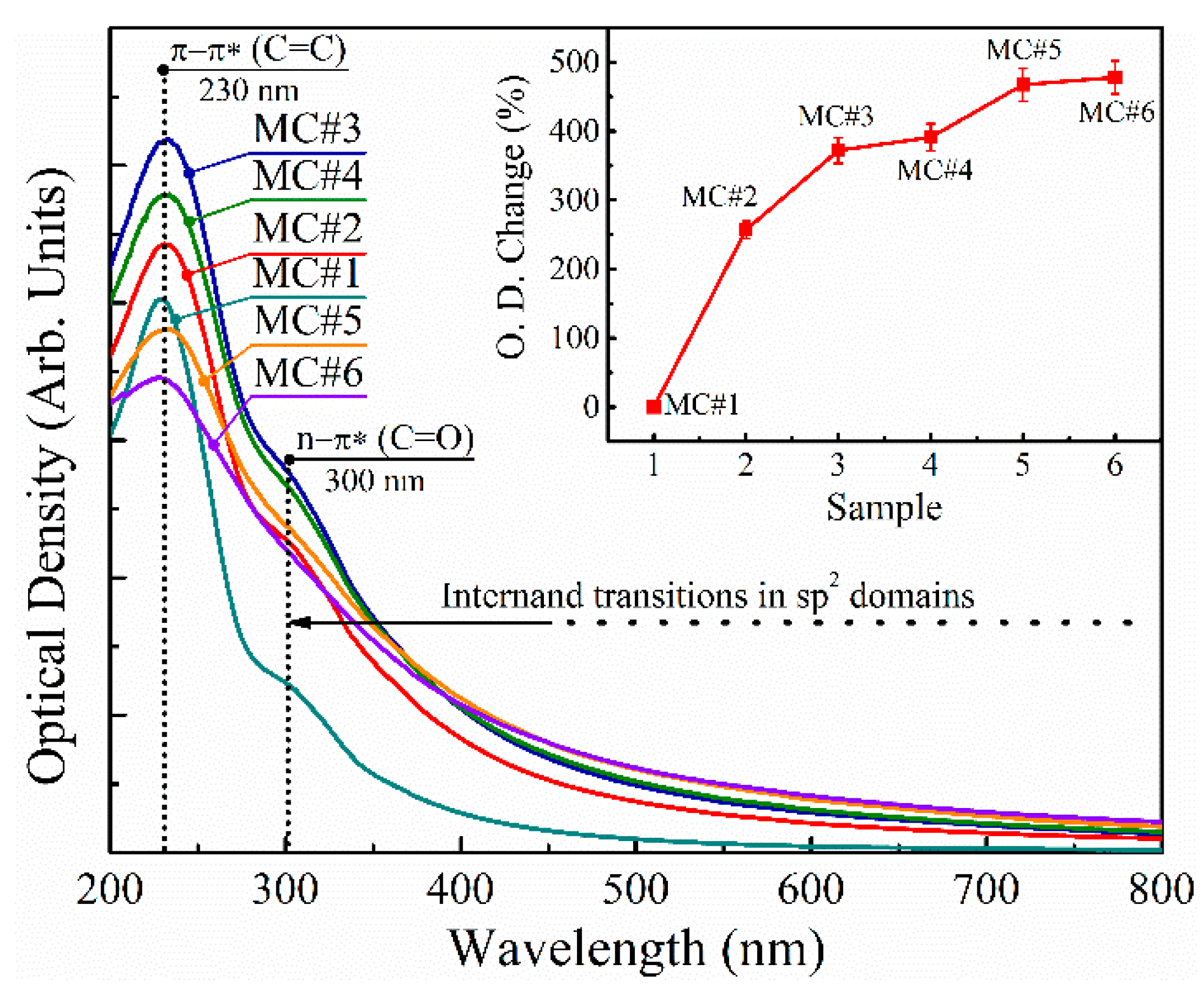
3.6. UV-Vis Spectroscopy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- McChesney, J.L.; Bostwick, A.; Ohta, T.; Seyller, T.; Horn, K.; González, J.; Rotenberg, E. Extended van Hove singularity and superconducting instability in doped graphene. Phys. Rev. Lett. 2010, 104, 136803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Skachko, I.; Duerr, F.; Luican, A.; Andrei, E.Y. Fractional quantum Hall effect and insulating phase of Dirac electrons in graphene. Nature 2009, 462, 192–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, C.; Song, Z.; Li, X.; Wu, X.; Brown, N.; Naud, C.; Mayou, D.; Li, T.; Hass, J.; Marchenkov, A.N.; et al. Electronic confinement and coherence in patterned epitaxial graphene. Science 2006, 312, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondavalli, P. Graphene, microelectronics/nanoelectronics, and more than moore. In Graphene and Related Nanomaterials; Elsevier: Cambridge, MA, USA, 2018; pp. 157–169. [Google Scholar]
- Sengupta, J.; Hussain, C.M. Graphene and its derivatives for analytical lab on Chip platforms. TrAC Trends Anal. Chem. 2019, 114, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Torrisi, F.; Carey, T. Graphene, related two-dimensional crystals and hybrid systems for printed and wearable electronics. Nano Today 2018, 23, 73–96. [Google Scholar] [CrossRef]
- Lin, S.; Lu, Y.; Xu, J.; Feng, S.; Li, J. High performance graphene/semiconductor van der Waals heterostructure optoelectronic devices. Nano Energy 2017, 40, 122–148. [Google Scholar] [CrossRef]
- Robinson, J.T.; Zalalutdinov, M.; Baldwin, J.W.; Snow, E.S.; Wei, Z.; Sheehan, P.; Houston, B.H. Wafer-scale reduced graphene oxide films for nanomechanical devices. Nano Lett. 2008, 8, 3441–3445. [Google Scholar] [CrossRef]
- Goyal, D.; Mittal, S.K.; Choudhary, A.; Dang, R.K. Graphene: A two dimensional super material for sensor applications. Mater. Today Proc. 2021, 43, 203–208. [Google Scholar] [CrossRef]
- Fowler, J.D.; Allen, M.J.; Tung, V.C.; Yang, Y.; Kaner, R.B.; Weiller, B.H. Practical chemical sensors from chemically derived graphene. ACS Nano 2009, 3, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedin, F.; Geim, A.K.; Morozov, S.V.; Hill, E.W.; Blake, P.; Katsnelson, M.I.; Novoselov, K.S. Detection of individual gas molecules adsorbed on graphene. Nat. Mater. 2007, 6, 652–655. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, L.; Wu, D.; Lv, W.; Wang, L. A high-sensitivity graphene ammonia sensor via aerosol jet printing. Sens. Actuators A Phys. 2021, 318, 112434. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, T.; Lee, J.; Choi, Y.S.; Moon, J.; Park, S.Y.; Lee, T.H.; Park, H.K.; A Lee, S.; Kwon, M.S.; et al. Tailored graphene micropatterns by wafer-scale direct transfer for flexible chemical sensor platform. Adv. Mater. 2021, 33, e2004827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Robinson, J.T.; Sun, X.; Dai, H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Zhang, Z.; Zhao, Y.; Qu, L. Graphene platforms for smart energy generation and storage. Joule 2018, 2, 245–268. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Wu, Z.-S.; Ren, W.; Cheng, H.-M.; Bao, X. Graphene: A promising 2D material for electrochemical energy storage. Sci. Bull. 2017, 62, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Lonkar, S.P.; Deshmukh, Y.S.; Abdala, A.A. Recent advances in chemical modifications of graphene. Nano Res. 2015, 8, 1039–1074. [Google Scholar] [CrossRef]
- Yu, W.; Sisi, L.; Haiyan, Y.; Jie, L. Progress in the functional modification of graphene/graphene oxide: A review. RSC Adv. 2020, 10, 15328–15345. [Google Scholar] [CrossRef]
- Rabchinskii, M.; Varezhnikov, A.; Sysoev, V.; Solomatin, M.; Ryzhkov, S.A.; Baidakova, M.; Stolyarova, D.Y.; Shnitov, V.; Pavlov, S.S.; Kirilenko, D.; et al. Hole-matrixed carbonylated graphene: Synthesis, properties, and highly-selective ammonia gas sensing. Carbon 2021, 172, 236–247. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Ryzhkov, S.A.; Kirilenko, D.A.; Ulin, N.V.; Baidakova, M.V.; Shnitov, V.V.; Pavlov, S.I.; Chumakov, R.G.; Stolyarova, D.Y.; Besedina, N.A.; et al. From graphene oxide towards aminated graphene: Facile synthesis, its structure and electronic properties. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, M.V.; Gorenberg, A.Y.; Shchegolikhin, A.N.; Shashkin, D.P.; Mel’Nikov, V.P. Explosive reduction of graphite oxide by hydrazine vapor at room temperature. Dokl. Phys. Chem. 2018, 478, 11–14. [Google Scholar] [CrossRef]
- Gudkov, M.V.; Bazhenov, S.L.; Bekhli, L.S.; Mel’Nikov, V.P. Explosive reduction of graphite oxide. Russ. J. Phys. Chem. B 2018, 12, 860–868. [Google Scholar] [CrossRef]
- Shin, H.; Kim, K.K.; Benayad, A.; Yoon, S.; Park, H.K.; Jung, I.; Jin, M.H.; Jeong, H.; Kim, J.M.; Choi, J.; et al. Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance. Adv. Funct. Mater. 2009, 19, 1987–1992. [Google Scholar] [CrossRef]
- Pei, S.; Zhao, J.; Du, J.; Ren, W.; Cheng, H.-M. Direct reduction of graphene oxide films into highly conductive and flexible graphene films by hydrohalic acids. Carbon 2010, 48, 4466–4474. [Google Scholar] [CrossRef]
- Moon, I.K.; Lee, J.; Ruoff, R.S.; Lee, H. Reduced graphene oxide by chemical graphitization. Nat. Commun. 2010, 1, 73. [Google Scholar] [CrossRef] [Green Version]
- Lee, R. XIII. On the atomic weight of graphite. Philos. Trans. R. Soc. Lond. 1859, 149, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Staudenmaier, L. Verfahren zur Darstellung der Graphitsäure. Berichte der Dtsch. Chem. Ges. 1898, 31, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Staudenmaier, L. Verfahren zur Darstellung der Graphitsäure. Berichte der Dtsch. Chem. Ges. 1899, 32, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.K.; Sofer, Z.; Pumera, M. Graphite oxides: Effects of permanganate and chlorate oxidants on the oxygen composition. Chem. A Eur. J. 2012, 18, 13453–13459. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Kovtyukhova, N.I.; Ollivier, P.J.; Martin, B.R.; Mallouk, T.E.; Chizhik, S.A.; Buzaneva, E.V.; Gorchinskiy, A.D. Layer-by-layer assembly of ultrathin composite films from micron-sized graphite oxide sheets and polycations. Chem. Mater. 1999, 11, 771–778. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Chandra, S.; Sahu, S.; Pramanik, P. A novel synthesis of graphene by dichromate oxidation. Mater. Sci. Eng. B 2010, 167, 133–136. [Google Scholar] [CrossRef]
- Rosillo-Lopez, M.; Salzmann, C.G. Detailed Investigation into the Preparation of Graphene Oxide by Dichromate Oxidation. ChemistrySelect 2018, 3, 6972–6978. [Google Scholar] [CrossRef]
- Díez-Pascual, A.M.; Sainz-Urruela, C.S.; Vallés, C.; Vera-López, S.; Andrés, M.P.S. Tailorable synthesis of highly oxidized graphene oxides via an environmentally-friendly electrochemical process. Nanomaterials 2020, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainz-Urruela, C.; Vera-López, S.; Andrés, M.P.S.; Díez-Pascual, A.M. Graphene oxides derivatives prepared by an electrochemical approach: Correlation between structure and properties. Nanomaterials 2020, 10, 2532. [Google Scholar] [CrossRef] [PubMed]
- Molodtsov, S.L.; Fedoseenko, S.I.; Vyalikh, D.V.; Iossifov, I.E.; Follath, R.; Gorovikov, S.A.; Brzhezinskaya, M.M.; Dedkov, Y.S.; Püttner, R.; Schmidt, J.-S.; et al. High-resolution Russian–German beamline at BESSY. Appl. Phys. A 2008, 94, 501–505. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy; Springer Series in Surface Sciences; Springer: Berlin/Heidelberg, Germany, 1992; Volume 25, ISBN 9783540544227. [Google Scholar]
- Rabchinskii, M.K.; Saveliev, S.D.; A Ryzhkov, S.; Nepomnyashchaya, E.K.; Pavlov, S.I.; Baidakova, M.V.; Brunkov, P.N. Establishing the applicability of the laser diffraction technique for the graphene oxide platelets lateral size measurements. J. Phys. Conf. Ser. 2020, 1695, 012070. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Pirkle, A.; Wallace, R.M.; Chhowalla, M.; Cho, K.; Chabal, Y. The Role of oxygen during thermal reduction of graphene oxide studied by infrared absorption spectroscopy. J. Phys. Chem. C 2011, 115, 19761–19781. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Cheng, Q.; Shi, G.; Jiang, L.; Qu, L. Self-Healing graphene oxide based functional architectures triggered by moisture. Adv. Funct. Mater. 2017, 27, 1703096. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Chhowalla, M.; Cho, K.; Chabal, Y.J. Unusual infrared-absorption mechanism in thermally reduced graphene oxide. Nat. Mater. 2010, 9, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Blume, R.; Kidambi, P.R.; Bayer, B.C.; Weatherup, R.S.; Wang, Z.-J.; Weinberg, G.; Willinger, M.-G.; Greiner, M.; Hofmann, S.; Knop-Gericke, A.; et al. The influence of intercalated oxygen on the properties of graphene on polycrystalline Cu under various environmental conditions. Phys. Chem. Chem. Phys. 2014, 16, 25989–26003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Peng, W.; Li, Y.; Li, X.; Wang, S.; Zhang, G.; Zhang, F. Deoxygenation of exfoliated graphite oxide under alkaline conditions: A green route to graphene preparation. Adv. Mater. 2008, 20, 4490–4493. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Voo, L.Y.; Ji, J.; Zhang, G.; Zhang, G.; Zhang, F.; Fan, X. Synthesis of partially hydrogenated graphene and brominated graphene. J. Mater. Chem. 2012, 22, 15021–15024. [Google Scholar] [CrossRef]
- Darmstadt, H.; Roy, C. Surface spectroscopic study of basic sites on carbon blacks. Carbon 2003, 41, 2662–2665. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; A Ryzhkov, S.; Gudkov, M.V.; Baidakova, M.V.; Saveliev, S.D.; Pavlov, S.I.; Shnitov, V.V.; A Kirilenko, D.; Stolyarova, D.Y.; Lebedev, A.M.; et al. Unveiling a facile approach for large-scale synthesis of N-doped graphene with tuned electrical properties. 2D Mater. 2020, 7, 045001. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Tour, J.M. Mechanism of graphene oxide formation. ACS Nano 2014, 8, 3060–3068. [Google Scholar] [CrossRef]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’Homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef]
- Sreeprasad, T.S.; Samal, A.K.; Pradeep, T. Tellurium nanowire-induced room temperature conversion of graphite oxide to leaf-like graphenic structures. J. Phys. Chem. C 2009, 113, 1727–1737. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Zhu, L.; Chen, H.; Qu, J.; Dai, L. Multiscale patterning of graphene oxide and reduced graphene oxide for flexible supercapacitors. Carbon 2015, 92, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Rabchinskii, M.K.; Dideikin, A.T.; Kirilenko, D.A.; Baidakova, M.V.; Shnitov, V.V.; Roth, F.; Konyakhin, S.V.; Besedina, N.A.; Pavlov, S.I.; Kuricyn, R.A.; et al. Facile reduction of graphene oxide suspensions and films using glass wafers. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Warren, B.E. X-ray diffraction in random layer lattices. Phys. Rev. 1941, 59, 693–698. [Google Scholar] [CrossRef]
- Ruland, W.; Smarsly, B. X-ray scattering of non-graphitic carbon: An improved method of evaluation. J. Appl. Crystallogr. 2002, 35, 624–633. [Google Scholar] [CrossRef]
- Kirilenko, D.A.; Dideykin, A.T.; Van Tendeloo, G. Measuring the corrugation amplitude of suspended and supported graphene. Phys. Rev. B Condens. Matter. Mater. Phys. 2011, 84, 235417. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Rios-Carvajal, T.; Ceccato, M.; Hassenkam, T. Nanoscale chemical mapping of oxygen functional groups on graphene oxide using atomic force microscopy-coupled infrared spectroscopy. J. Colloid Interface Sci. 2019, 556, 458–465. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.-J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Liu, Z.; Nørgaard, K.; Overgaard, M.H.; Ceccato, M.; Mackenzie, D.M.; Stenger, N.; Stipp, S.L.; Hassenkam, T. Direct observation of oxygen configuration on individual graphene oxide sheets. Carbon 2018, 127, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Eda, G.; Chhowalla, M. Chemically derived graphene oxide: Towards large-area thin-film electronics and optoelectronics. Adv. Mater. 2010, 22, 2392–2415. [Google Scholar] [CrossRef]
- Kumar, P.V.; Bardhan, N.M.; Tongay, S.; Wu, J.; Belcher, A.M.; Grossman, J.C. Scalable enhancement of graphene oxide properties by thermally driven phase transformation. Nat. Chem. 2014, 6, 151–158. [Google Scholar] [CrossRef]
- Chien, C.-T.; Li, S.-S.; Lai, W.-J.; Yeh, Y.-C.; Chen, H.-A.; Chen, I.-S.; Chen, L.-C.; Chen, K.-H.; Nemoto, T.; Isoda, S.; et al. Tunable photoluminescence from graphene oxide. Angew. Chem. Int. Ed. 2012, 51, 6662–6666. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.A.; Denisov, N.N. Photoreduction of graphite oxide. In Innovative Graphene Technologies: Developments, Characterization and Evaluation; Rapra-Smithers Press: Shawbury, UK, 2013; pp. 43–54. ISBN 1847359663. [Google Scholar]
- Robertson, J.; O’Reilly, E.P. Electronic and atomic structure of amorphous carbon. Phys. Rev. B 1987, 35, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Su, H. Exciton characteristics in graphene epoxide. ACS Nano 2014, 8, 1284–1289. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | K2Cr2O7, g | KMnO4, g | K2Cr2O7:KMnO4 |
|---|---|---|---|
| MC#1 | 0 | 3 | 0:100 |
| MC#2 | 1.5 | 2.4 | 20:80 |
| MC#3 | 3 | 1.8 | 40:60 |
| MC#4 | 4.5 | 1.2 | 60:40 |
| MC#5 | 6 | 0.6 | 80:20 |
| MC#6 | 7.5 | 0 | 100:0 |
| Component | C-V | C=C | C-C | C-OH & C-O-C | >C=O | COOH | C/O Ratio |
|---|---|---|---|---|---|---|---|
| Binding Energy (eV) | 283.7 | 284.6 | 285.1 | 286.8 | 288.2 | 289.0 | |
| MC#1 | 5.83 | 47.9 | 2.11 | 40.69 | 3.47 | <0.10 | 2.26 |
| MC#2 | 2.55 | 49.94 | 4.59 | 35.30 | 6.98 | 0.64 | 2.29 |
| MC#3 | 2.58 | 56.26 | 4.73 | 29.41 | 5.18 | 1.84 | 2.61 |
| MC#4 | 3.02 | 57.84 | 4.93 | 27.77 | 4.74 | 1.70 | 2.81 |
| MC#5 | 2.83 | 58.09 | 8.09 | 22.38 | 5.29 | 3.32 | 2.92 |
| MC#6 | 5.99 | 52.44 | 8.34 | 21.87 | 6.42 | 4.94 | 2.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiyanova, K.A.; Gudkov, M.V.; Rabchinskii, M.K.; Sokura, L.A.; Stolyarova, D.Y.; Baidakova, M.V.; Shashkin, D.P.; Trofimuk, A.D.; Smirnov, D.A.; Komarov, I.A.; et al. Graphene Oxide Chemistry Management via the Use of KMnO4/K2Cr2O7 Oxidizing Agents. Nanomaterials 2021, 11, 915. https://doi.org/10.3390/nano11040915
Shiyanova KA, Gudkov MV, Rabchinskii MK, Sokura LA, Stolyarova DY, Baidakova MV, Shashkin DP, Trofimuk AD, Smirnov DA, Komarov IA, et al. Graphene Oxide Chemistry Management via the Use of KMnO4/K2Cr2O7 Oxidizing Agents. Nanomaterials. 2021; 11(4):915. https://doi.org/10.3390/nano11040915
Chicago/Turabian StyleShiyanova, Kseniya A., Maksim V. Gudkov, Maxim K. Rabchinskii, Liliia A. Sokura, Dina Y. Stolyarova, Marina V. Baidakova, Dmitriy P. Shashkin, Andrei D. Trofimuk, Dmitry A. Smirnov, Ivan A. Komarov, and et al. 2021. "Graphene Oxide Chemistry Management via the Use of KMnO4/K2Cr2O7 Oxidizing Agents" Nanomaterials 11, no. 4: 915. https://doi.org/10.3390/nano11040915