
Quantum Chemistry Study on the Structures and Electronic Properties of Bimetallic Ca2-Doped Magnesium Ca2Mgn (n = 1–15) Clusters

 ,
,
Abstract
:1. Introduction
2. Computational Detail
3. Results and Discussions
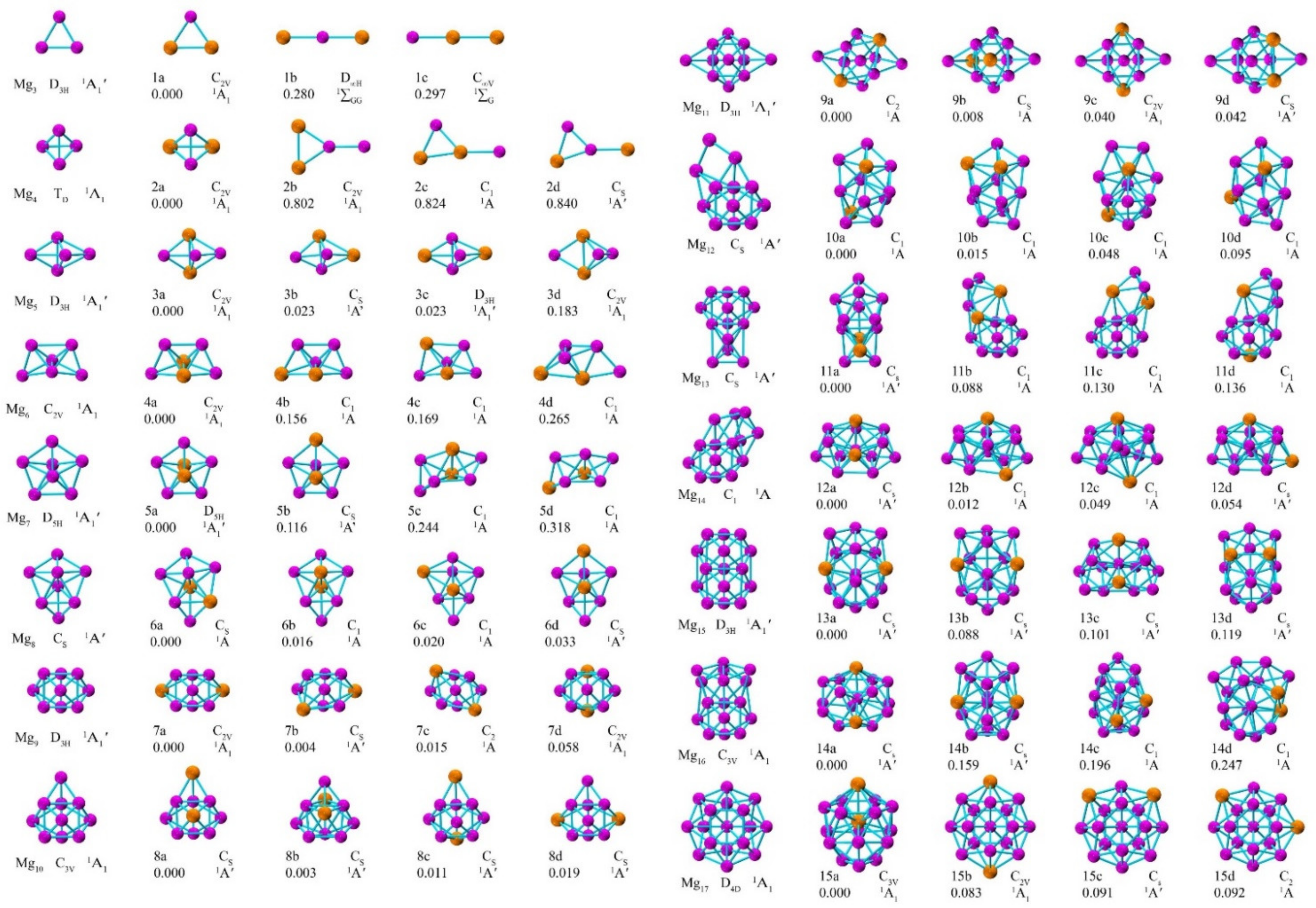
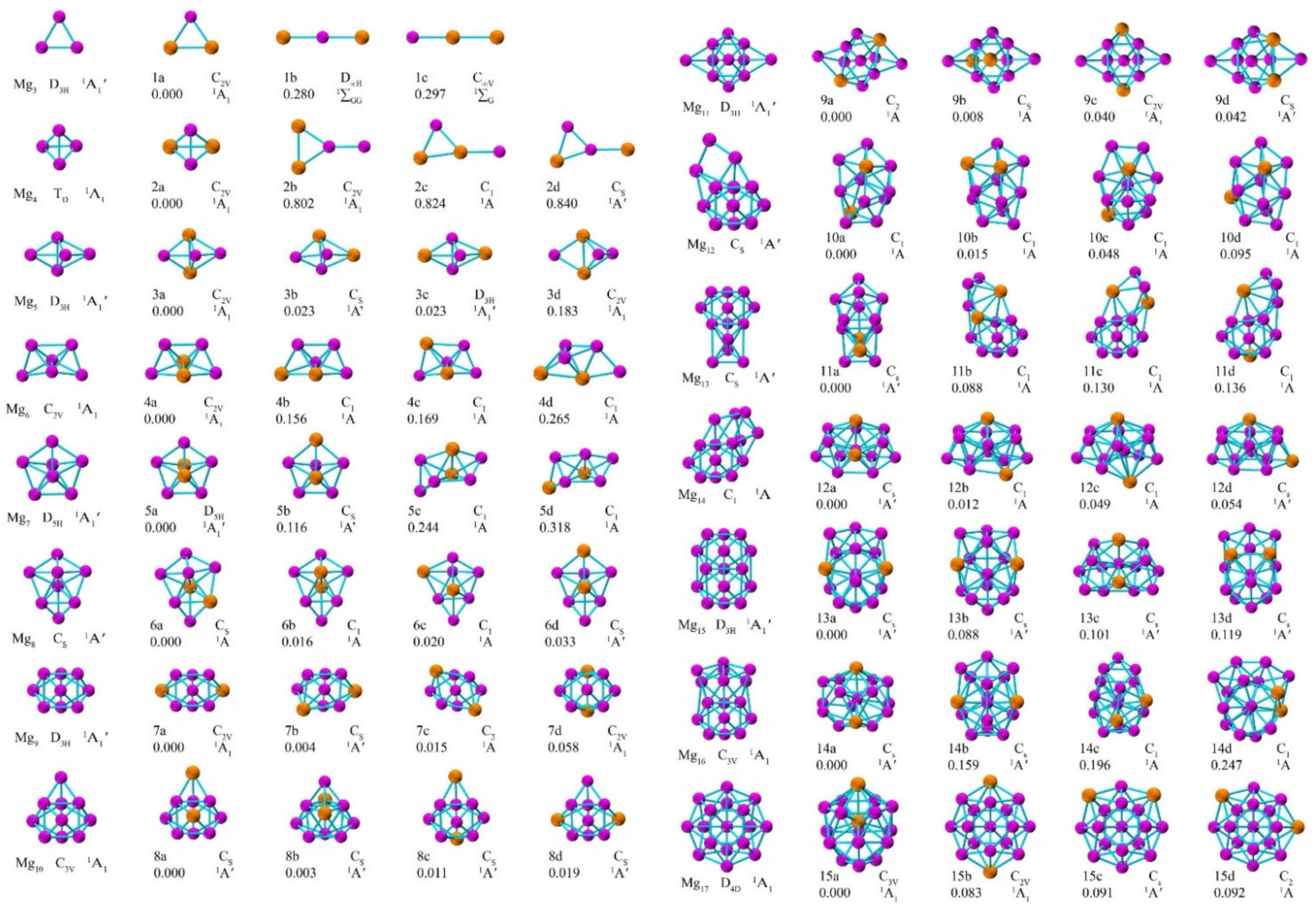
3.1. Geometric Structures
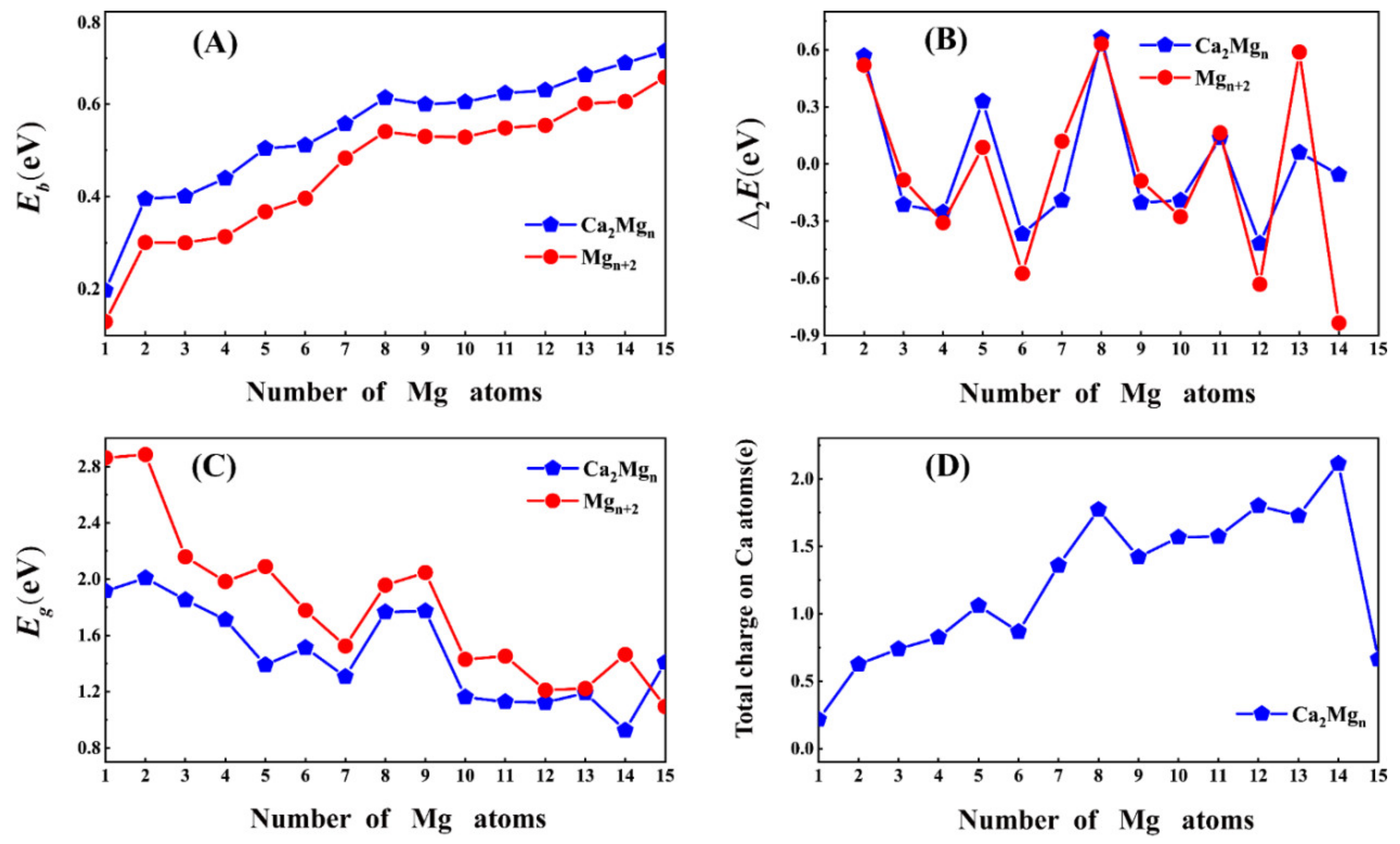
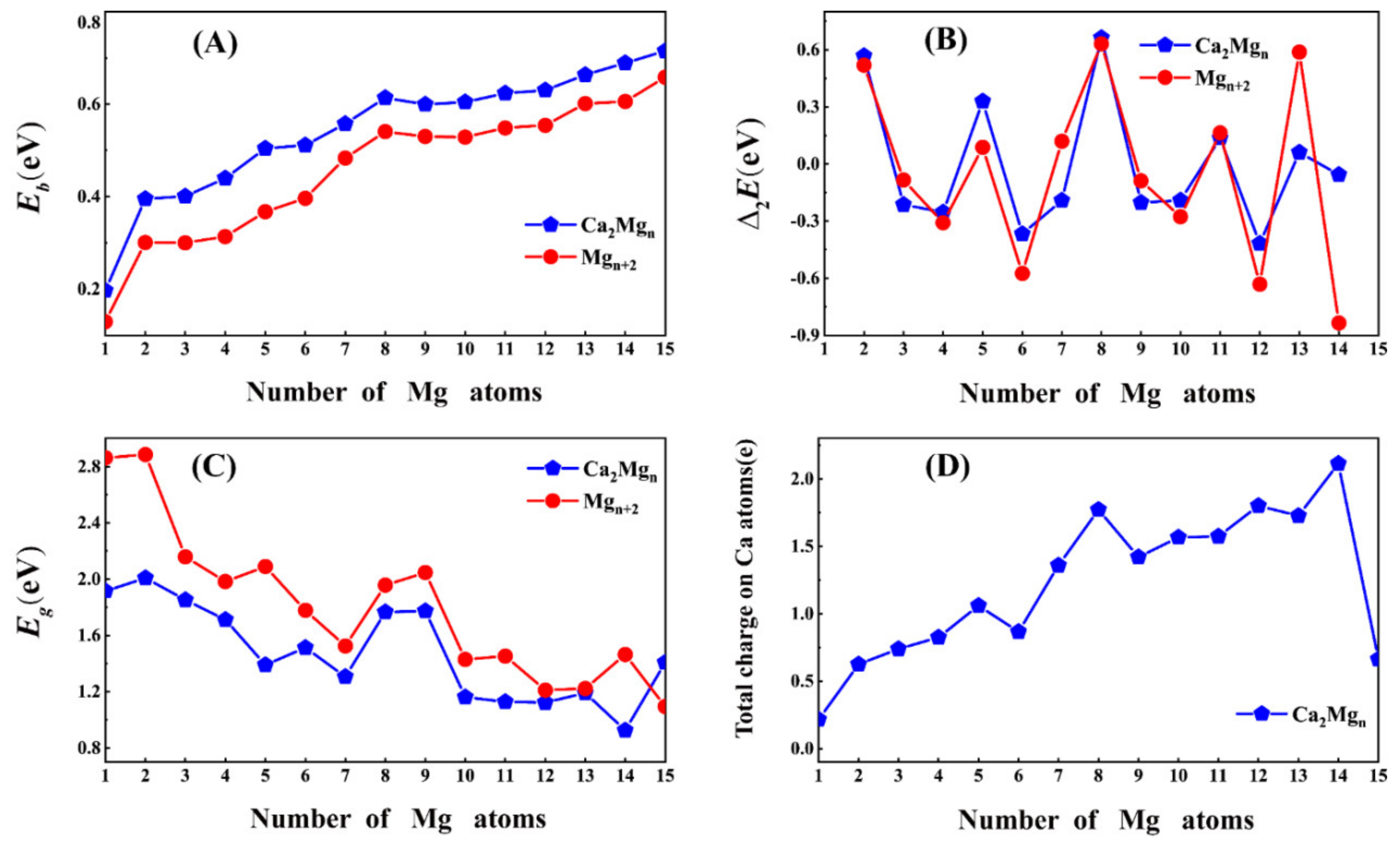
3.2. Relative Stability
3.3. Charge Transfer
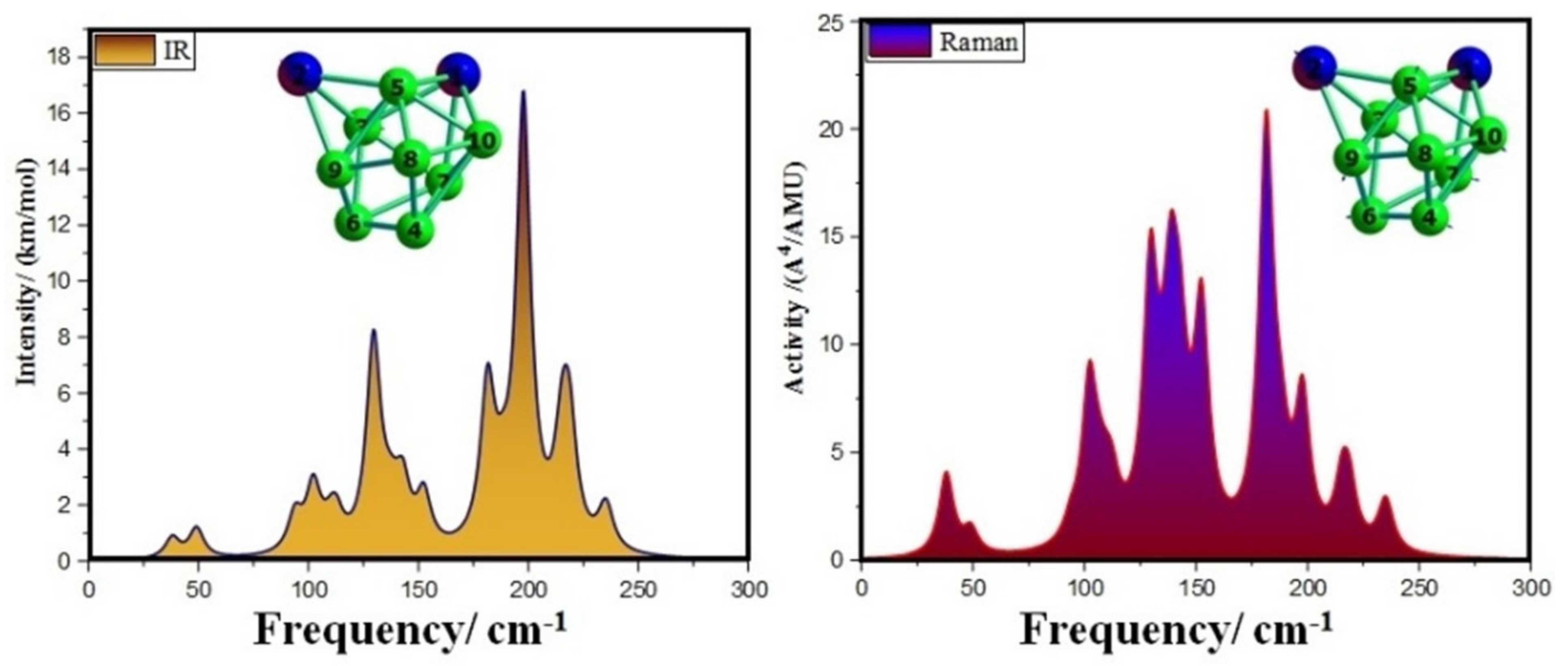
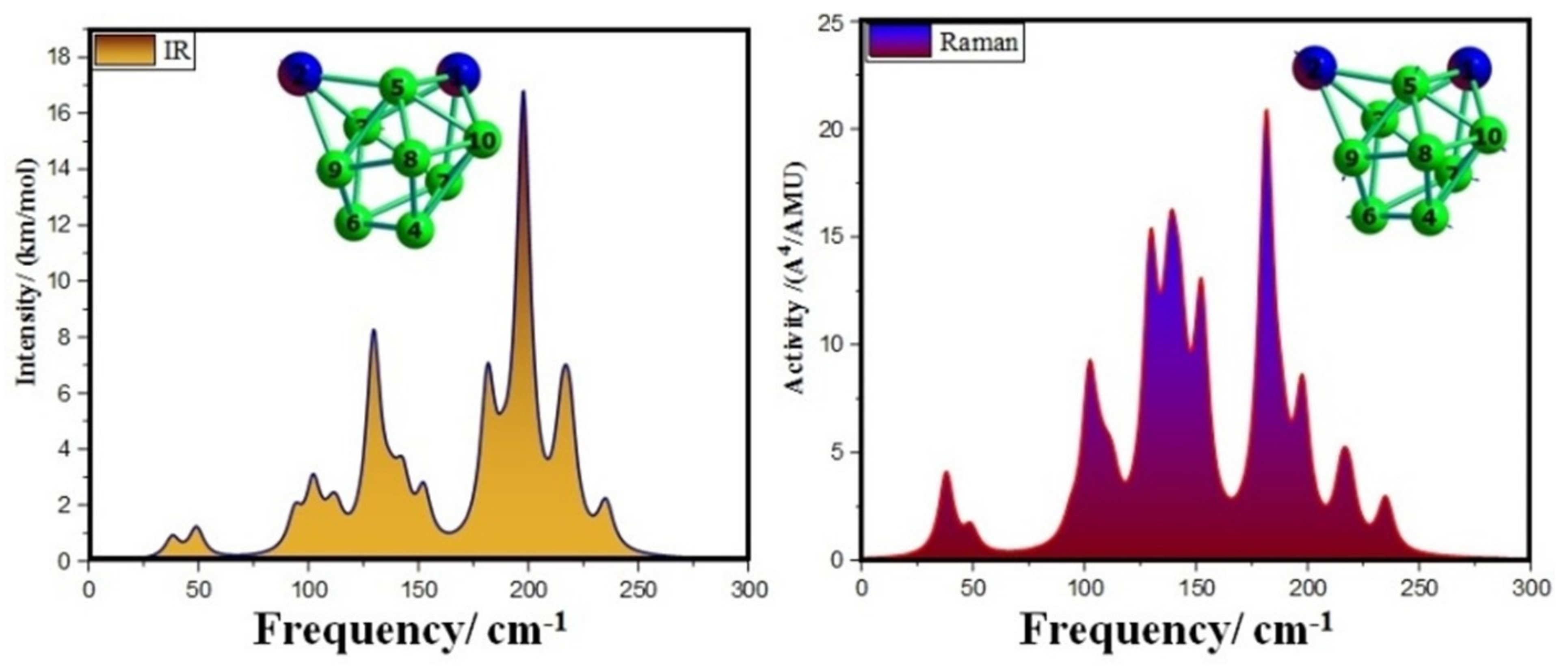
3.4. Infrared (IR) and Raman Spectra
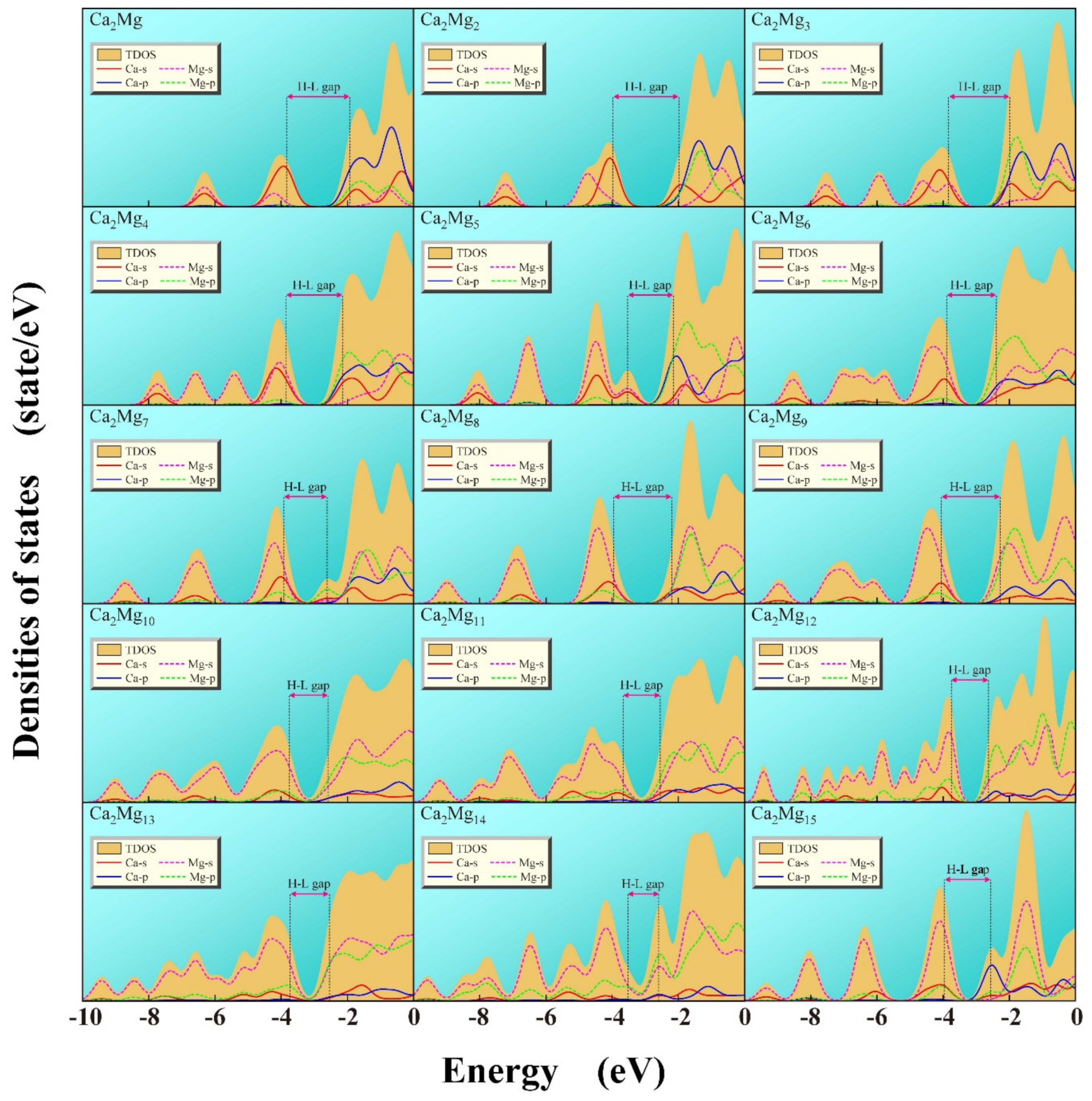
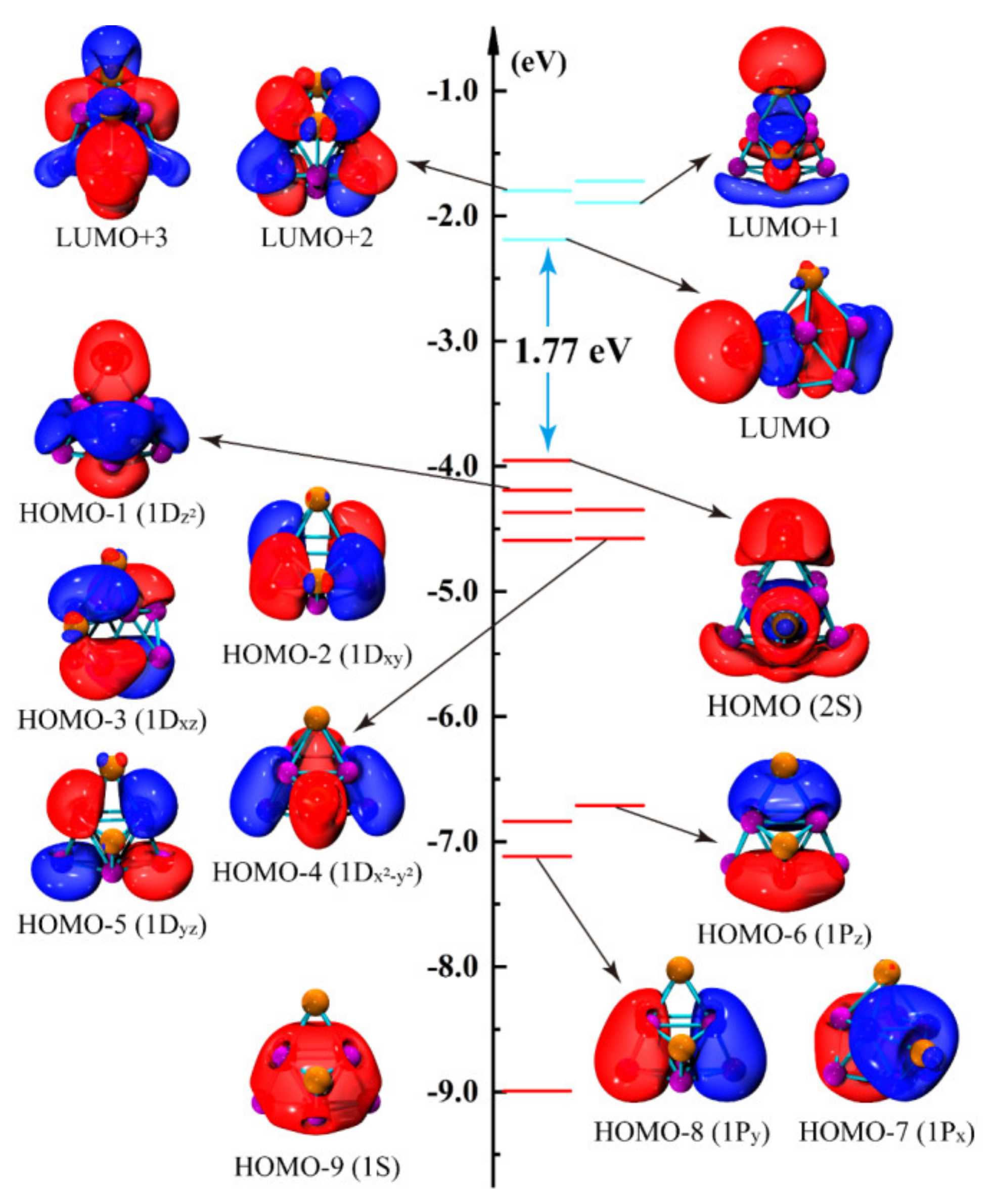
3.5. The Density of States
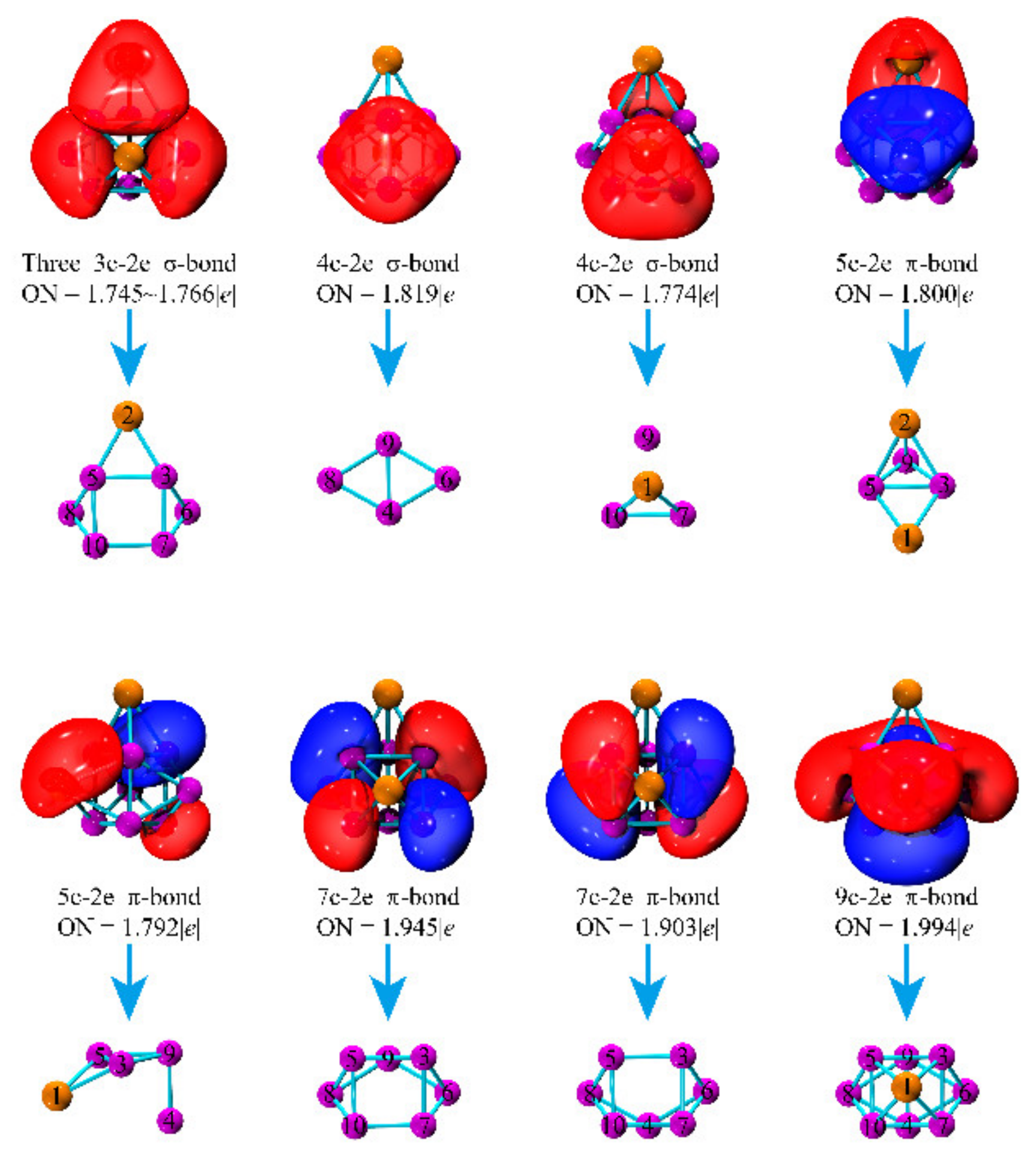
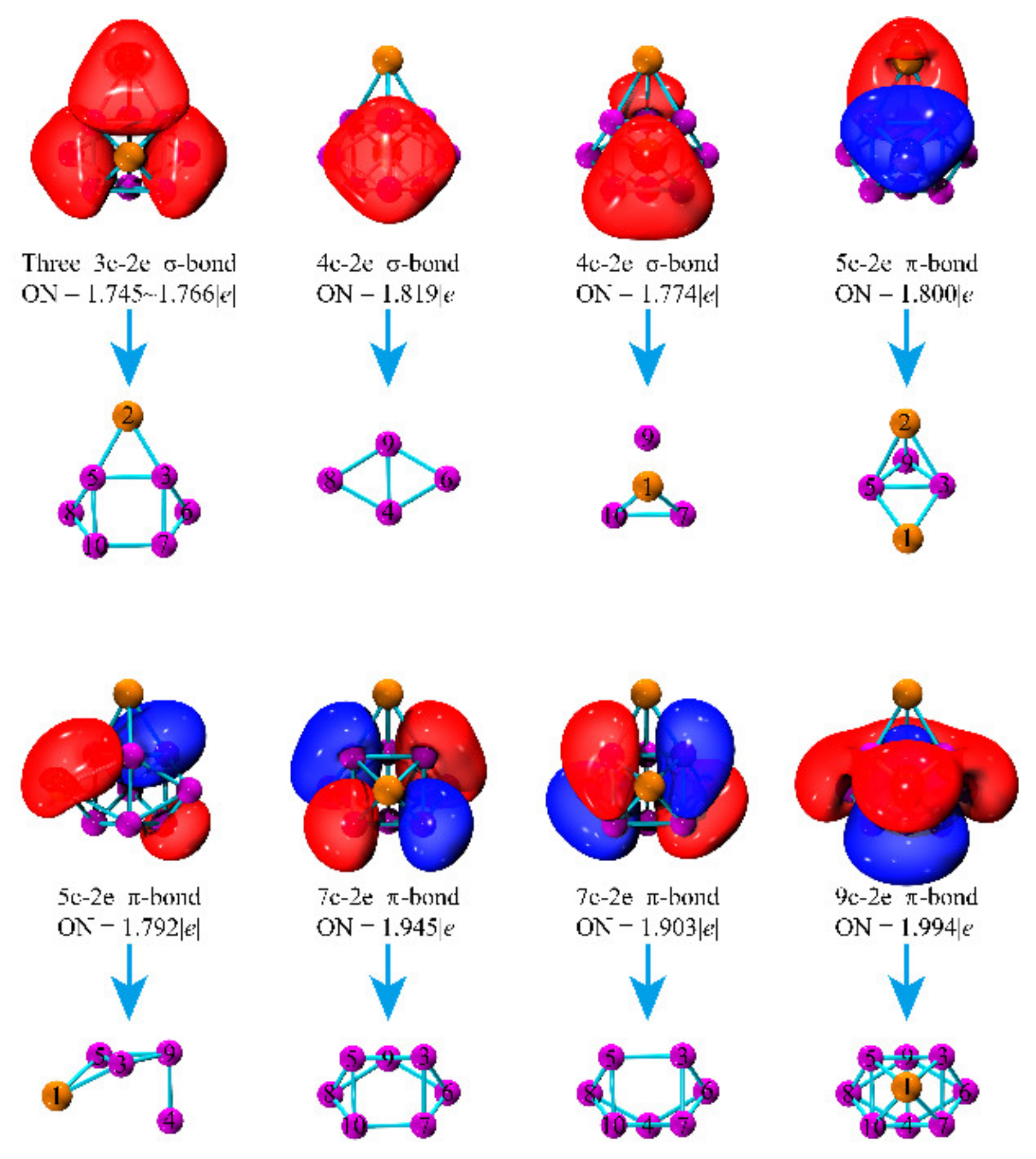
3.6. Bonding Characters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nanan, M.E.; Bierwagen, G.P. Mg-Rich coatings: A new paradigm for Cr-free corrosion protection of Al arrospece alloy. JCT Res. 2014, 1, 69–80. [Google Scholar]
- Yoo, J.; Aksimentiev, A. Improved parametrization of Li+, Na+, K+ and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar] [CrossRef]
- Pollock, T.M. Weight loss with magnesium alloys. Science 2010, 328, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; You, B.S.; Yim, C.D. Texture and microstructure changes in asymmetrically hot rolled Az31 magnesium alloy sheets. Mater. Lett. 2005, 59, 3876–3880. [Google Scholar] [CrossRef]
- Diederich, T.; Doppner, T.; Braune, J.; Tiggesbaumker, J.; Meiwes-Broer, K.H. Electron delocalization in magnesium clusters grown in superclod helium drolpets. Phys. Rev. Lett. 2001, 86, 4807–4810. [Google Scholar] [CrossRef]
- Thomas, O.C.; Zheng, W.J.; Xu, S.J.; Bowen, K.H. Onset of metallic behavior in magnesium clusters. Phys. Rev. Lett. 2002, 89, 213403. [Google Scholar] [CrossRef]
- Li, P.; Xie, W.; Tang, K.T. The van der Waals potential of the magnesium dimer. J. Chem. Phys. 2010, 133, 084308. [Google Scholar] [CrossRef]
- Janecek, S.; Krotscheck, E.; Liebrecht, M.; Wahl, R. Structure of Mgn and Mgn+ clusters up to n = 30. Eur. Phys. J. D-At. Mol. Opt. Plasma Phys. 2011, 63, 377–390. [Google Scholar] [CrossRef]
- Kaplan, I.G.; Diaz, C.C. Comparative study of the electron affinities of beryllium and magnesium dimmers and trimers. Int. J. Quantum Chem. 2005, 104, 468–474. [Google Scholar] [CrossRef]
- Gong, X.G.; Zheng, Q.Q.; He, Y.Z. Electronic structures of magnesium clusters. Phys. Lett. A 1993, 181, 459–464. [Google Scholar] [CrossRef]
- Akola, J.; Rytkonen, K.; Manninen, M. Metallic evolution of small magnesium clusters. Eur. Phys. J. D 2001, 16, 21–24. [Google Scholar] [CrossRef]
- Jellinek, J.; Acioli, P.H. Magnesium Clusters: Structural and Electronic Properties and the Size-Induced Nonmetal-to-Metal Transition. J. Phys. Chem. A 2002, 106, 10919–10925. [Google Scholar] [CrossRef]
- Acioli, P.H.; Jellinek, J. Electron binding energies of anionic magnesium clusters and the nonmetal-to-metal transition. Phys. Rev. Lett. 2002, 89, 213402. [Google Scholar] [CrossRef]
- Lyalin, A.; Solov’yov, I.A.; Solov’yov, A.V.; Greiner, W. Evolution of the electronic and ionic structure of Mg clusters with increase in cluster size. Phys. Rev. A 2003, 67, 063203. [Google Scholar] [CrossRef] [Green Version]
- Kohn, A.; Weigend, F.; Ahlrichs, R. Theoretical study on clusters of magnesium. Phys. Chem. Chem. Phys. 2001, 23, 711–719. [Google Scholar] [CrossRef]
- Herdari, I.; Ghazi, S.M.; Goedecker, S.; Kanhere, D.G. Growth and structural properties of Mgn (n = 10–56) clusters: Density functional theory study. J. Phys. Chem. A 2011, 115, 12307–12314. [Google Scholar] [CrossRef]
- Xia, X.X.; Kuang, X.Y.; Lu, C.; Jin, Y.Y.; Xing, X.D.; Merino, G. Deciphering the structural evolution and electronic properties of magnesium clusters: An aromatic homonuclear metal. J. Phys. Chem. A 2016, 120, 7947–7954. [Google Scholar] [CrossRef] [Green Version]
- Duanmu, K.; Roberto-Neto, O.; Machado, F.B.C.; Hansen, J.A.; Shen, J.; Piecuch, P.; Truhlar, D.G. Geometries, binding energies, ionization potentials, and electron affinities of metal clusters: Mgn 0,±1, n = 1–7. J. Phys. Chem. C 2016, 120, 13275. [Google Scholar] [CrossRef]
- Duanmu, K.; Friedrich, J.; Truhlar, D.G. Thermodynamics of metal nonparties: Energies and enthalpies of formation of magnesium clusters and nanoparticles as large as 1.3 nm. J. Phys. Chem. C 2016, 120, 26110. [Google Scholar] [CrossRef]
- Kong, F.J.; Hu, Y.F. Density functional theory study of small X-doped Mgn (X = Fe, Co, and Ni, n = 1–9) bimetallic clusters: Equilibrium structures, stabilities, electronic and magnetic properties. J. Mol. Model. 2014, 20, 2087. [Google Scholar] [CrossRef]
- Ge, G.X.; Han, Y.; Wan, J.G.; Zhao, J.J.; Wang, G.H. First-principles prediction of magnetic superatoms in 4d-transition-metal-doped magnesium clusters. J. Chem. Phys. 2013, 139, 174309. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, Z.; Zhou, Z.H.; Wang, H.B.; Li, S.L. First-principles calculations on small MgnZn and Mgn−1Zn2 clusters: Structures, stabilities, and electronic properties. Mater. Chem. Phys. 2017, 199, 585–590. [Google Scholar] [CrossRef]
- Xing, X.D.; Wang, J.J.; Kuang, X.Y.; Xia, X.X.; Lu, C.; Maroulis, G. Probing the low-energy structures of aluminum-magnesium alloy clusters: A detailed study. Phys. Chem. Chem. Phys. 2016, 18, 26177–26183. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Bandyopadhyay, D. Study of absorption and dissociation pathway of H2 molecule on MgnRh (n = 1–10) clusters: A first principle investigation. Int. J. Hydrogen Energy 2016, 41, 20113–20121. [Google Scholar] [CrossRef]
- Afshar, M.; Chegeni, M.H. Spin and orbital magnesium in XMg8 (X = Sc-Ni): A relativistic density functional theory study. Mol. Phys. 2016, 114, 3620–3628. [Google Scholar] [CrossRef]
- Medel, V.M.; Ulises, J.; Khanna, S.N.; Chauhan, V.; Sen, P.; Castleman, A.W. Hund’s rule in superatoms with transition metal impurities. Proc. Natl. Acad. Sci. USA 2011, 108, 10062–10066. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.C.; Zhang, S.; Zeng, L. The effect of silicon doping on the geometrical structures, stability, and electronic and spectral properties of magnesium clusters: DFT study of SiMgn (n = 1–12) clusters. Int. J. Quantum Chem. 2020, 120, e26143. [Google Scholar] [CrossRef]
- Medel, V.M.; Reber, R.C.; Reveles, J.U.; Khanna, S.N. Metallic and molecular orbital concepts in XMg8 clusters, X = Be-F. J. Chem. Phys. 2012, 136, 134311. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Liang, M.K.; Wei, X.F.; Guo, J.; Zhu, B.C. New potential stable structures of XMgn (X = Ge, C, Sn; n = 1–12) clusters: XMg8 with high stability. J. Phys. Condens. Matter 2021, 33, 065302. [Google Scholar] [CrossRef]
- Trivedi, R.; Bandyopadhyay, D. Hydrogen storage in small size MgnCo clusters: A density functional study. Int. J. Hydrogen Energy 2015, 40, 12727–12735. [Google Scholar] [CrossRef]
- Lu, C.; Ma, Y.L.; Li, F.; Zhu, H.; Zeng, X.Q.; Ding, W.J.; Deng, T.; Wu, J.B.; Zhou, J.X. Visualization of fast “hydrogen pump” in core–shell nanostructure Mg@Pt through hydrogen-stabilized Mg3Pt. J. Mater. Chem. A 2019, 7, 14629–14637. [Google Scholar] [CrossRef]
- Zeng, L.; Wei, X.F.; Liang, M.K.; Zhao, J.; Zhu, B.C. Probing on the Stable Structure of Silicon-Doped Charged Magnesium Nanomaterial Sensor: SiMgn±1 (n = 2−12) Clusters DFT Study. Front. Mater. 2020, 7, 221. [Google Scholar] [CrossRef]
- Zeng, L.; Liang, M.K.; Wei, X.F.; Guo, J.; Zhang, S.; Bi, J.; Dai, W.; Zhu, B.C. Probing the structural evolution, electronic and spectral properties of beryllium doped magnesium and their ions clusters. New J. Chem. 2020, 44, 16929–16940. [Google Scholar] [CrossRef]
- Zhao, Y.R.; Xu, Y.Q.; Chen, P.; Yuan, Y.Q.; Qian, Y.; Li, Q. Structural and electronic properties of medium-sized beryllium doped magnesium BeMgn clusters and their anions. Results Phys. 2021, 26, 104341. [Google Scholar] [CrossRef]
- Zhang, F.G.; Zhang, H.R.; Xin, W.; Chen, P.; Hu, Y.F.; Zhang, X.Y.; Zhao, Y.R. Probing the structural evolution and electronic properties of divalent metal Be2Mgn clusters from small to medium-size. Sci. Rep. 2020, 10, 6052. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Y.; Xi, S.G.; Hu, Y.F.; Yuan, Y.Q.; Zhao, Y.R.; Li, M.C.; Yuan, J.J.; Yang, Y.J. Probing the structural and electronic properties of neutral and anionic strontium-doped magnesium clusters. Comput. Mater. Sci. 2021, 197, 110605. [Google Scholar] [CrossRef]
- Li, Q.Y.; Hu, Y.F.; Xi, S.G.; Li, Y.Y.; Yang, H.; Yuan, Y.Q.; Yang, J.; Li, M.C. Structural and electronic properties of neutral and anionic magnesium clusters doped with two barium atoms. J. Mol. Liq. 2021, 343, 117622. [Google Scholar] [CrossRef]
- Li, C.G.; Cui, Y.Q.; Tian, H.; Shao, Q.Q.; Zhang, J.; Ren, B.Z.; Yuan, Y.Q. Systematic investigation of geometric structures and electronic properties of lithium doped magnesium clusters. Comput. Mater. Sci. 2021, 200, 110800. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. Crystal structure prediction via particle swarm optimization. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 094116. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Wang, Y.C.; Zhu, L.; Ma, Y.M. Particle swarm structure prediction on clusters. J. Chem. Phys. 2012, 137, 084104. [Google Scholar] [CrossRef]
- Chen, B.L.; Sun, W.G.; Kuang, X.Y.; Lu, C.; Xia, X.X.; Shi, H.X.; Maroulis, G. Structural Stability and Evolution of Medium-Sized Tantalum-Doped Boron Clusters: A Half-Sandwich-Structured TaB12− Cluster. Inorg. Chem. 2018, 57, 343–350. [Google Scholar] [CrossRef]
- Li, C.G.; Li, H.J.; Cui, Y.Q.; Tian, H.; Shao, Q.Q.; Zhang, J.; Zhao, G.; Ren, B.Z.; Hu, Y.F. A density functional investigation on the structures, electronic, spectral and fluxional properties of VB20− cluster. J. Mol. Liq. 2021, 339, 116764. [Google Scholar] [CrossRef]
- Zhao, Y.R.; Bai, T.T.; Jia, L.N.; Xin, W.; Hu, Y.H.; Zheng, X.S. Probing the Structural and Electronic Properties of Neutral and Anionic Lanthanum-Doped Silicon Clusters. J. Phys. Chem. C 2019, 123, 28561–28568. [Google Scholar] [CrossRef]
- Lai, L.; Die, D.; Zheng, B.X.; Du, Q. Growth mechanism and electronic and magnetic properties of AgnTi alloy clusters. J. Phys. Chem. Solids 2021, 148, 109757. [Google Scholar] [CrossRef]
- Jin, S.Y.; Chen, B.L.; Kuang, X.Y.; Lu, C.; Sun, W.G.; Xia, X.X.; Gutsev, G.L. Structural and Electronic Properties of Medium-Sized Aluminum-Doped Boron Clusters AlBn and Their Anions. J. Phys. Chem. C 2019, 123, 6276–6283. [Google Scholar] [CrossRef]
- Li, C.G.; Shen, Z.G.; Zhang, J.; Cui, Y.Q.; Li, J.J.; Xue, H.Y.; Li, H.F.; Ren, B.Z.; Hu, Y.F. Analysis of the structures, stabilities and electronic properties of MB16– (M = V, Cr, Mn, Fe, Co, Ni) clusters and assemblies. New J. Chem. 2020, 44, 5109–5119. [Google Scholar] [CrossRef]
- Lu, C.; Gong, W.G.; Li, Q.; Chen, C.F. Elucidating stress-strain of ZrB12 from first-priciples studies. J. Phys. Chem. Lett. 2020, 11, 9165–9170. [Google Scholar] [CrossRef] [PubMed]
- Die, D.; Zheng, B.X.; Yue, J.Y.; Guo, J.J.; Du, Q. The ground-state structure, optical-absorption and photoelectron spectrum of silver clusters. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 117, 113805. [Google Scholar] [CrossRef]
- Li, C.G.; Gao, J.H.; Zhang, J.; Song, W.T.; Liu, S.Q.; Gao, S.Z.; Ren, B.Z.; Hu, Y.F. Structures, stabilities and electronic properties of boron-doped silicon clusters B3Sin (n = 1–17) and their anions. Mol. Phys. 2019, 117, 382. [Google Scholar] [CrossRef]
- Krishnan, R. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401–146405. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Fox, Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. Comput. Phys. Commun. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ruette, F.; Sanchez, M.; Anez, R.; Bermudez, A.; Sierraalta, A. Diatomic molecule data for parametric methods. I. J. Mol. Struct. THEOCHEM 2005, 729, 19–37. [Google Scholar] [CrossRef]
- Soltani, A.; Boudjahem, A.G.; Bettahar, M. Electronic and magnetic properties of small RhnCa (n = 1–9) clusters: A DFT Study. Int. J. Quantum Chem. 2016, 116, 346–356. [Google Scholar] [CrossRef]
- Zhou, G.D.; Duan, L.Y. Free Movement in the EU: The Case of Great Britain Structural Chemistry Basis; Peking University Press: Beijing, China, 2002; pp. 1–10. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clusters | State | Sym. | Eb (eV) | Δ2E (eV) | Eg (eV) | Charge (e) | |
|---|---|---|---|---|---|---|---|
| Ca1 | Ca2 | ||||||
| Ca2Mg | 1A1 | C2V | 0.20 | - | 1.91 | 0.11 | 0.11 |
| Ca2Mg2 | 1A1 | C2V | 0.40 | 0.57 | 2.01 | 0.31 | 0.31 |
| Ca2Mg3 | 1A1 | C2V | 0.40 | −0.21 | 1.85 | 0.37 | 0.37 |
| Ca2Mg4 | 1A1 | C2V | 0.44 | −0.25 | 1.71 | 0.41 | 0.41 |
| Ca2Mg5 | 1A1’ | D5H | 0.50 | 0.33 | 1.39 | 0.53 | 0.53 |
| Ca2Mg6 | 1A | D5H | 0.51 | −0.37 | 1.51 | 0.37 | 0.37 |
| Ca2Mg7 | 1A1 | C2V | 0.56 | −0.19 | 1.31 | 0.68 | 0.68 |
| Ca2Mg8 | 1A’ | CS | 0.61 | 0.66 | 1.77 | 0.84 | 0.94 |
| Ca2Mg9 | 1A | C2 | 0.60 | −0.20 | 1.77 | 0.71 | 0.71 |
| Ca2Mg10 | 1A | C1 | 0.60 | −0.19 | 1.16 | 0.86 | 0.71 |
| Ca2Mg11 | 1A | CS | 0.62 | 0.14 | 1.13 | 0.87 | 0.71 |
| Ca2Mg12 | 1A’ | CS | 0.63 | −0.42 | 1.12 | 0.85 | 0.85 |
| Ca2Mg13 | 1A’ | CS | 0.66 | 0.06 | 1.19 | 0.86 | 0.86 |
| Ca2Mg14 | 1A’ | CS | 0.69 | −0.06 | 0.93 | 1.06 | 1.06 |
| Ca2Mg15 | 1A1 | C3V | 0.71 | - | 1.41 | −0.18 | 0.85 |
| Clusters | HOMO (eV) | LUMO (eV) | Clusters | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|---|
| Ca2Mg | −3.834 | −1.920 | Mg3 | −4.799 | −1.937 |
| Ca2Mg2 | −3.981 | −1.973 | Mg4 | −4.961 | −2.076 |
| Ca2Mg3 | −3.831 | −1.979 | Mg5 | −4.258 | −2.099 |
| Ca2Mg4 | −3.860 | −2.148 | Mg6 | −4.294 | −2.311 |
| Ca2Mg5 | −3.531 | −2.140 | Mg7 | −4.436 | −2.347 |
| Ca2Mg6 | −3.897 | −2.384 | Mg8 | −4.284 | −2.507 |
| Ca2Mg7 | −3.917 | −2.611 | Mg9 | −4.393 | −2.868 |
| Ca2Mg8 | −3.954 | −2.188 | Mg10 | −4.306 | −2.350 |
| Ca2Mg9 | −4.048 | −2.275 | Mg11 | −4.385 | −2.338 |
| Ca2Mg10 | −3.750 | −2.588 | Mg12 | −4.012 | −2.583 |
| Ca2Mg11 | −3.671 | −2.543 | Mg13 | −4.105 | −2.652 |
| Ca2Mg12 | −3.746 | −2.623 | Mg14 | −4.092 | −2.881 |
| Ca2Mg13 | −3.730 | −2.539 | Mg15 | −3.973 | −2.750 |
| Ca2Mg14 | −3.518 | −2.592 | Mg16 | −3.985 | −2.521 |
| Ca2Mg15 | −3.965 | −2.558 | Mg17 | −3.917 | −2.823 |
| Atoms | Ca-1 | Ca-2 | Mg-3 | Mg-4 | Mg-5 | Mg-6 | Mg-7 | Mg-8 | Mg-9 |
|---|---|---|---|---|---|---|---|---|---|
| Ca-2 | 0.130 | ||||||||
| Mg-3 | 0.383 | 0.462 | |||||||
| Mg-4 | 0.151 | 0.143 | 0.233 | ||||||
| Mg-5 | 0.383 | 0.462 | 0.524 | 0.233 | |||||
| Mg-6 | 0.139 | 0.131 | 0.517 | 0.529 | 0.228 | ||||
| Mg-7 | 0.398 | 0.149 | 0.528 | 0.439 | 0.237 | 0.523 | |||
| Mg-8 | 0.139 | 0.131 | 0.228 | 0.529 | 0.517 | 0.182 | 0.199 | ||
| Mg-9 | 0.179 | 0.410 | 0.465 | 0.458 | 0.465 | 0.487 | 0.231 | 0.487 | |
| Mg-10 | 0.398 | 0.149 | 0.237 | 0.439 | 0.528 | 0.199 | 0.523 | 0.523 | 0.231 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Cui, Y.; Tian, H.; Ren, B.; Li, Q.; Li, Y.; Yang, H. Quantum Chemistry Study on the Structures and Electronic Properties of Bimetallic Ca2-Doped Magnesium Ca2Mgn (n = 1–15) Clusters. Nanomaterials 2022, 12, 1654. https://doi.org/10.3390/nano12101654
Li C, Cui Y, Tian H, Ren B, Li Q, Li Y, Yang H. Quantum Chemistry Study on the Structures and Electronic Properties of Bimetallic Ca2-Doped Magnesium Ca2Mgn (n = 1–15) Clusters. Nanomaterials. 2022; 12(10):1654. https://doi.org/10.3390/nano12101654
Chicago/Turabian StyleLi, Chenggang, Yingqi Cui, Hao Tian, Baozeng Ren, Qingyang Li, Yuanyuan Li, and Hang Yang. 2022. "Quantum Chemistry Study on the Structures and Electronic Properties of Bimetallic Ca2-Doped Magnesium Ca2Mgn (n = 1–15) Clusters" Nanomaterials 12, no. 10: 1654. https://doi.org/10.3390/nano12101654
APA StyleLi, C., Cui, Y., Tian, H., Ren, B., Li, Q., Li, Y., & Yang, H. (2022). Quantum Chemistry Study on the Structures and Electronic Properties of Bimetallic Ca2-Doped Magnesium Ca2Mgn (n = 1–15) Clusters. Nanomaterials, 12(10), 1654. https://doi.org/10.3390/nano12101654