
Innovative Microstructural Transformation upon CO2 Supercritical Conditions on Metal-Nucleobase Aerogel and Its Use as Effective Filler for HPLC Biomolecules Separation

 ,
,  , ,
, ,  ,
,
Abstract
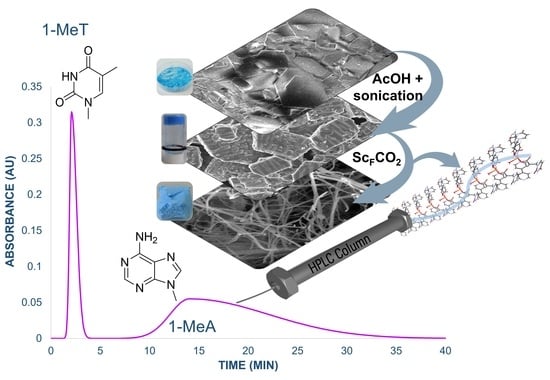
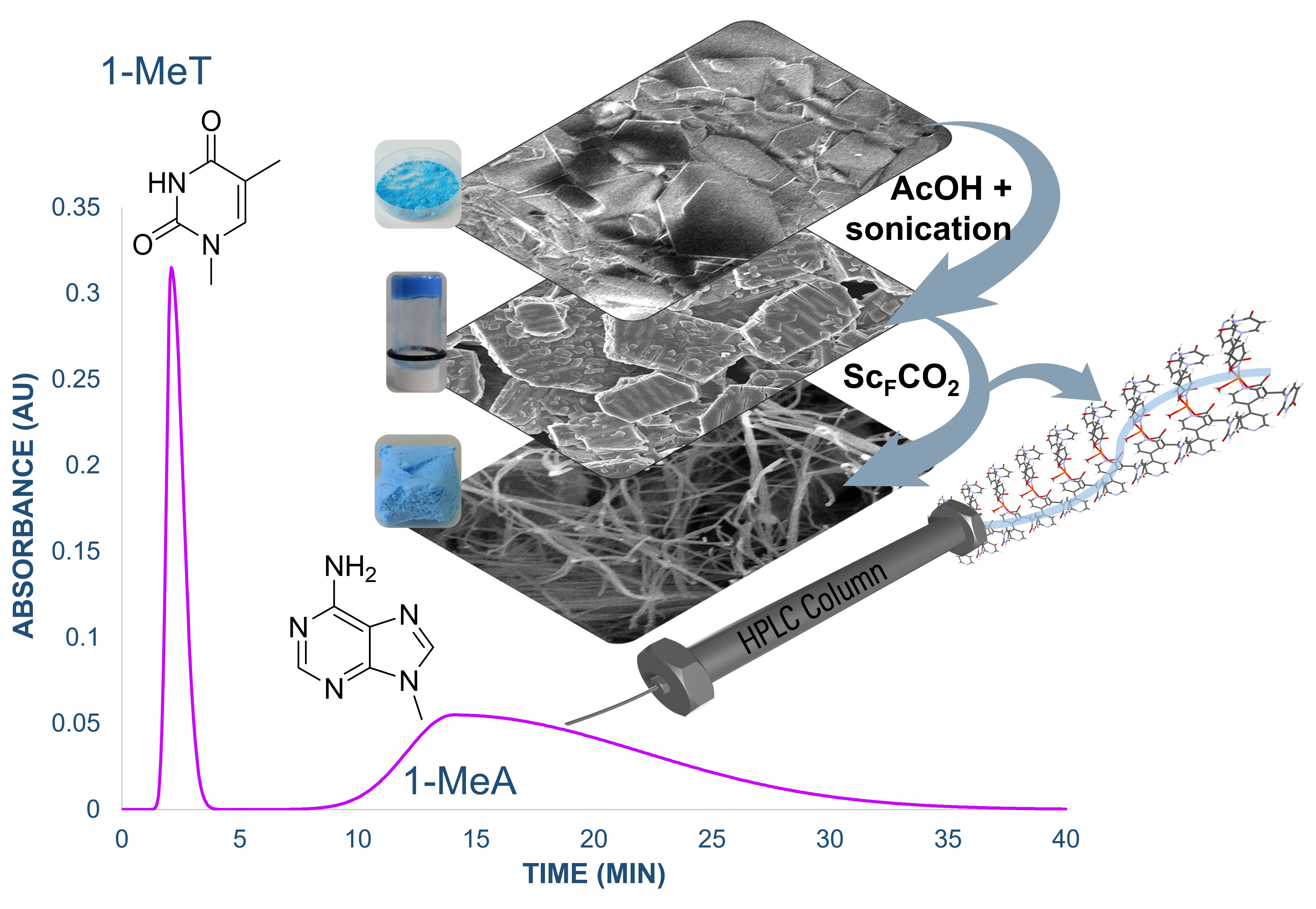
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Characterization Techniques
2.3. Theoretical Methodology and Computational Details
2.4. Synthesis
2.4.1. Synthesis of [Cu(UPrO)2(4,4′-bipy)2(H2O)] (CuUPrO) and [Cu(SO4)(4,4′-bipy)]n·2nH2O Single-Crystals
2.4.2. Synthesis of [Cu(UPrO)2(4,4′-bipy)2(H2O)] (CuUPrO) in Bulk
2.4.3. Synthesis of CuUPrO@MOG and CuUPrO@MOA
2.5. HPLC Column Design
3. Results and Discussion
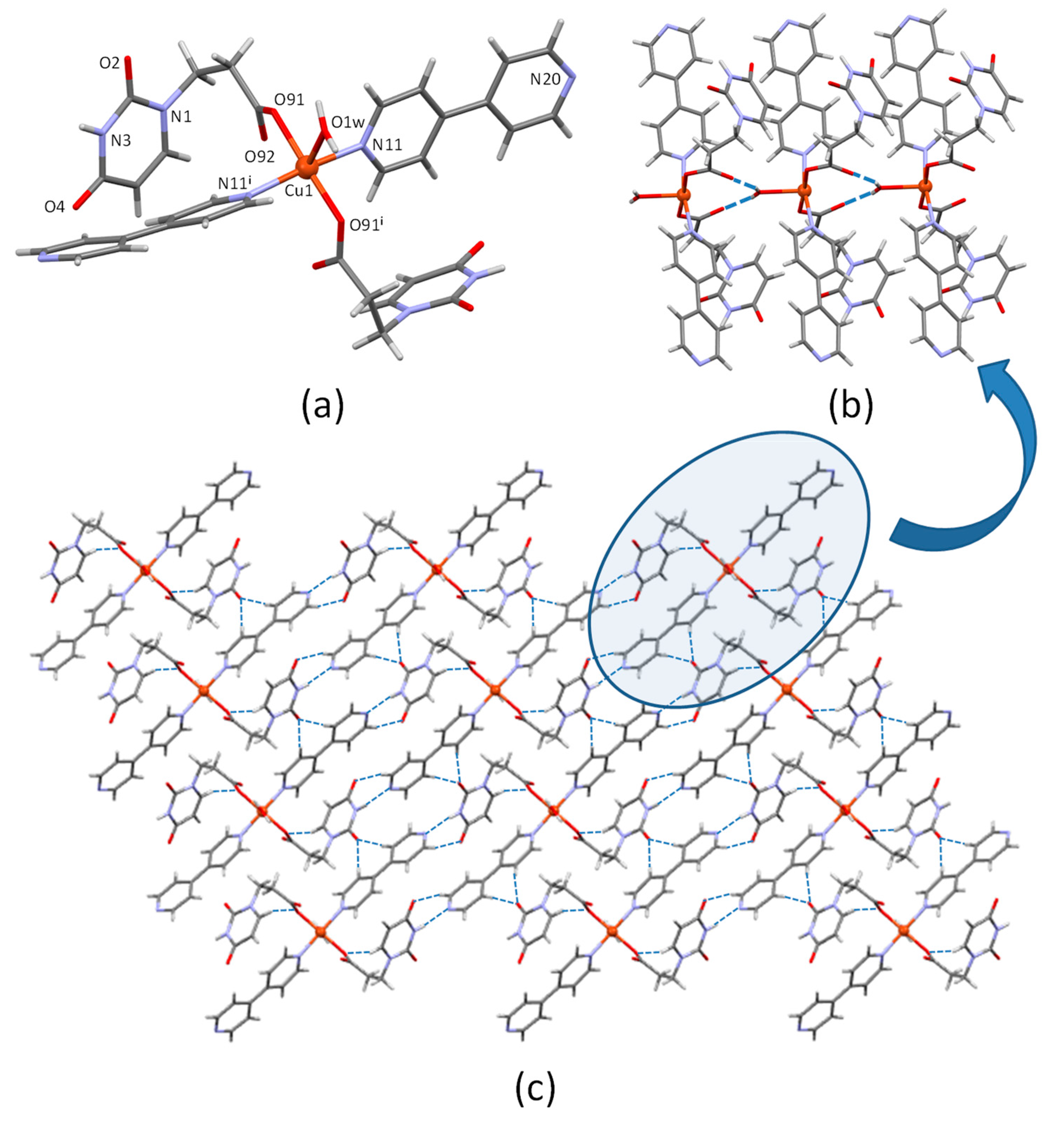
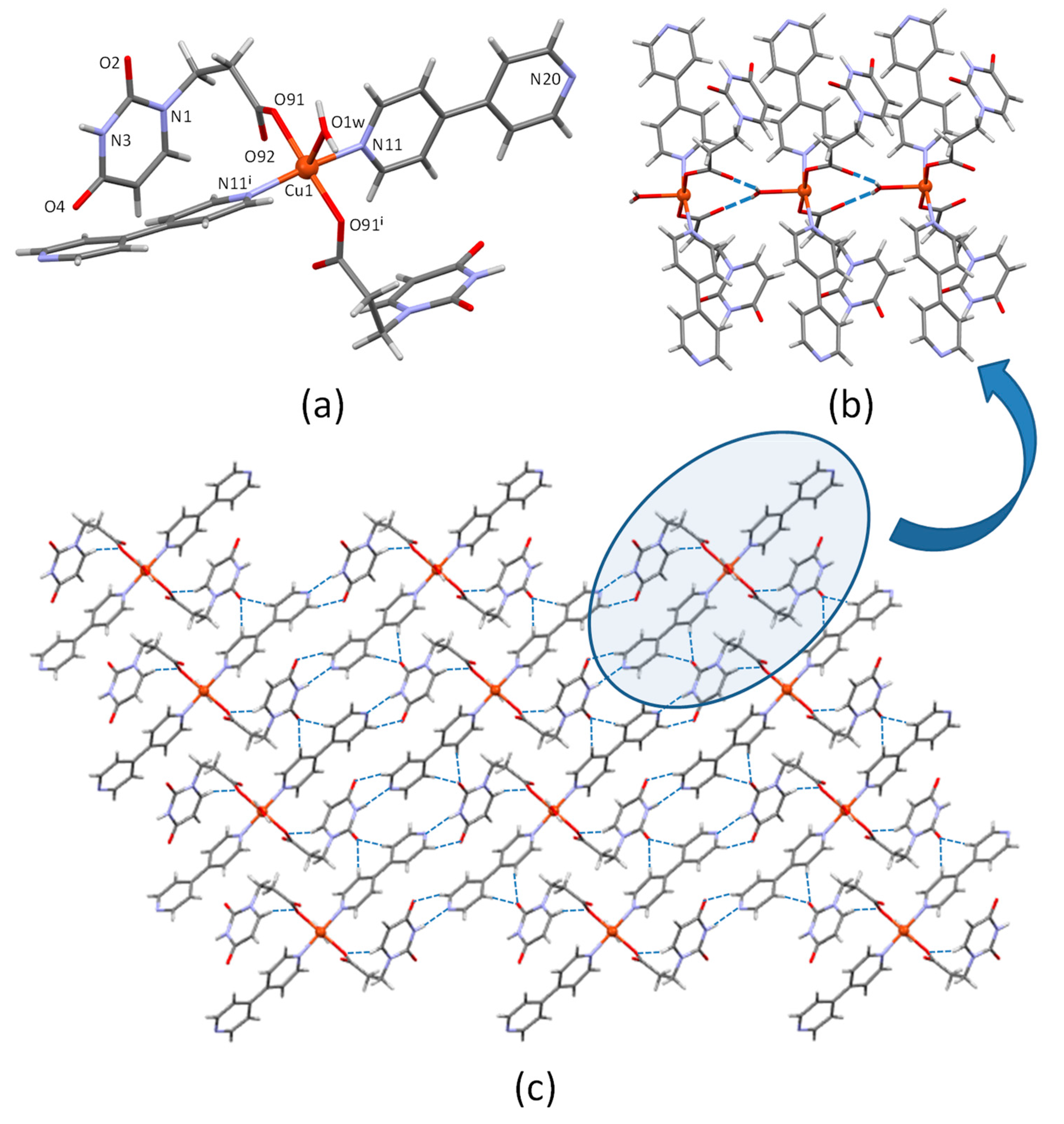
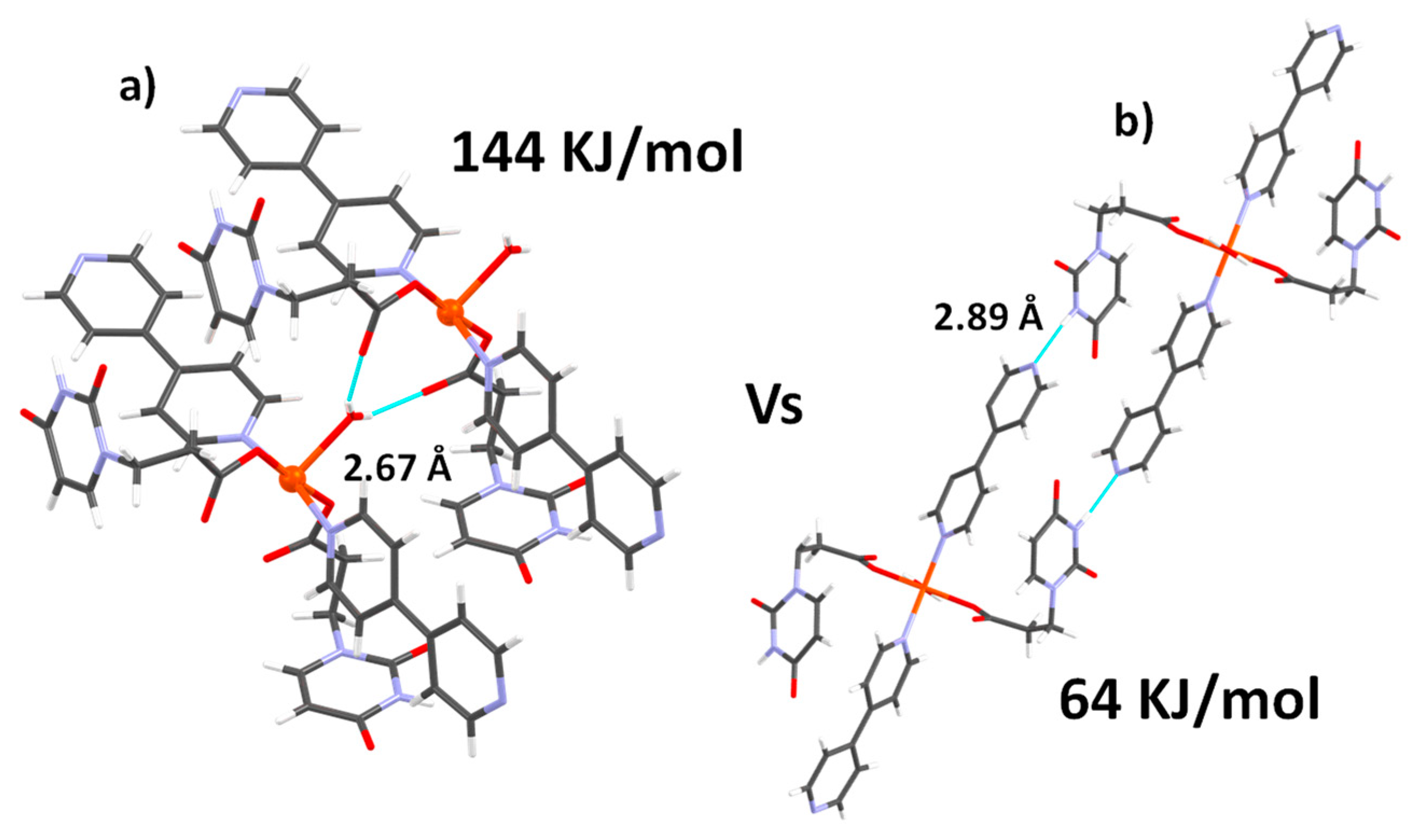
3.1. Crystal Structure Description of CuUPrO
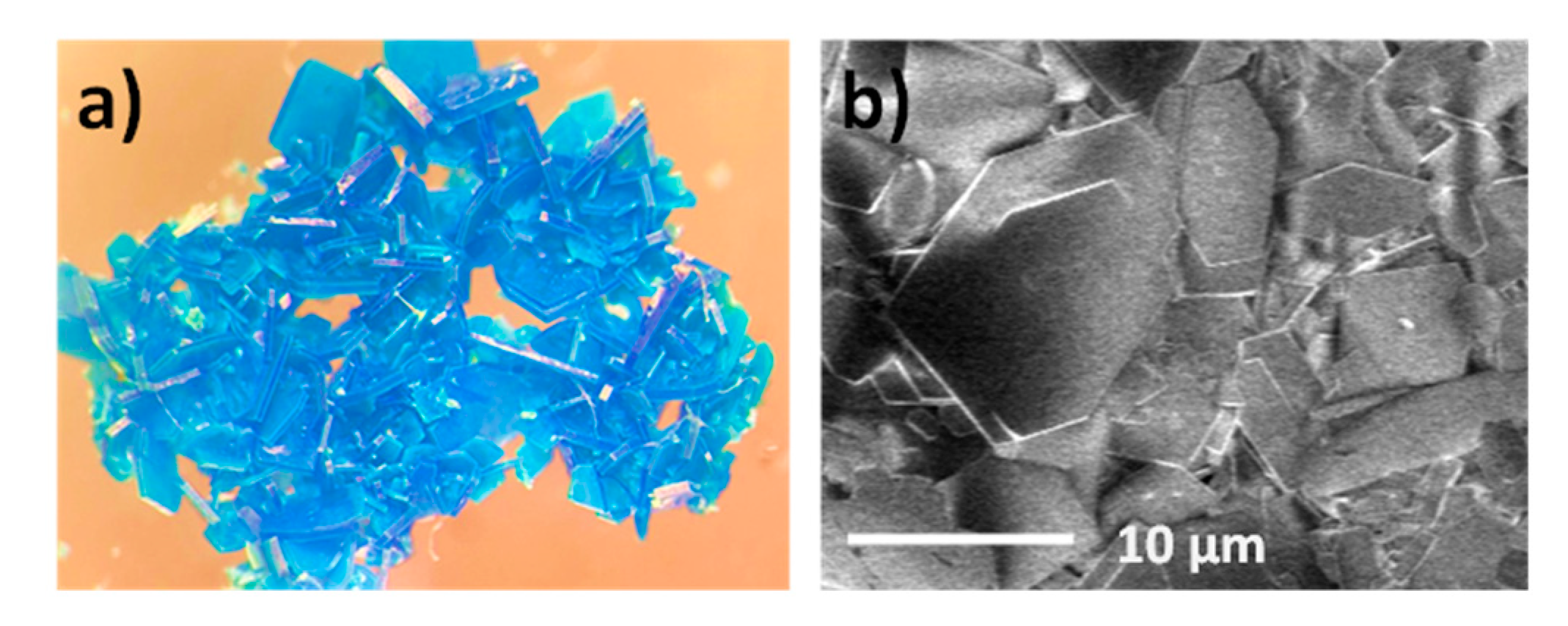
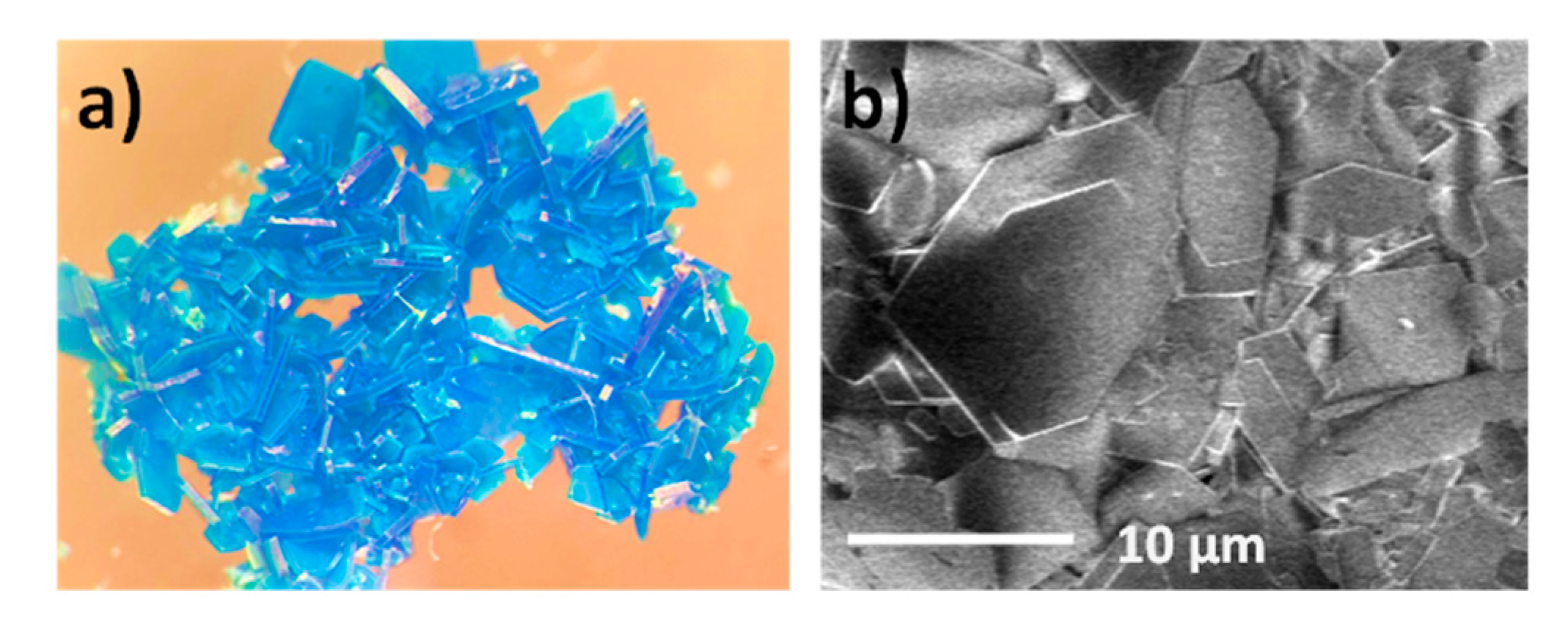
3.2. Preparation of MOG and MOA
3.3. Analysis of CuUPrO@MOA Adsorption Properties
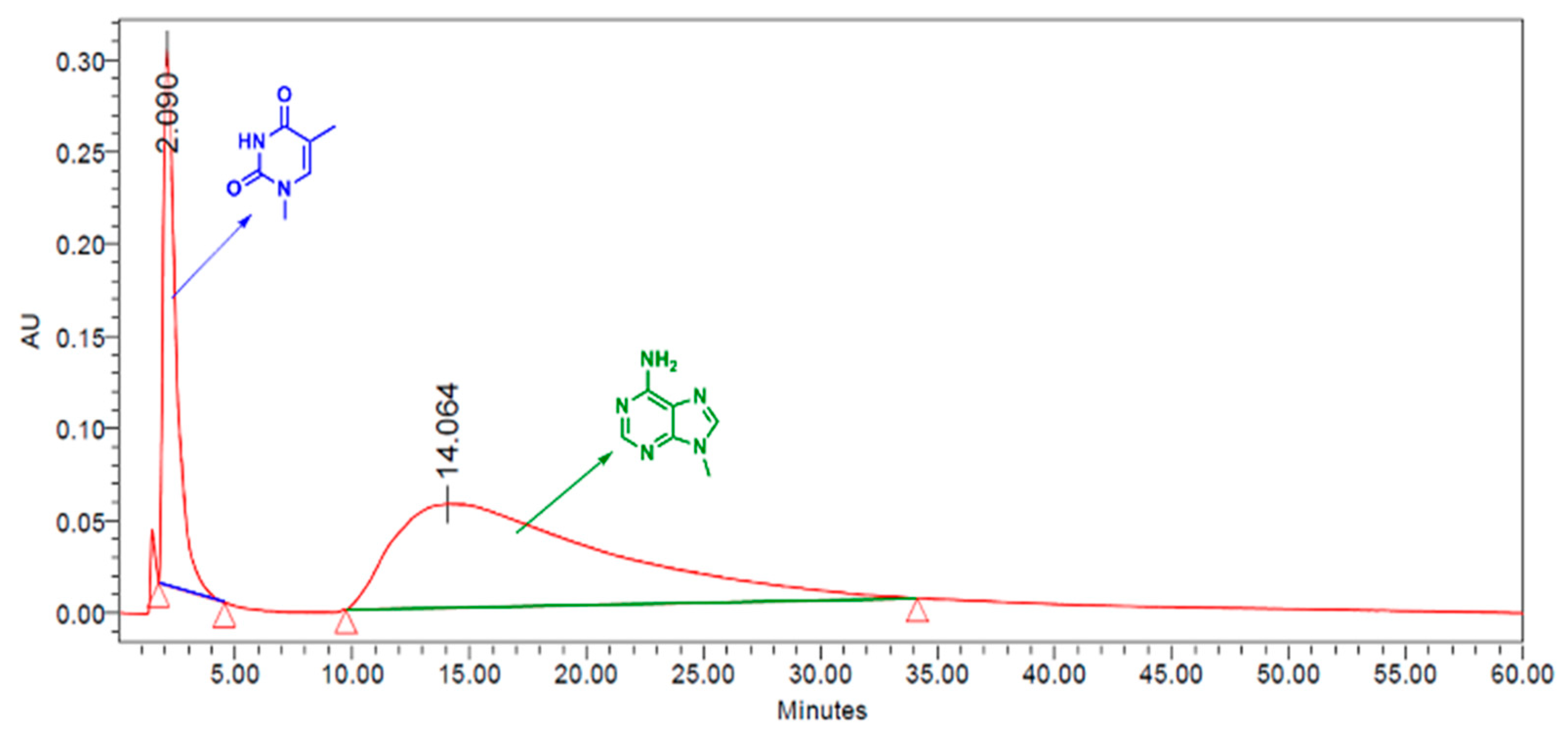
3.4. CuUPrO@MOA as Stationary Phase for HPLC Column Design
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batten, S.R.; Neville, S.M.; Turner, D. Coordination Polymers: Design, Analysis and Applications; RSC Publishing: Cambridge, UK, 2009. [Google Scholar]
- Amo-Ochoa, P.; Zamora, F. Coordination polymers with nucleobases: From structural aspects to potential applications. Coord. Chem. Rev. 2014, 276, 34–58. [Google Scholar] [CrossRef]
- Batten, S.R.; Chen, B.; Vittal, J.J. Coordination Polymers/MOFs: Structures, Properties and Applications. ChemPlusChem 2016, 81, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herrero, J.; Zamora, F. Coordination polymers for nanoelectronics. Adv. Mater. 2011, 23, 5311–5317. [Google Scholar] [CrossRef] [PubMed]
- Amo-Ochoa, P.; Alexandre, S.S.; Hribesh, S.; Galindo, M.A.; Castillo, O.; Gomez Garcia, C.J.; Pike, A.R.; Soler, J.M.; Houlton, A.R.; Harrington, W.; et al. Coordination Chemistry of 6-Thioguanine Derivatives with Cobalt: Toward Formation of Electrical Conductive One-Dimensional Coordination Polymers. Inorg. Chem. 2013, 52, 5290–5299. [Google Scholar] [CrossRef] [PubMed]
- Amo-Ochoa, P.; Castillo, O.; Alexandre, S.S.; Welte, L.; de Pablo, P.J.; Rodriguez-Tapiador, M.I.; Gomez-Herrero, J.; Zamora, F. Synthesis of designed conductive one-dimensional coordination polymers of Ni (II) with 6-mercaptopurine and 6-thioguanine. Inorg. Chem. 2009, 48, 7931–7936. [Google Scholar] [CrossRef]
- Cepeda, J.; Castillo, O.; García-Terán, J.P.; Luque, A.; Pérez-Yáñez, S.; Román, P. Supramolecular architectures and magnetic properties of self-assembled windmill-like dinuclear copper (II) complexes with purine ligands. Eur. J. Inorg. Chem. 2009, 2009, 2344–2353. [Google Scholar] [CrossRef]
- Garcia-Teran, J.P.; Castillo, O.; Luque, A.; Garcia-Couceiro, U.; Beobide, G.; Roman, P. Supramolecular architectures assembled by the interaction of purine nucleobases with metal-oxalato frameworks. Non-covalent stabilization of the 7H-adenine tautomer in the solid-state. Dalton Trans. 2006, 7, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Amo-Ochoa, P.; Castillo, O.; Guijarro, A.; Sanz Miguel, P.J.; Zamora, F. Supramolecular architectures based on 6-purinethione complexes. Inorg. Chim. Acta 2014, 417, 142–147. [Google Scholar] [CrossRef]
- Pérez-Yáñez, S.; Castillo, O.; Cepeda, J.; García-Terán, J.P.; Luque, A.; Román, P. Supramolecular architectures of metal–oxalato complexes containing purine nucleobases. Inorg. Chim. Acta 2011, 365, 211–219. [Google Scholar] [CrossRef]
- Hassanein, K.; Zamora, F.; Castillo, O.; Amo-Ochoa, P. Supramolecular interactions in Cobalt (II)–nucleobases complexes: A methyl matter. Inorg. Chim. Acta 2016, 452, 251–257. [Google Scholar] [CrossRef]
- Haldar, S.; Karmakar, K. Concentration dependent morphological transition of nanostructured self-assembly towards hydrogelation seeding from micellar aggregates through stereochemically optimized H-bonding network of amino acid derived cationic amphiphiles. Colloid Surf. A 2017, 516, 394–404. [Google Scholar] [CrossRef]
- Vasu, A.K.; Khurana, R.; Mohanty, J.; Kanvah, S. pH-responsive molecular assemblies of pyridylbutadiene derivative with cucurbit[7]uril. RSC Adv. 2018, 8, 16738–16745. [Google Scholar] [CrossRef] [Green Version]
- Moriche, R.; Prolongo, S.G.; Sanchez, M.; Jimenez-Suarez, A.; Sayagues, M.J.; Urena, A. Morphological changes on graphene nanoplatelets induced during dispersion into an epoxy resin by different methods. Comp. B Eng. 2015, 72, 199–205. [Google Scholar] [CrossRef]
- Basak, S.; Falcone, N.; Ferranco, A.; Kraatz, H.-B. Remarkable Morphology Transformation from Fiber to Nanotube of a Histidine Organogel in Presence of a Binuclear Iron (III)–Sulfur Complex. J. Inorg. Organomet. Polym. Mater. 2020, 30, 121–130. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, S.; Lan, J.; Tang, Y.; Xue, Y.; You, J. Ultrasound-induced switching of sheetlike coordination polymer microparticles to nanofibers capable of gelating solvents. J. Amer. Chem. Soc. 2009, 131, 1689–1691. [Google Scholar] [CrossRef]
- Ye, F.; Chen, S.; Tang, G.; Wang, X. Sonication induced morphological transformation between 3D gel network and globular structure in a two-component gelation system. Colloids Surf. A-Physicochem. Eng. Asp. 2014, 452, 165–172. [Google Scholar] [CrossRef]
- Higashiguchi, K.; Matsuda, K. Photoinduced LCST Behavior of Amphiphilic Diarylethene Assemblies: Phototransformative Supramolecular Architectures and Photodriven Actuation. J. Syn. Org. Chem. Jpn. 2019, 77, 236–245. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.D.; Praveen, V.K.; Hazra, A.T.; Maji, K.; Ajayaghosh, A. Light driven mesoscale assembly of a coordination polymeric gelator into flowers and stars with distinct properties. Chem. Sci. 2015, 6, 6583–6591. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Levy, A.; Liu, C.; Olivier, J.-H. Tuning Structure–Function Properties of π-Conjugated Superstructures by Redox-Assisted Self-Assembly. Chem. Mater. 2018, 30, 2143–2150. [Google Scholar] [CrossRef]
- Vegas, V.G.; Lorca, R.; Latorre, A.; Hassanein, K.; Gómez-García, C.J.; Castillo, O.; Somoza, Á.; Zamora, F.; Amo-Ochoa, P. Copper (II)–Thymine Coordination Polymer Nanoribbons as Potential Oligonucleotide Nanocarriers. Angew. Chem. Int. Ed. 2017, 56, 987–991. [Google Scholar] [CrossRef]
- Vegas, V.G.; Beobide, G.; Castillo, O.; Reyes, E.; Gomez-Garcia, C.J.; Zamora, F.; Amo-Ochoa, P. A bioinspired metal–organic approach to cross-linked functional 3D nanofibrous hydro- and aero-gels with effective mixture separation of nucleobases by molecular recognition. Nanoscale 2020, 12, 14699–14707. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; He, L.; Chen, L.; Su, C.-Y.; James, S.L. Metal–organic gels as functionalisable supports for catalysis. New J. Chem. 2009, 33, 1070–1075. [Google Scholar] [CrossRef]
- Das, D.; Biradha, K. Metal–organic gels of silver salts with an α, β-unsaturated ketone: The influence of anions and solvents on gelation. Inorg. Chem. Front. 2017, 4, 1365–1373. [Google Scholar] [CrossRef]
- Vallejo-Sánchez, D.; Amo-Ochoa, P.; Beobide, G.; Castillo, O.; Fröba, M.; Hoffmann, F.; Luque, A.; Ocón, P.; Pérez-Yáñez, S. Chemically Resistant, Shapeable, and Conducting Metal-Organic Gels and Aerogels Built from Dithiooxamidato Ligand. Adv. Funct. Mater. 2017, 27, 1605448. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Li, L.; Wu, R.; Liu, D.; Wu, J.; Wu, W. High-temperature-resistant polymer gel system with metal–organic mixed cross-linking agents. J. Appl. Polym. Sci. 2015, 132, 42261. [Google Scholar] [CrossRef]
- Pandey, V.K.; Dixit, M.K.; Manneville, S.; Bucher, C.; Dubey, M. A multi-stimuli responsive conductive sonometallogel: A mechanistic insight into the role of ultrasound in gelation. J. Mater. Chem. A 2017, 5, 6211–6218. [Google Scholar] [CrossRef]
- Das Mahapatra, R.; Dey, J. Ultrasound-Induced Gelation of Organic Liquids by l-Cysteine-Derived Amphiphile Containing Poly(ethylene glycol) Tail. Langmuir 2015, 31, 8703–8709. [Google Scholar] [CrossRef]
- Thakur, N.; Sharma, B.; Bishnoi, S.; Mishra, S.K.; Nayak, D.; Kumar, A.; Sarma, T.K. Multifunctional Inosine Monophosphate Coordinated Metal–Organic Hydrogel: Multistimuli Responsiveness, Self-Healing Properties, and Separation of Water from Organic Solvents. ACS Sustain. Chem. Eng. 2018, 6, 8659–8671. [Google Scholar] [CrossRef]
- Zou, D.-H.; Wang, P.; Luo, W.; Hou, J.-L.; Zhu, Q.-Y.; Dai, J. Fluorescent Hydrogel Generated Conveniently from a Perylene Tetracarboxylate Derivative of Titanium(IV) Alkoxide. Inorg. Chem. 2018, 57, 1623–1629. [Google Scholar] [CrossRef]
- Wang, R.; Geiger, C.; Chen, L.; Swanson, B.; Whitten, D.G. Direct Observation of sol−gel conversion: The role of the solvent in organogel formation. J. Amer. Chem. Soc. 2000, 122, 2399–2400. [Google Scholar] [CrossRef]
- Wang, X.; He, T.; Yang, L.; Wu, H.; Zhang, R.; Zhang, Z.; Shen, R.; Xiang, J.; Zhang, Y.; Wei, C. A Co2+-selective and chirality-sensitive supermolecular metallohydrogel with a nanofiber network skeleton. Nanoscale 2016, 8, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Maleki, H.; Durães, L.; García-González, C.A.; del Gaudio, P.; Portugal, A.; Mahmoudi, M. Synthesis and biomedical applications of aerogels: Possibilities and challenges. Adv. Colloid Interface Sci. 2016, 236, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Yavvari, P.S.; Pal, S.; Kumar, S.; Kar, A.; Awasthi, A.K.; Naaz, A.; Srivastava, A.; Bajaj, A. Injectable, self-healing chimeric catechol-Fe (III) hydrogel for localized combination cancer therapy. ACS Biomater. Sci. Eng. 2017, 3, 3404–3413. [Google Scholar] [CrossRef]
- Sharma, G.; Thakur, B.; Naushad, M.; Kumar, A.; Stadler, F.J.; Alfadul, S.M.; Mola, G.T. Applications of nanocomposite hydrogels for biomedical engineering and environmental protection. Environ. Chem. Lett. 2018, 16, 113–146. [Google Scholar] [CrossRef]
- Vegas, V.G.; Villar-Alonso, M.; Gomez-Garcia, C.J.; Zamora, F.; Amo-Ochoa, P. Direct Formation of Sub-Micron and Nanoparticles of a Bioinspired Coordination Polymer Based on Copper with Adenine. Polymers 2017, 9, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, H. Recent advances in aerogels for environmental remediation applications: A review. Chem. Eng. J. 2016, 300, 98–118. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97. In Program for Crystal Structure Refinement; University Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- SMART and SAINT. Area Detector Software Package and SAX Area-Detector Integration Program; Bruker Analytical X-ray Instruments: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. Structure Determination Package; Bruker Analytical X-ray Instruments: Madison, WI, USA, 2000. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Hagrman, D.; Hammond, R.P.; Haushalter, R.; Zubieta, J. Organic/Inorganic Composite Materials: Hydrothermal Syntheses and Structures of the One-, Two-, and Three-Dimensional Copper(II) Sulfate−Organodiamine Phases [Cu(H2O)3(4,4′-bipyridine)(SO4)]·2H2O, [Cu(bpe)2][Cu(bpe)(H2O)2(SO4)2]·2H2O, and [Cu(bpe)(H2O)(SO4)] (bpe = trans-1,2-Bis(4-pyridyl)ethylene). Chem. Mater. 1998, 10, 2091–2100. [Google Scholar]
- Torres, O.; Tena, N.M.; Murray, B.; Sarkar, A. Novel starch based emulsion gels and emulsion microgel particles: Design, structure and rheology. Carbohydr. Polym. 2017, 178, 86–94. [Google Scholar] [CrossRef]
- Sutar, P.; Maji, T.K. Recent advances in coordination-driven polymeric gel materials: Design and applications. Dalton Trans. 2020, 49, 7658–7672. [Google Scholar] [CrossRef]
- Jung, S.; Oh, M. Monitoring shape transformation from nanowires to nanocubes and size-controlled formation of coordination polymer particles. Angew. Chem. Int. Ed. 2008, 47, 2049–2051. [Google Scholar] [CrossRef]
- Piepenbrock, M.-O.M.; Clarke, N.; Steed, J.W. Shear induced gelation in a copper (II) metallogel: New aspects of ion-tunable rheology and gel-reformation by external chemical stimuli. Soft Matter 2010, 6, 3541–3547. [Google Scholar] [CrossRef]
- Yu, X.; Chen, L.; Zhang, M.; Yi, T. Low-molecular-mass gels responding to ultrasound and mechanical stress: Towards self-healing materials. Chem. Soc. Rev. 2014, 43, 5346–5371. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.L.; Monson, P.A. Adsorption in porous materials at high pressure: Theory and experiment. Langmuir 2002, 18, 10261–10273. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon dioxide capture in metal–organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Liu, H.; Lin, M.; Li, S.; Ouyang, G.; Chen, L.; Su, C.-Y. Highly porous aerogels based on imine chemistry: Syntheses and sorption properties. J. Mater. Chem. A 2015, 3, 10990–10998. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Shen, Z.-R.; Li, Y.; Han, S.-D.; Hu, T.-L.; Zhang, D.-S.; Bu, X.-H.; Ruan, W.-J. Solvent induced rapid modulation of micro/nano structures of metal carboxylates coordination polymers: Mechanism and morphology dependent magnetism. Sci. Rep. 2014, 4, srep06023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, M.; Liu, H.; Williams, T.; Ma, J.; Wang, H.; Zhang, X. Temperature-induced oriented growth of large area, few-layer 2D metal–organic framework nanosheets. Chem. Commun. 2017, 53, 13161–13164. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-H···A [b] | H···A | D···A | D-H···A |
|---|---|---|---|
| N3-H···N20i | 2.05 | 2.900(4) | 172.5 |
| O1w-H···O92ii | 1.91 | 2.677(3) | 156.0 |
| C6-H···O91iii | 2.42 | 3.279(4) | 153.0 |
| C15-H···O2iv | 2.38 | 3.302(4) | 170.1 |
| C22-H···O2iv | 2.27 | 3.200(5) | 174.4 |
| C19-H···O4v | 2.41 | 3.333(5) | 169.4 |
| Sample | SBET a (m2 g−1) | Smicro b (m2 g−1) | Vmicro b (cm3 g−1) | VT c (cm3 g−1) |
|---|---|---|---|---|
| CuUPrO@MOA | 67.1 | 47.1 | 0.019 | 0.065 |
| Composition | tR (min) | wh/2 (s) | Rs | ||||
|---|---|---|---|---|---|---|---|
| SiO2 Grinding | MOA (%) | Toluene | Binol | Toluene | Binol | ||
| A | manually | 5.0 | 1.525 | 2.690 | 15 | 37 | 1.59 |
| B | ball-miller | 2.5 | 1.516 | 2.627 | 23 | 43 | 1.19 |
| C | ball-miller | 5.0 | 1.414 | 3.185 | 15 | 50 | 1.93 |
| D | ball-miller | 7.5 | 1.598 | 4.180 | 36 | 98 | 1.36 |
| E b | ball-miller | 5.0 | 1.597 | 2.022 | 10 | 11 | 1.43 |
| Composition | tR (min) | wh/2 (s) | α a | Rs b | ||||
|---|---|---|---|---|---|---|---|---|
| SiO2 Grinding | MOA (%) | 2-Naphthol | Binol | 2-Naphthol | Binol | |||
| B | ball-miller | 2.5 | 1.696 | 2.433 | 49 | 27 | 5.09 | 0.60 |
| C | ball-miller | 5.0 | 1.686 | 3.045 | 70 | 28 | 6.00 | 1.00 |
| D | ball-miller | 7.5 | 2.045 | 3.843 | 81 | 47 | 5.02 | 0.95 |
| Configuration I (Eint/molecule, KJ/mol) | Configuration II (Eint/molecule, KJ/mol) | |
|---|---|---|
| 9-MeA | 59.7 | 42.4 |
| 1-MeT | 30.8 | 20.2 |
| 1-MeU | 31.8 | 22.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, N.; Beobide, G.; Reyes, E.; Martínez, J.I.; Gómez-García, C.J.; Castillo, O.; Amo-Ochoa, P. Innovative Microstructural Transformation upon CO2 Supercritical Conditions on Metal-Nucleobase Aerogel and Its Use as Effective Filler for HPLC Biomolecules Separation. Nanomaterials 2022, 12, 675. https://doi.org/10.3390/nano12040675
Maldonado N, Beobide G, Reyes E, Martínez JI, Gómez-García CJ, Castillo O, Amo-Ochoa P. Innovative Microstructural Transformation upon CO2 Supercritical Conditions on Metal-Nucleobase Aerogel and Its Use as Effective Filler for HPLC Biomolecules Separation. Nanomaterials. 2022; 12(4):675. https://doi.org/10.3390/nano12040675
Chicago/Turabian StyleMaldonado, Noelia, Garikoitz Beobide, Efraim Reyes, José Ignacio Martínez, Carlos J. Gómez-García, Oscar Castillo, and Pilar Amo-Ochoa. 2022. "Innovative Microstructural Transformation upon CO2 Supercritical Conditions on Metal-Nucleobase Aerogel and Its Use as Effective Filler for HPLC Biomolecules Separation" Nanomaterials 12, no. 4: 675. https://doi.org/10.3390/nano12040675
APA StyleMaldonado, N., Beobide, G., Reyes, E., Martínez, J. I., Gómez-García, C. J., Castillo, O., & Amo-Ochoa, P. (2022). Innovative Microstructural Transformation upon CO2 Supercritical Conditions on Metal-Nucleobase Aerogel and Its Use as Effective Filler for HPLC Biomolecules Separation. Nanomaterials, 12(4), 675. https://doi.org/10.3390/nano12040675