1. Introduction
Carbon dioxide emission reduction has become the consensus of many countries. Therefore, the development of effective adsorbents for CO
2 capture is a top priority. It is known that good CO
2 adsorbents are dependent on pore volumes of sizes less than 0.7 nm and the presence of surface functional groups [
1,
2,
3,
4,
5]. Various porous materials have been intensively studied, such as carbon nanofibers, zeolitic imidazolate frameworks (ZIFs), metal organic frameworks, carbon nanotubes, silica, activated carbon fibers, zeolites, and activated carbons.
ZIFs are a subgroup of MOFs, with perpetual porosity consisting of metals bridging to imidazolate-like ligands, with the tetrahedral arrangement as molecular structural blocks [
6,
7]. The bond angles are 145° [
6], similar to the Si–O–Si bond angle in zeolites [
8,
9,
10,
11]. These building units are congregated and assemble in large molecular configurations. They have emerged as novel crystalline porous materials. Due to possessing features of both MOFs and zeolites, ZIFs have several unique characteristics, including unimodal micropores, high surface areas, abundant functionalities, high thermal stability, and remarkable chemical robustness. [
12]. Therefore, ZIFs have gained a large amount of attention for many potential applications, such as gas separation/adsorption [
13,
14], water treatment [
15], catalysis [
16], chemical sensing [
17], and biomedicine [
18].
Among the various ZIFs, ZIF-8 is one of the most widely investigated, having a sodalite (SOD) topology and displaying a three-dimensional structure composed of zinc ions and 2-methylimidazole (2-MeIM) linkers. ZIF-8 has large pores, with diameters of 1.16 nm, which are accessible through small apertures with diameters of 0.34 nm, and they have a cubic space group with unit cell dimensions of 1.632 nm [
19]. ZIF-8 possesses high thermal stability to 550 °C in N
2, a large surface area (1630–1700 m
2/g), and exceptional chemical resistance to various solvents [
20]. The crystal sizes of ZIF-8 depend on several synthesis conditions. ZIF-8 can be prepared using several different synthesis routines in water or organic solvents, such as conventional solvothermal [
21] or hydrothermal [
22], colloidal precipitation [
23,
24], microwave-assisted [
25], mechanochemical [
25], sonochemical [
25], dry-gel [
25], electrochemical [
26], microfluidic [
25], or template [
27] methods. The hydrothermal or solvothermal processes are generally conducted with a reaction temperature from room temperature up to 200 °C and a reaction duration from hours to days. Banerjee et al. [
21] found that a metal precursor concentration of 0.20 M, a temperature of 85 or 100 °C, and a reaction time of 72 h were optimal conditions for preparing ZIF nanoparticles. The colloidal precipitation method is a facile route [
23] for the mass production of nano- to micro-meter scale ZIF-8 crystals with controlled particle sizes, and has been widely used in the literature.
Mphuthi et al. [
16] reported that the average powder size of ZIF-8 was 65 nm (synthesized in water) and 52 nm (prepared in methanol). Zheng et al. [
28] found that ZIF-8 crystals with sizes of 40~ 110 nm could be obtained in microemulsions, by controlling the ratio of Zn
2+ to 2-MeIM at 1:16~1:2. Venna et al. [
19] found that the structural evolution of ZIF-8 is a function of synthesis time, which can be divided into three stages: (I) the nucleation stage (
t < 10 min); (II) the growth stage (a maximum in crystallinity is achieved at ~60 min); and (III) the stationary phase (
t > 60 min). The relative crystallinity of ZIF-8 increased slowly in the initial stage of synthesis. Next, a rapid increase was reported in the 30–40 min range. The relative crystallinity of ZIF-8 was almost unchanged after 50 min (above 90%), and the relative crystallinity of ZIF-8 reached a maximum at 24 h [
19]. Different zinc salts with a methanol-based recipe have been reported to have different effects on the crystal sizes of ZIF-8. It has been observed that greater nucleation rates led to smaller crystal sizes for zinc nitrate, while the reverse was true for ZIF-8 crystals from zinc acetate [
23,
29]. Ramu et al. [
30] found that zinc nitrate was a more effective zinc source, probably due to the fast nucleation of zinc nitrate compared to other zinc salts.
In order to improve the chemical stability of ZIF-8, nitrogen-functionalized and Zn-decorated microporous carbon materials can be obtained via the carbonization of ZIF-8 with imidazolate framework ligands and Zn metal centers (melting point of 419.53 °C and boiling point of 907 °C) [
31]. Torad et al. [
23] synthesized nanoporous carbon by the direct carbonization of ZIF-8 crystals, and observed that the products showed a large adsorption uptake and a faster sensor response for toluene vapors. A systematic study was conducted by Gadipelli and Guo [
31], where the carbonization process of ZIF-8 was reported. The results showed that the ligand decomposition and metal evaporation were highly associated with the carbonization temperature, reaction time, and heating rate. As the temperature was above 800 °C, this would result in the development of porosity at the expense of Zn and N atoms, as well as the yield of the products. The mass loss could be attributed to the dissociation of -CH
3 groups (commencing at approximately 600 °C), the ligand decomposition and liberation of –C-N-H groups (between 600 and 900 °C), and the evaporation of Zn metal (above 900 °C). Nevertheless, the actual framework decomposition and subsequent carbonization were relatively slow compared with the rapid decomposition of other MOF structures with carboxylate ligands. When ZIF-8 was heat treated at a temperature of 1000 °C, nitrogen-doped carbon nanopolyhedra were obtained with a highly graphitic carbon skeleton, during which Zn ions in ZIF-8 were reduced to Zn metal and vaporized. The abundant nitrogen in ZIF-8 was directly incorporated into an aromatic methyl-imidazole ligand, which could promote the combination of enriched nitrogen-containing active sites into carbon structures. The resultant carbons had a hierarchical porous structure and uniform distribution of active sites [
32]. Therefore, the carbonization of ZIF-8 at different temperatures (from 600 to 1100 °C) yielded different carbon structures.
MOF-derived nanoporous carbons have been used for electrochemical double-layered capacitors; though they had a high specific surface area, their many micropores led to poor conductivity and resulted in a weak capacitance [
33,
34]. Two Zr-based MOFs were doped into the polyimide-based nanofibers to form nanocomposites, and these nanocomposites could remove non-polar and polar volatile organic compounds from the air. The results showed that the introduction of MOFs powders significantly improved the adsorption performance of the nanofibers [
35]. MOFs can be synthesized in situ on electrospun PAN-based nanofibers using two processes: electrospinning and aqueous synthesis, where MOFs/PAN composites served as the SO
2 gas sensing layer. The sensors comprised MOFs/PAN composites, and carbon nanotubes possessed remarkable sensitivity and long-term stability to SO
2 gas [
36]. Wang et al. [
37] reported that when ZIF-67 was cast on carbon cloth and then polyaniline was electrodeposited on composites, they exhibited a good capacitance. A ZIF-based carbon fiber for supercapacitors was prepared via the pyrolysis of ZIF-8-based polyacrylonitrile (ZIF-8/PAN) nanofibers, where ZIF-8: PAN precursor = 3:4 (
w/w). In this study, after ZIF-8 was carbonized at 800 °C for 3 h, the Zn totally disappeared in the ZIF-8/PAN fibers [
38].
Wang et al. [
39] utilized an in situ growth method, composed of electrospinning and a hydrothermal treatment, to prepare a ZIF-8/PAN fibrous filter, and reported that U(VI) chelated with the N atoms of 2-MeIM was the major adsorption mechanism, while in situ ZIF-8/PAN fibers exhibited a high adsorption capacity for applications to nuclear wastewater. The in situ growth method was used to prepare ZIF-8/cellulose nanofiber composites [
40]. Due to the electrostatic forces between Zn metal in ZIF-8 and –COO- groups on nanofibers, ZIF-8 crystals could be homogenously deposited on nanofibers. The composites showed good CO
2/N
2 selectivity. A novel structure of PAN nanofibers was fabricated using an electrospinning technique, decorated with ZIF-8 using a phase conversion growth method, and investigated for CO
2 capture [
41]. The results indicated that electrostatic interaction and hydrogen bonds existed between the PAN matrix and the ZIF-8 nanocrystals confined to the PAN nanofiber surface. However, the CO
2 uptakes were only 7.0 cm
3/g at 25 °C, 1 bar. Recently, a methodology was proposed to predict the purity, yield, and crystallinity of ZIF-8 crystals prepared via electrochemical routes, and which used the application of artificial intelligence, involving the design of experiments with a machine learning module [
42]. This could be a feasible route towards the sustainable design and development of advanced materials and chemicals. Before it is practically applied, the collection of baseline information is important.
Several synthesis techniques or novel structures have been reported for the growing applications of ZIF-8. ZIF-8-based nanomaterials were selected because they are one of the most investigated MOFs and as they have various applications, especially based on adsorption. The interactions between CO
2 and ZIF-8 can enhance the storage of CO
2 in the framework, and the storage performance of CO
2 on ZIF-8 was strongly pressure dependent [
43]. At present, the CO
2 adsorption capacities of pristine ZIFs crystals were not distinct enough at, or under, atmospheric pressure [
6,
41,
44]. Thus, several studies were focused on applications with high pressure [
14,
43]. Most studies on ZIFs have focused on the selectivity of CO
2 from various gas mixtures [
6,
8,
45]. However, currently, studies on the high-temperature activation of ZIF/PAN nanocomposites have not been published in the literature. Here, for the first time, PAN-based activated nanofiber composites embedded with ZIF-8 crystals or ZIF-8-derived carbons (ZDC) were fabricated using successive processes, comprising electrospinning, stabilization, carbonization, and activation. The properties of the adsorbents were characterized thoroughly, and the CO
2 adsorption performance of the adsorbents was investigated. It was intended to incorporate the advantages of carbon nanofibers with the benefits of ZIF-8 based nanoparticles, to develop high-performance CO
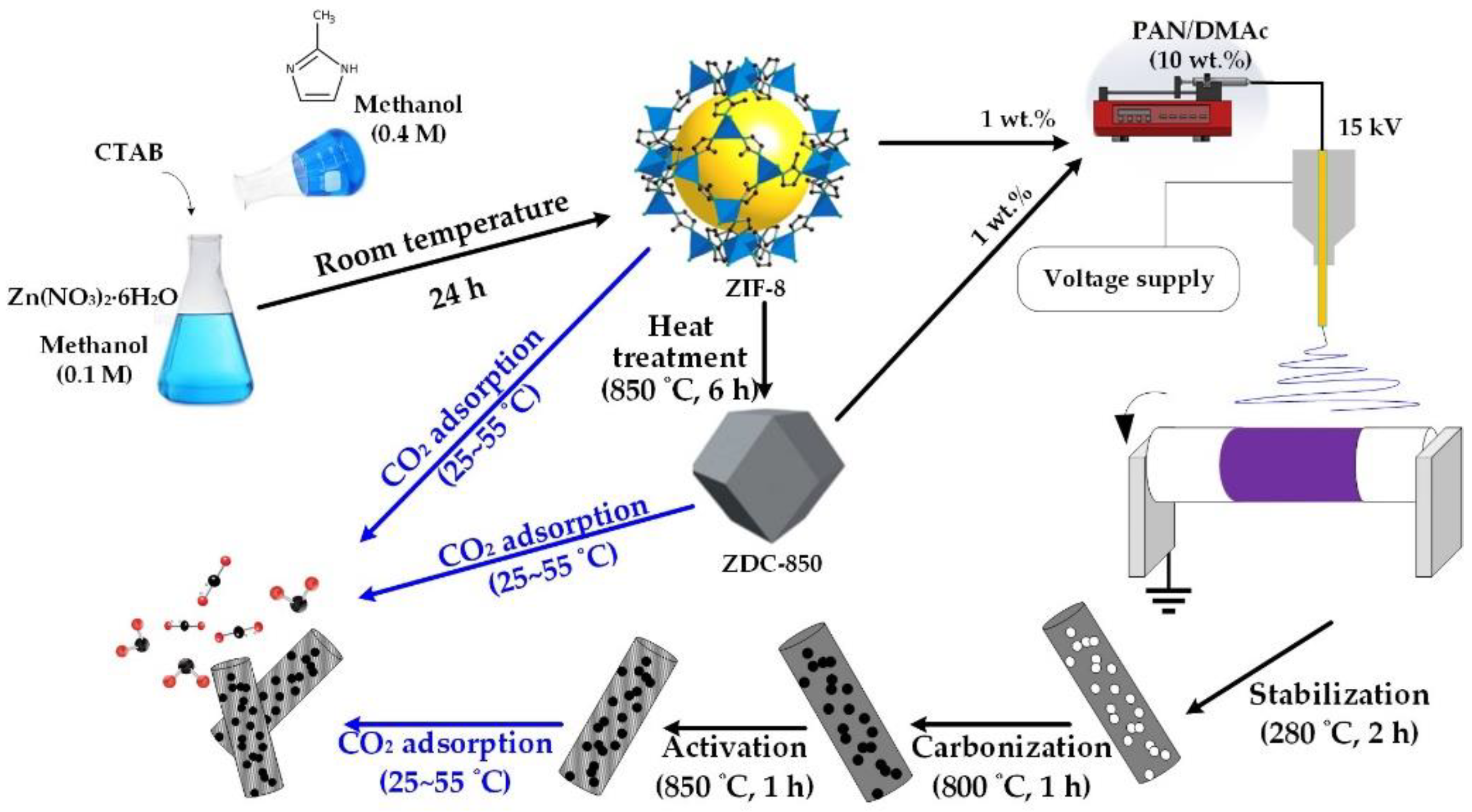
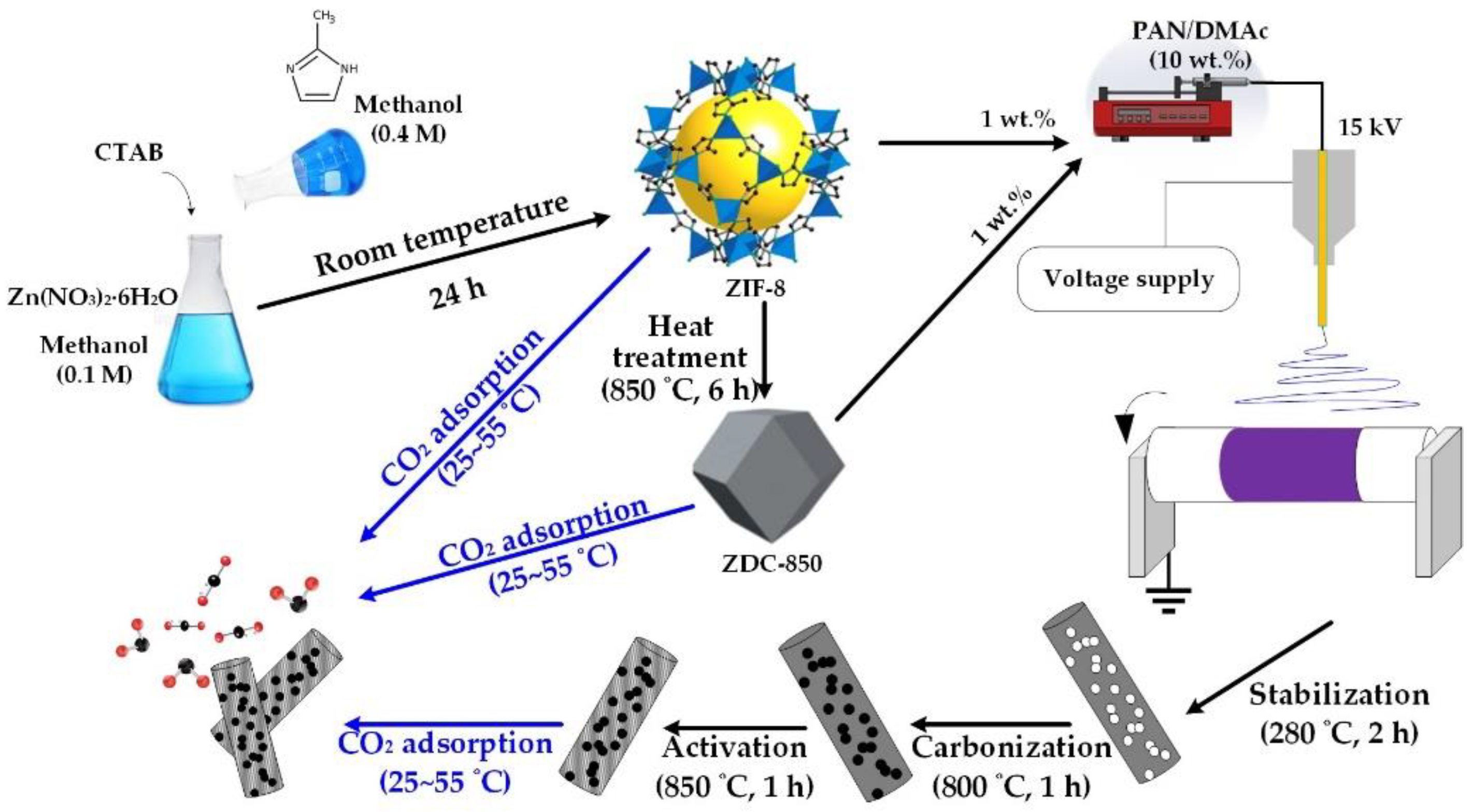
2 adsorbents. A schematic diagram of this study is shown in
Scheme 1.
2. Materials and Methods
2.1. Chemicals
Zinc nitrate hexahydrate (≥99%) was purchased from J.T. Baker (Radnor, PA, USA); 2-methylimidazole (2-MeIM, 99%) and polyacrylonitrile (PAN) (Mw = 150 kDa) were obtained from Sigma-Aldrich (St. Louis, MO, USA); cetyltrimethylammonium bromide (CTAB, 98%) and N, N-dimethylacetamide (DMAc, 99%) were purchased from Alfa Aesar (Sisli-Istanbul, Turkey); methanol (≥99.8%) was purchased from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). All the chemicals were used without further purification.
2.2. Preparation of ZIF-8
A rapid colloidal chemistry route was used to produce ZIF-8 nanocrystals at room temperature [
24,
46]. The sizes of nanocrystals were highly related to the molar ratio of 2-MeIM to zinc salt [
28]. In a typical process, 0.74 g of zinc nitrate hexahydrate and 19 mg CTAB were dissolved in 25 mL methanol, and 0.82 g of 2-MeIM was dissolved in 25 mL methanol, respectively. CTAB was used as a surfactant. Additionally, the methanol solution of zinc nitrate hexahydrate was quickly poured into the methanol solution of 2-MeIM under stirring, at room temperature. After rigorous and continuous stirring for 30 min, the mixture solution was placed at room temperature to settle for 24 h [
19]. Finally, the white precipitate was collected and thoroughly washed with methanol using centrifugation to remove the excess 2-MeIM. The ZIF-8 powders were obtained after drying at 80 °C for 24 h in an oven.
2.3. Heat Treatment of ZIF-8
In order to improve the chemical stability of ZIF-8, carbonization of ZIF-8 powders was conducted. In a typical carbonization process, 0.5 g of ZIF-8 was placed in a porcelain combustion boat and then transferred into a tubular furnace. The samples were heated in a nitrogen atmosphere with a heating rate of 5 °C/min, and carbonized at 850 °C for 6 h. The sample was named ZDC-850.
2.4. Fabrication of Electrospun Nanofiber Composites
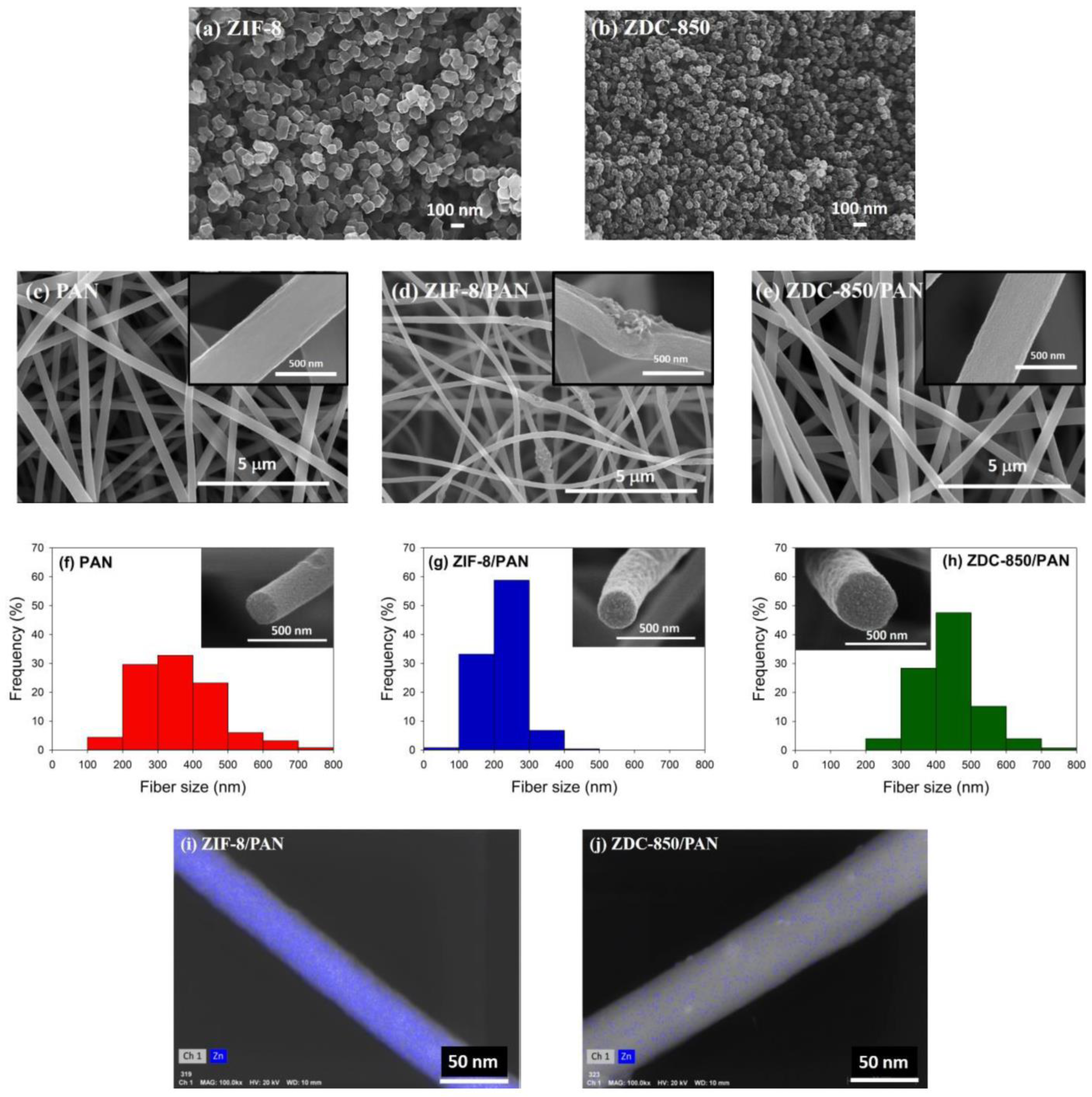
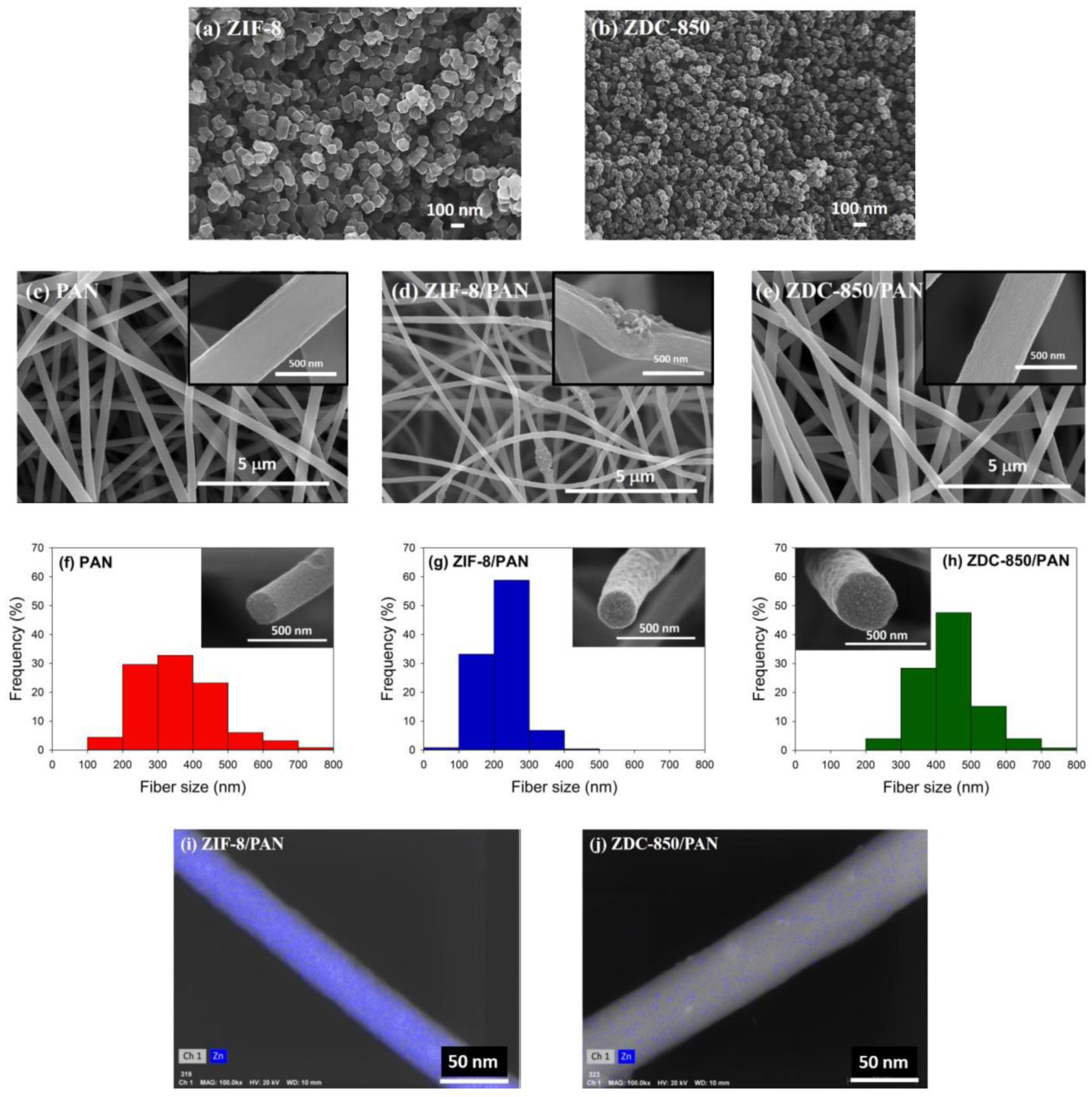
Nanofiber composites were prepared using an electrospinning process. Initially, PAN was dissolved in DMAc to form a polymer solution with 10 wt.% PAN. The resulting solutions were subjected to continuous magnetic stirring at 60 °C for 24 h in order to obtain a homogeneous polymer solution. Next, ZIF-8 (or ZDC-850) with a ratio of 10 wt.% of ZIF-8 (or ZDC-800) to PAN was added into the polymer solution and continuously stirred for 24 h at room temperature. Electrospinning equipment (FES-COE, Falco Tech Enterprise Co., Ltd., Taiwan) was used for fiber electrospinning. The electrospinning was carried out under an applied electrical voltage of 15 kV onto a metal drum collector (∅ 15 cm) rotated at 300 rpm and covered with aluminum foil, and a tip-to-collector distance of 15 cm was used. The mixture solution was pumped at a flow rate of 1.0 mL/h, pushed by a syringe pump (NE-1000, New Era Pump Systems, Inc., Farmingdale, NY, USA). A 21-gauge needle (inner diameter: 0.52 mm) was selected.
The electrospun fibers were subjected to the following post-treatments. First, the fibers were stabilized in air, from room temperature to 280 °C at a heating rate of 1 °C/min; then, the temperature was held at 280 °C for 2 h to complete the cyclization and dehydrogenation reactions for converting PAN from a thermoplastic to a non-plastic compound [
47,
48,
49]. The stabilized nanofibers were cooled down to room temperature and then carbonized at 800 °C with a heating rate of 5 °C/min, and maintained for 1 h, before the sample was cooled down to room temperature, in a nitrogen atmosphere with a flow of 100 sccm in a tubular furnace. After carbonization, an activation process was conducted by raising the temperature to 850 °C at a rate of 10 °C/min under flowing nitrogen. Then, CO
2 was switched in and held for 1 h, which was used as the activation agent with a flow rate of 100 sccm. The product was denoted as ZIF-8/PAN (or ZDC-850/PAN). For comparison, the pure PAN carbon nanofibers were prepared and labeled as PAN.
2.5. Characterizations
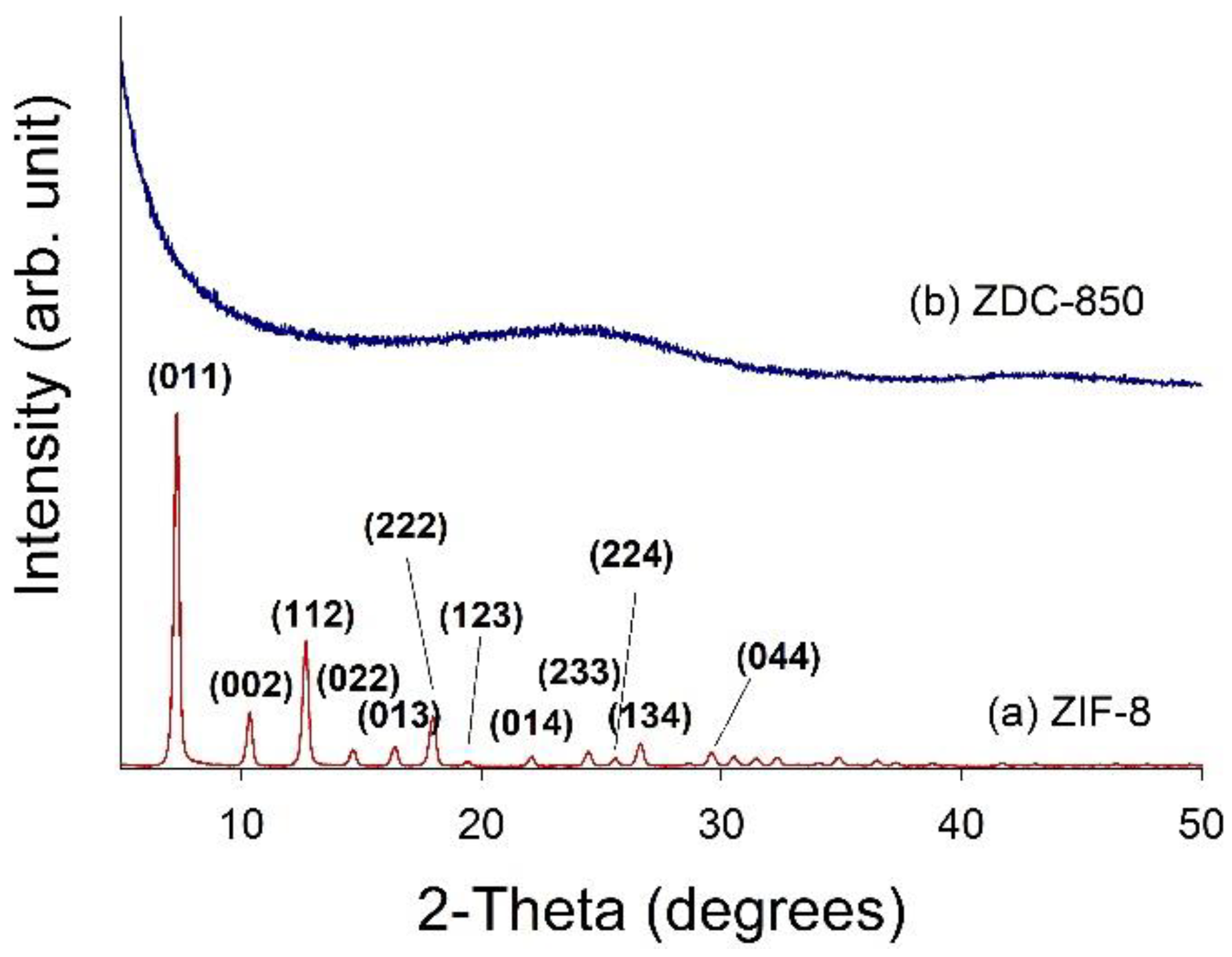
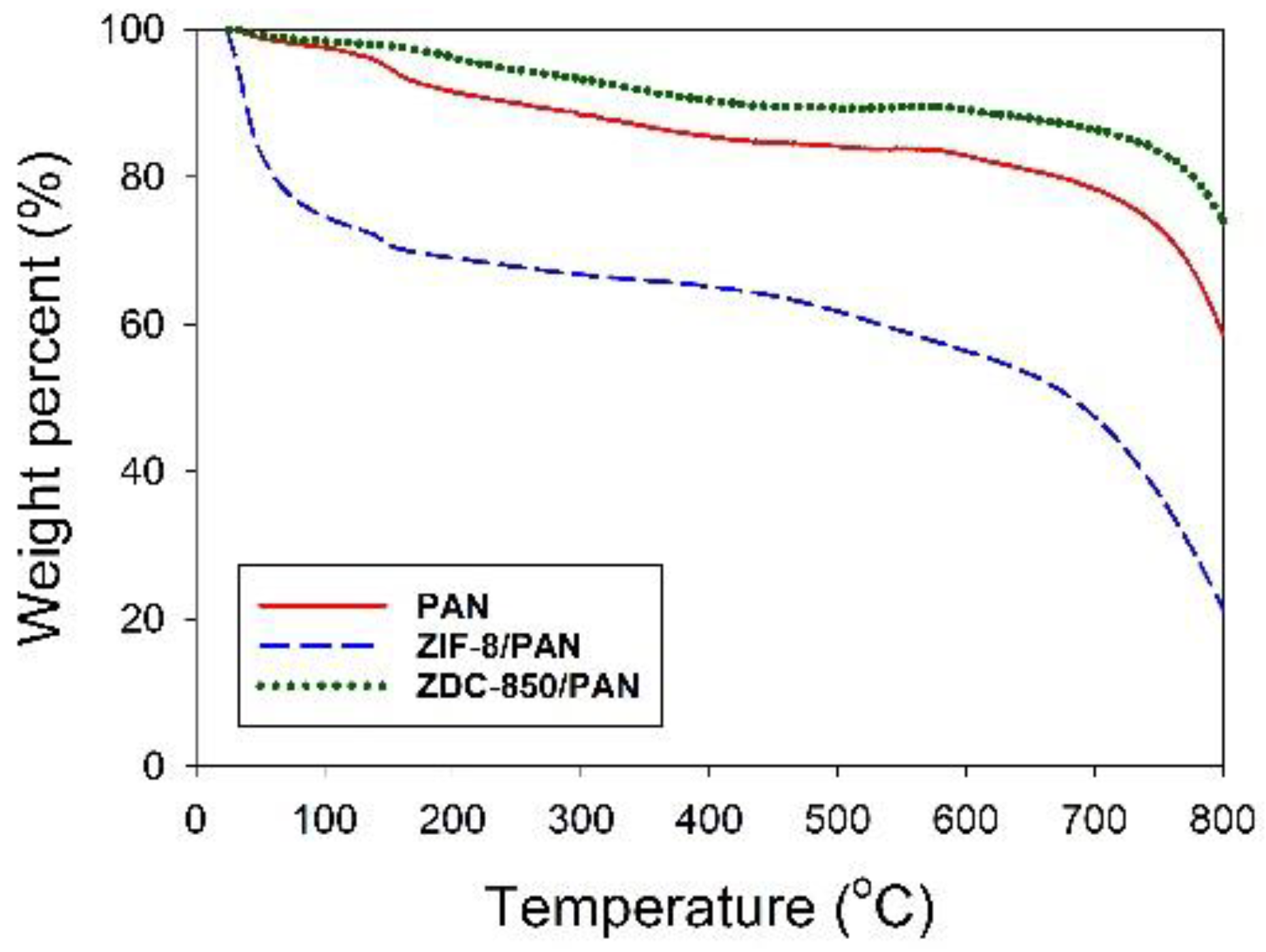
Field emission scanning electron microscopy (FESEM) with a microscope (S-4700, Hitachi, Krefeld, Germany) was utilized to probe the morphology of the samples. High-resolution transmission electron microscopy (HRTEM) images were obtained using a Philips/FEI Tecnai 20 G2 S-Twin Transmission Electron* Microscope (Hillsboro, OR, USA), to observe the morphology of the distribution of Zn metals deposited in the nanofiber composites. X-ray diffraction (XRD) was performed using a powder diffractometer (Rigaku TTRAX III, Tokyo, Japan) equipped with Cu-Kα radiation (λ = 0.15418 nm) at 30 kV and 20 mA. The XRD patterns were collected over 5–90° in 2θ mode at a rate speed of 4°/min. Thermogravimetric analysis (TGA) was used to determine the thermal stability of the samples in flowing nitrogen (60 cm3/min) with a heating rate of 10 °C/min. A thermogravimetric analyzer (Dynamic TGA Q500 in TA Instrument 5100) (TA Instrument, New Castle, DE, USA) was employed to measure any changes in the weight of the sample, as a function of temperature.
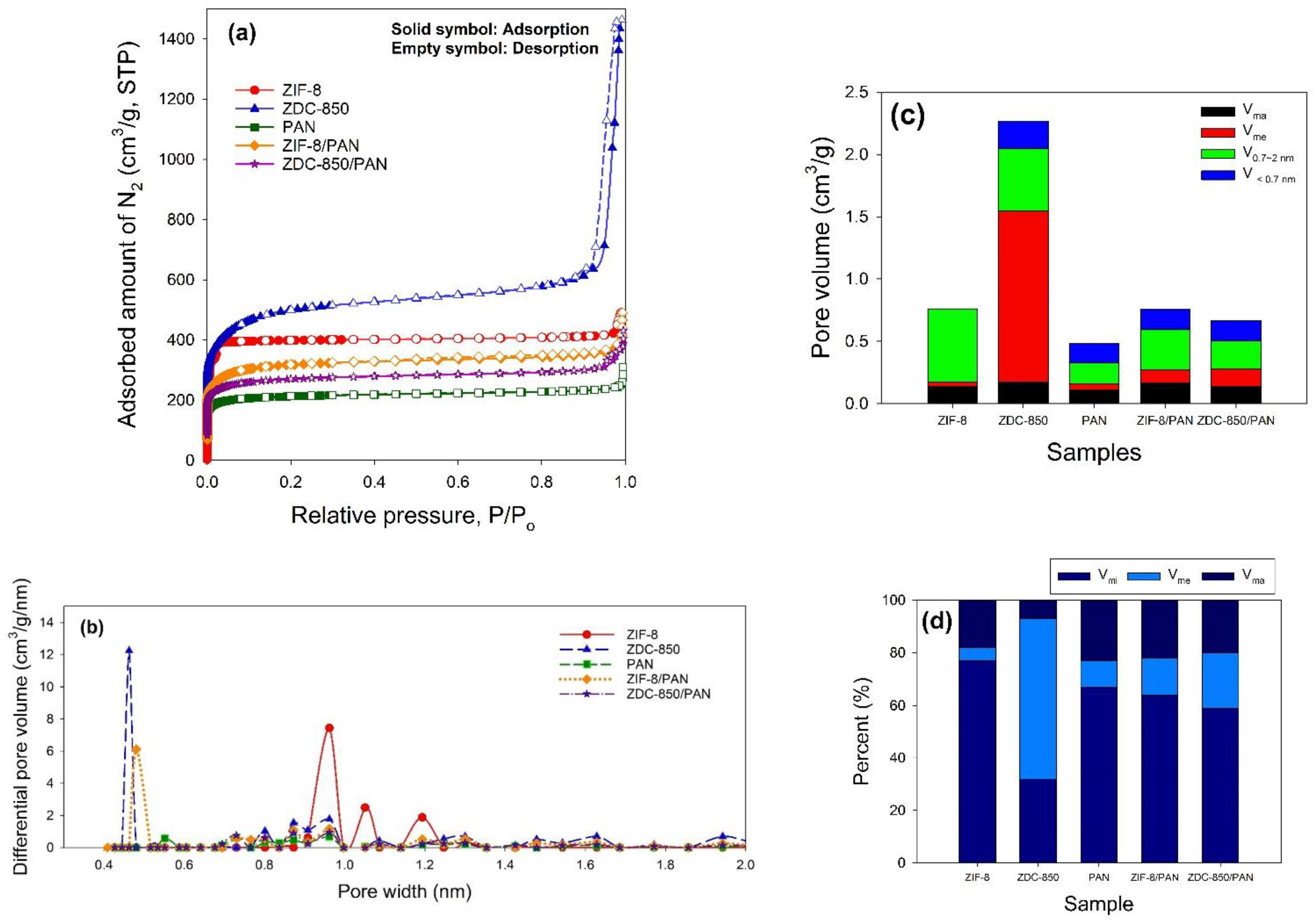
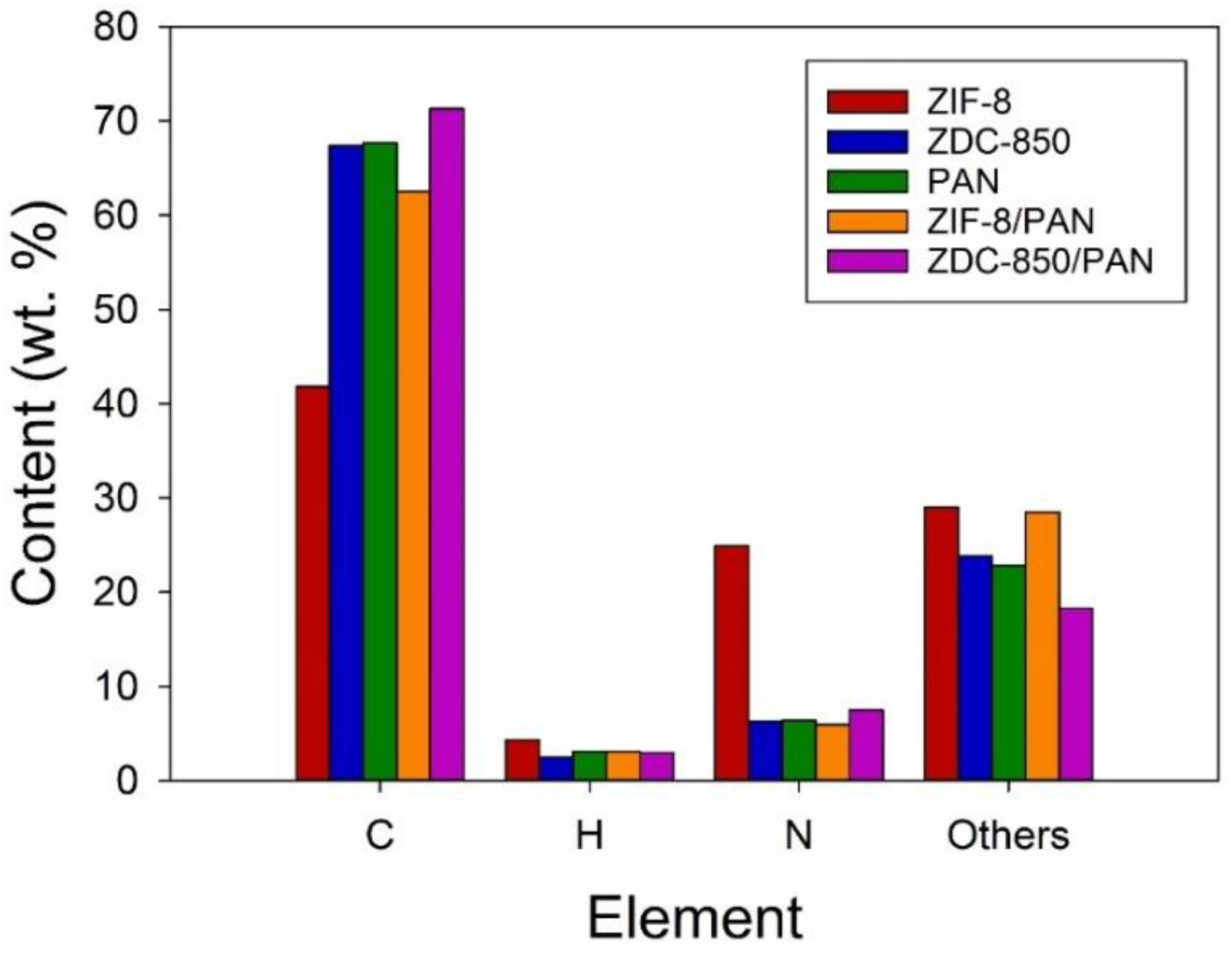
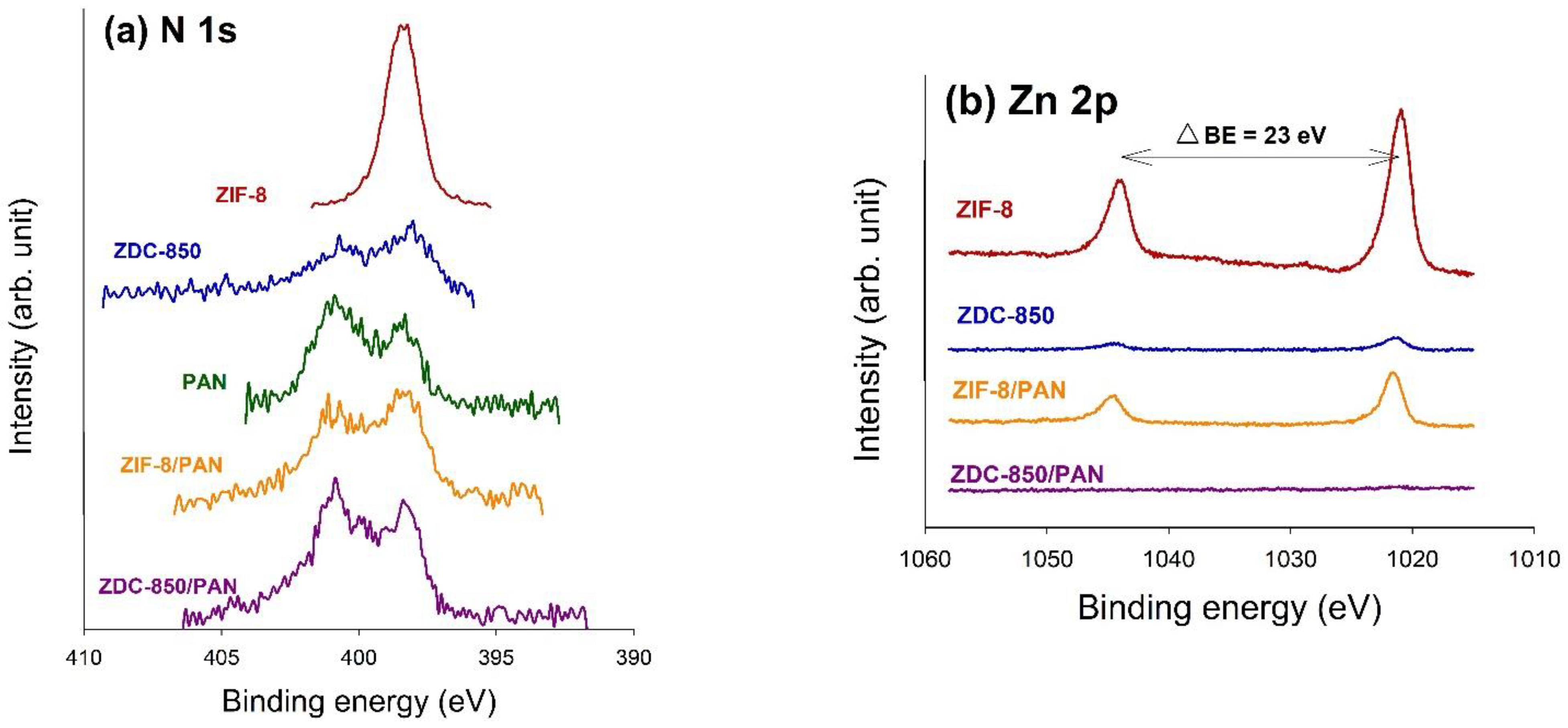
The surface properties of the samples were estimated from N2 adsorption–desorption isotherms measured at −196 °C, using an ASAP 2020 accelerated surface area and porosimetry system (Micromeritics, Norcross, GA, USA). Prior to the adsorption measurements, the samples were outgassed at 350 °C for 24 h. One exception was ZIF-8, which was degassed at 90 °C for 24 h, to prevent changes in the structure. The specific surface areas (SSAs) of the samples were measured in P/Po = 0.05~0.3 using the Brunauer–Emmett–Teller (BET) method. The micropore (<2.0 nm) surface area (Smi) was determined using the t-plot method. The single point total pore volume (Vt) was obtained at P/Po ≈ 0.99. The mesopore volume (Vme), micropore volume (Vmi), and ultra-micropore (<0.7 nm) volume (V<0.7nm) were obtained by applying a non-local density functional theory (NLDFT) model, where V<0.7nm is part of Vmi. Pore size distribution curves were also acquired with an NLDFT model. Elemental analysis (EA) was used to analyze the elemental compositions in the samples, where the weight percent (wt.%) of the C, H, and N elements was measured using an elemental analyzer (Elementar vario EL cube, Langenselbold, Germany). The chemical states on the surface of the samples were determined by X-ray photoelectron spectroscopy (XPS). The XPS spectra were collected using a spectrophotometer (PHI 5000 VersaProbe II, ULVAC-PHI, Kanagawa, Japan), in which a scanning X-ray monochromator (Al Anode, hν = 1401 eV) was used, and information on the elements within a few nanometers of the sample surface could be acquired. For calibration purposes, the C 1s electron binding energy (B.E.) that corresponds to graphitic carbon was set at 285 eV. The deconvolution of the XPS spectra was carried out using a nonlinear least squares curve-fitting program (XPSPEAK software, version 4.1, The Chinese University of Hong Kong, Hong Kong, China).
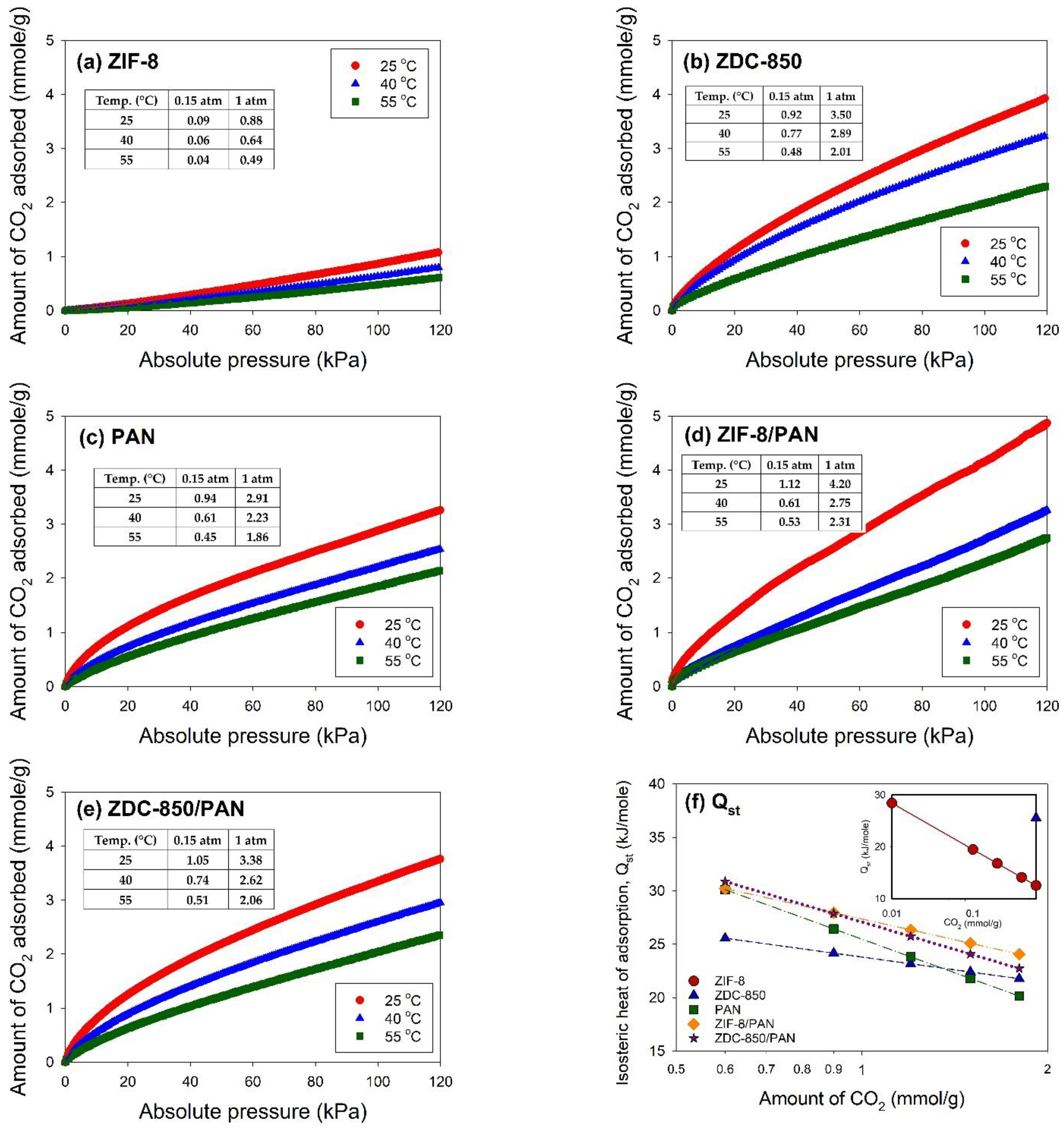
2.6. CO2 Adsorption Experiments
The CO2 adsorption isotherms on the samples were measured using the Micromeritics ASAP 2020 system. The samples were degassed at 350 °C for 24 h for removal of adsorbed contaminants prior to the measurement. For ZIF-8, the degas process was conducted at 90 °C for 24 h. The equilibration time for each pressure point was 45 s. An ISO Controller (Micromeritics), a thermoelectric cooled dewar, was utilized for temperature control during the CO2 adsorption process.
Based on our previous studies [
50,
51], the Freundlich equation [
52] was used for adsorption data fitting. The Freundlich adsorption isotherm (Equation (1)), an empirical model, assumes heterogeneous adsorption, due to the diversity of adsorption active sites:
where
(mmol/g) is the equilibrium adsorption capacity,
[(mmol/g)(1/kPa)
1/n] is the Freundlich adsorption coefficient,
P (kPa) is the gas pressure, and
n is a constant indicating the isotherm curvature.
When adsorbate molecules are adsorbed from the bulk gas phase to the adsorbed phase, the change in enthalpy can be measured by the isosteric heat of adsorption (
) [
52].
denotes the interactions between the gas molecules and the adsorbent lattice atoms and provides a measure of energetic heterogeneity to the gas–solid interfaces [
53]. The Clausius–Clapeyron equation (Equation (2)) was used to determine the values of
:
where
(kJ/mol) is the isosteric heat of adsorption,
R (= 8.314 J/mol/K) is the gas constant,
P (kPa) is the CO
2 pressure, and
T (K) is the adsorption temperature.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}