
Constructing Randomly Lamellar HKUST–1@Clinoptilolite through Polyethylene Glycol—Assisted Hydrothermal Method and Coordinated Complexation for Enhanced Adsorptive Separation for CO2 and CH4

Abstract
:1. Introduction
2. Experimental Methods
2.1. Materials
2.2. Synthesis of CP
2.3. Delamination, Ammonization and Ion Exchange of CP-X
2.4. Synthesis of HKUST-1
2.5. Synthesis of HKUST-1@CP Composites
2.6. Characterizations
2.7. Breakthrough Experiments
3. Results and Discussion
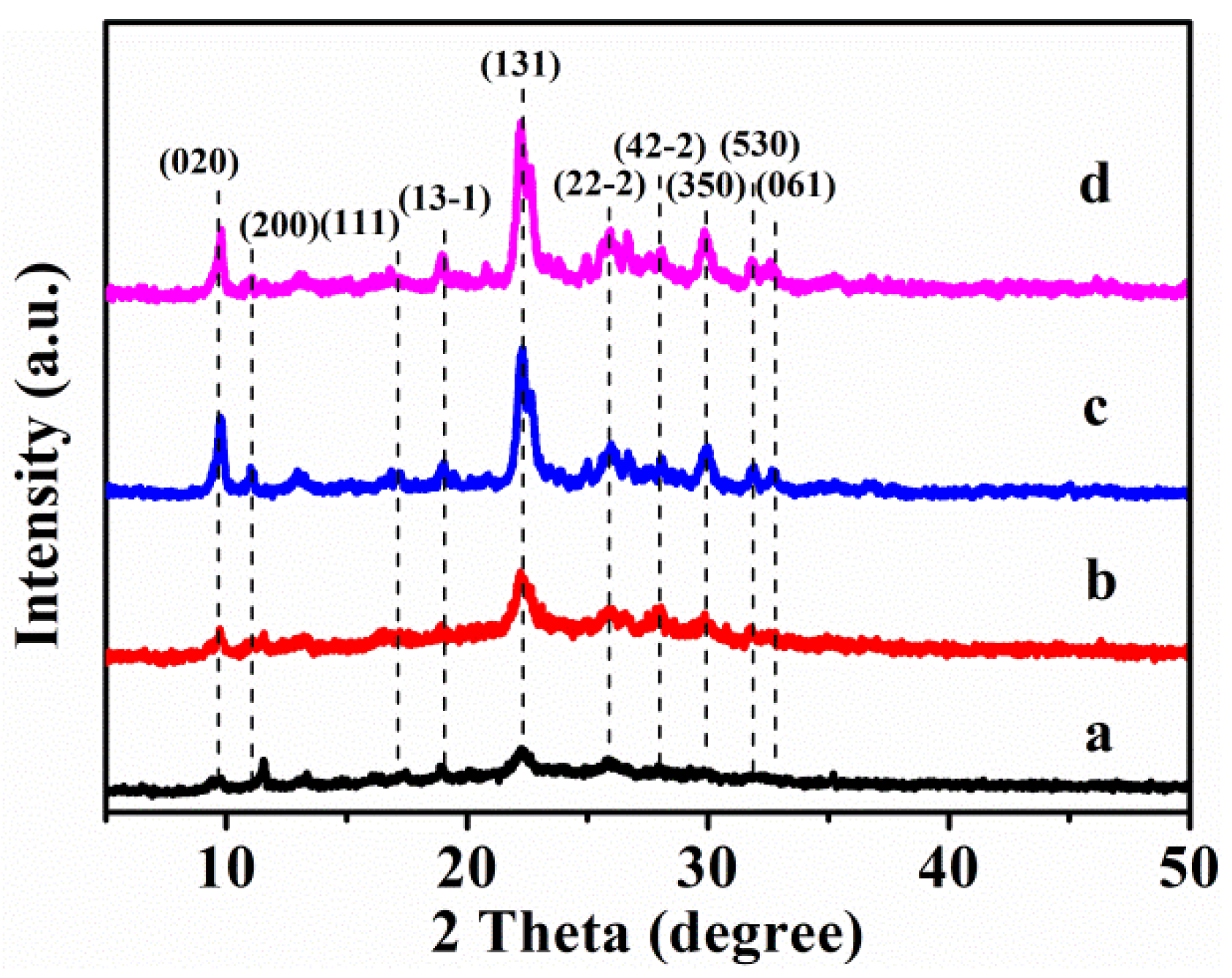
3.1. Structure Characterizations
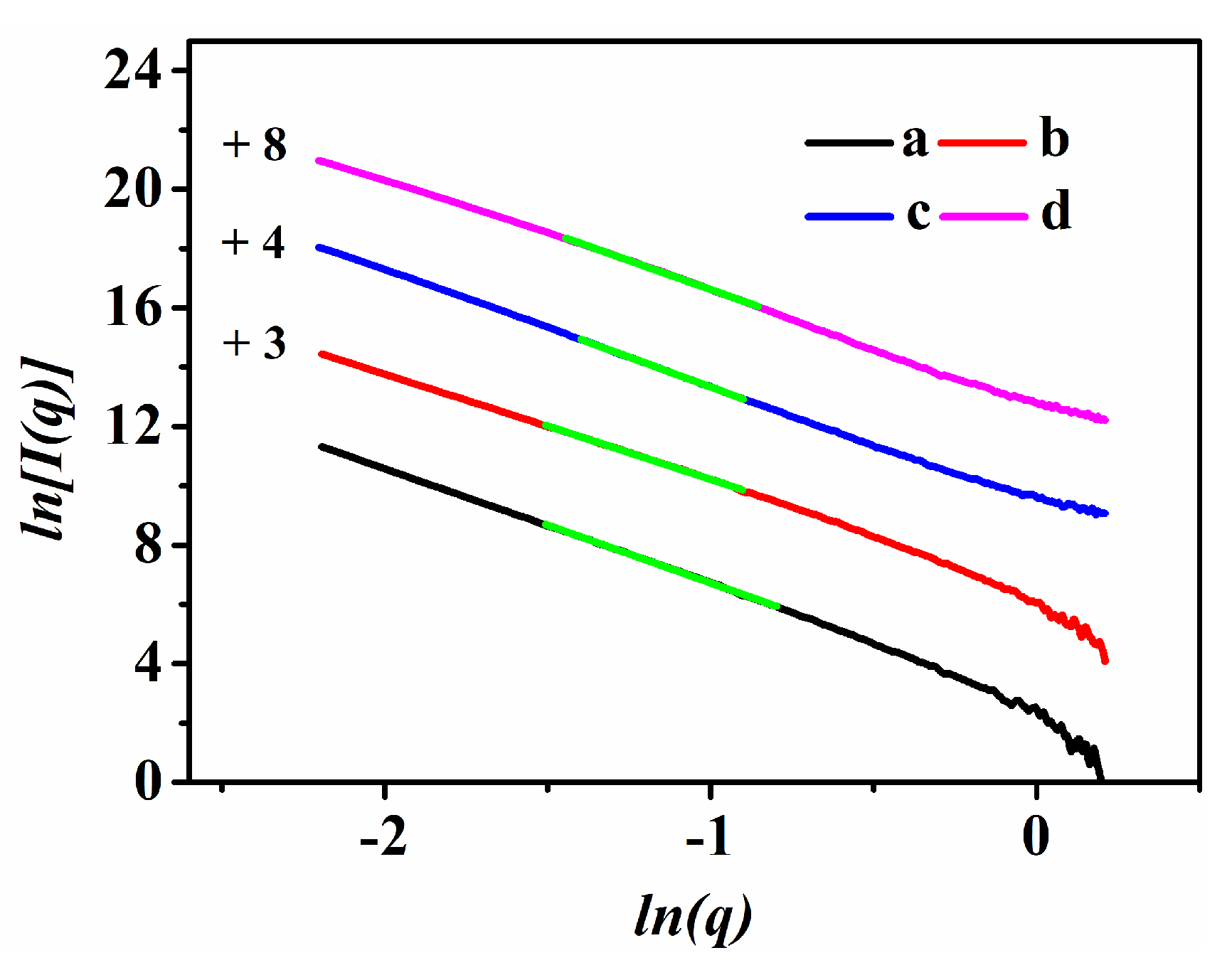
3.2. Crystallization Kinetics of the Synthesized CP with the Additive PEG
3.3. Morphologies and Thermal Analysis
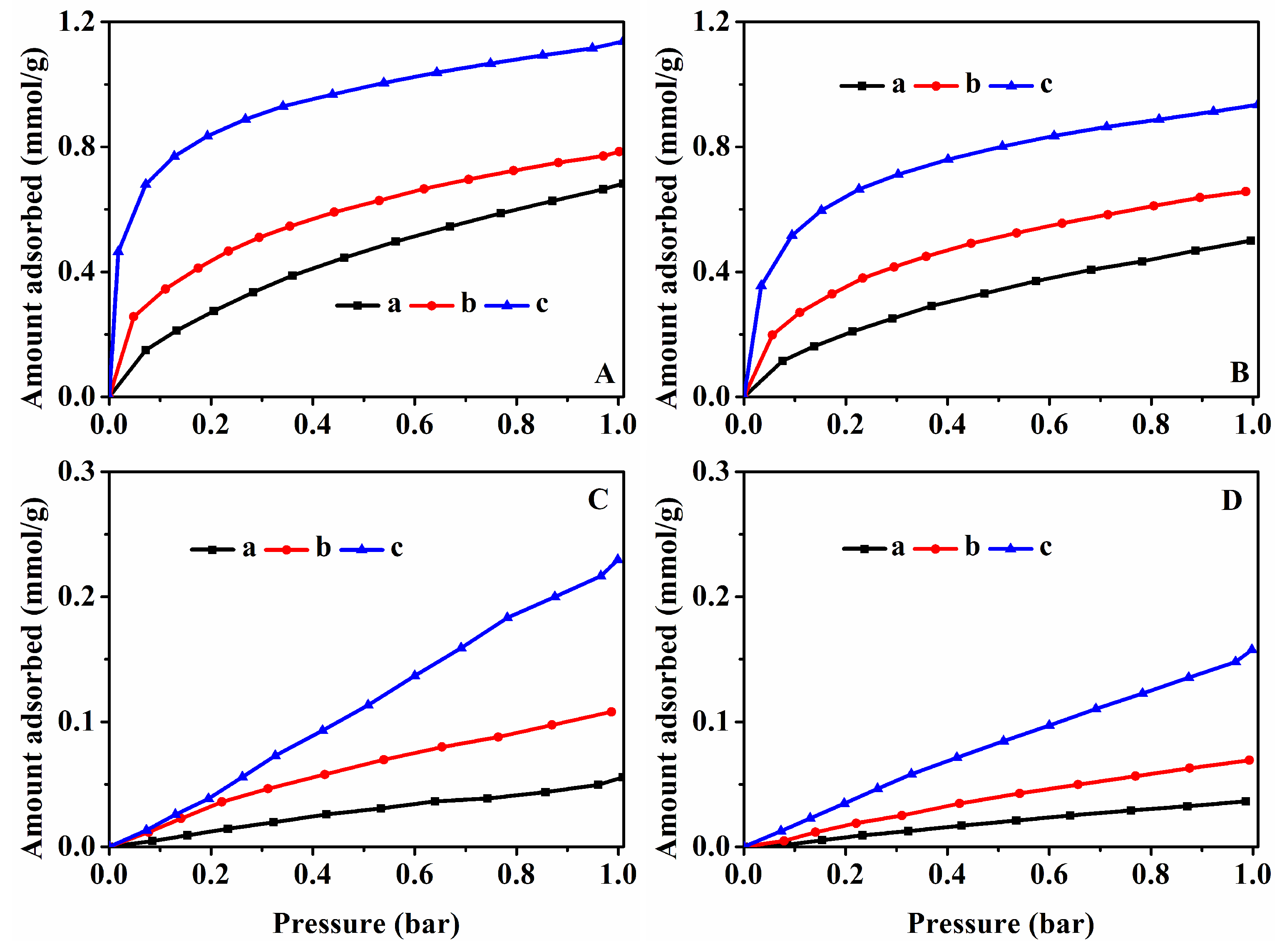
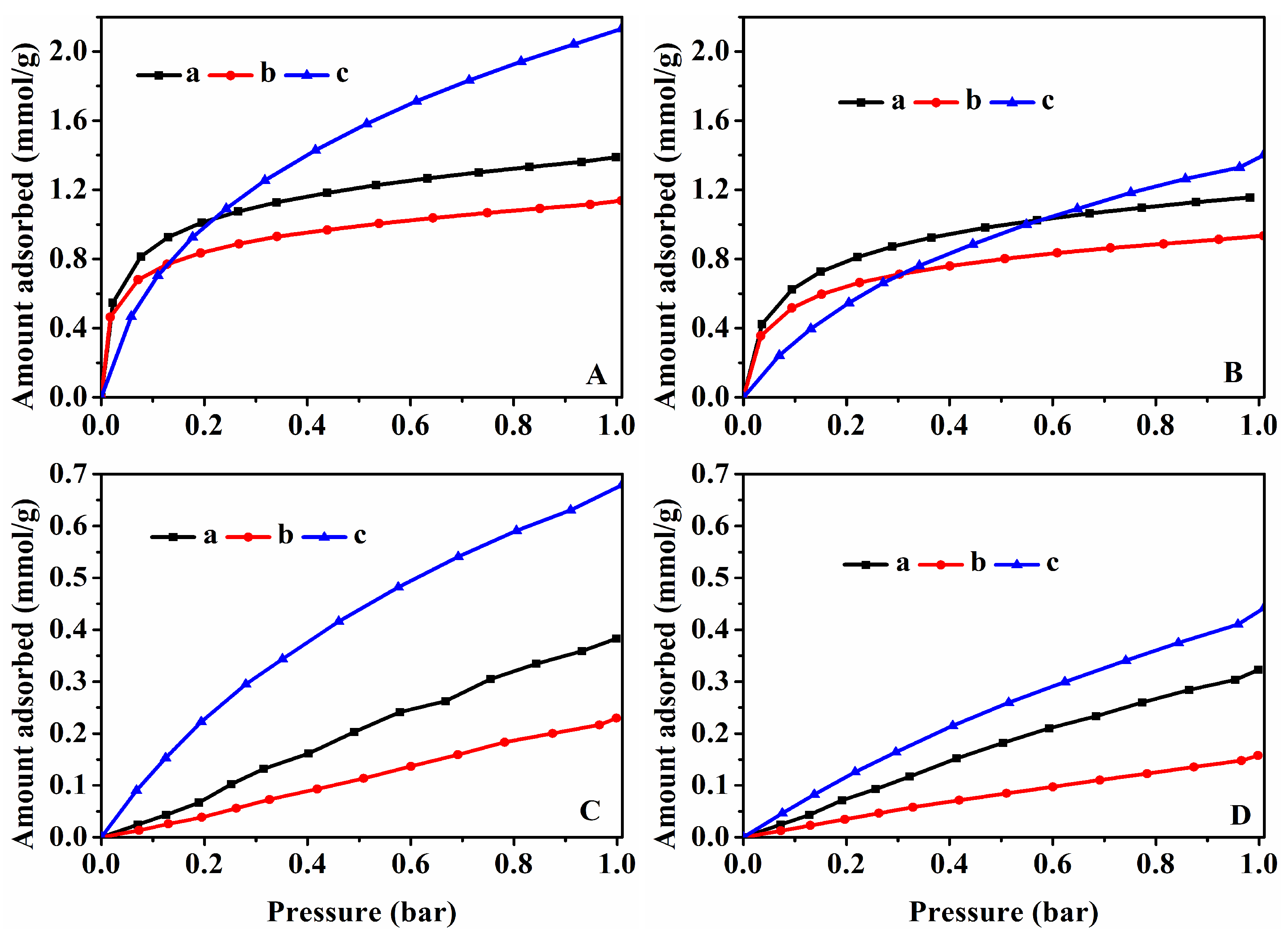
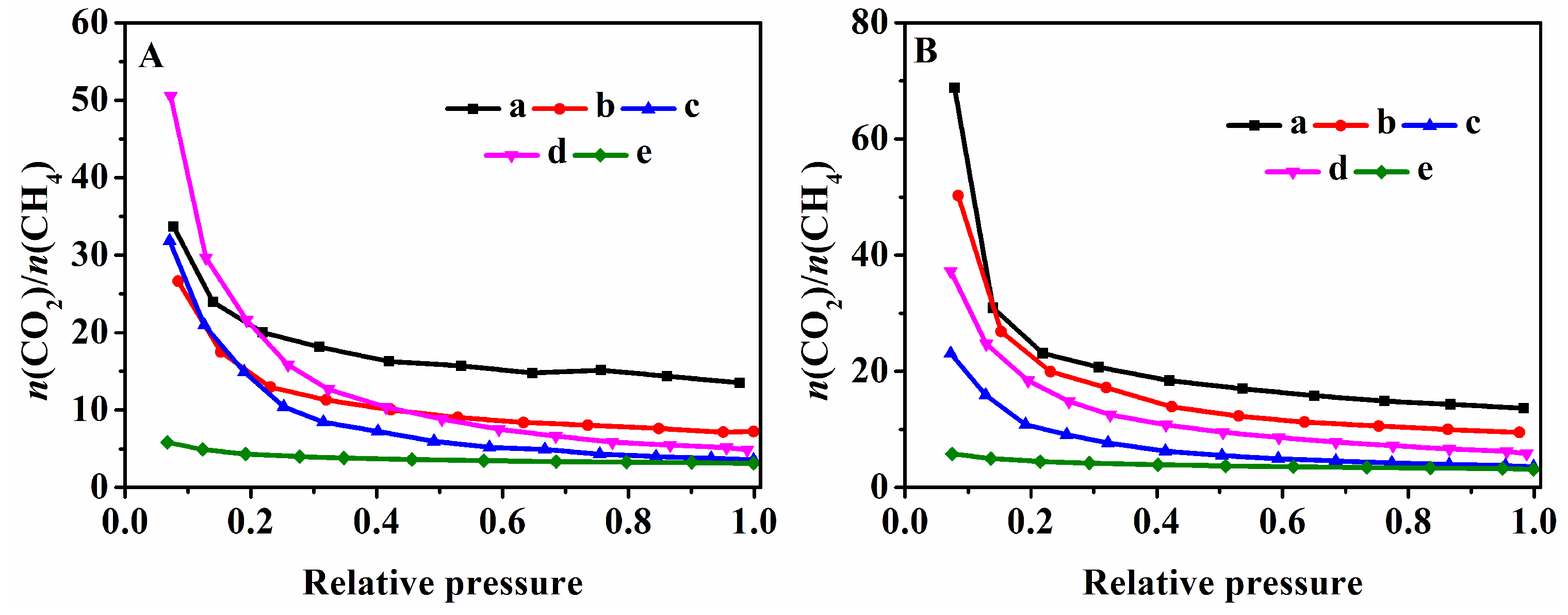
3.4. CO2 and CH4 Adsorption Performances
3.5. Breakthrough Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cakicioglu-Ozkan, F.; Ulku, S. The effect of HCl treatment on water vapor adsorption characteristics of clinoptilolite rich natural zeolite. Microporous Mesoporous Mater. 2005, 77, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Leggom, P.J.; Ledésert, B.A.; Day, J. Organo-zeolitic treatment of mine waste to enhance the growth of vegetation. Eur. J. Miner. 2010, 22, 813–822. [Google Scholar] [CrossRef]
- Zaremotlagh, S.; Hezarkhani, A. Removal of textile dyes from aqueous solution by conducting polymer modified clinoptilolite. Environ. Earth Sci. 2014, 71, 2999–3006. [Google Scholar] [CrossRef]
- Kouvelos, E.; Kesore, K.; Steriotis, T.; Grigoropoulou, H.; Bouloubasi, D.; Theophilou, N.; Tzintzos, S.; Kanelopoulos, N. High pressure N2/CH4 adsorption measurements in clinoptilolites. Microporous Mesoporous Mater. 2007, 99, 106–111. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, F.; Pang, W. Ion exchange of ammonium in natural and synthesized zeolites. J. Hazard. Mater. 2008, 160, 371–375. [Google Scholar] [CrossRef]
- Du, Q.; Liu, S.; Cao, Z.; Wang, Y. Ammonia removal from aqueous solution using natural Chinese clinoptilolite. Sep. Purif. Technol. 2005, 44, 229–234. [Google Scholar] [CrossRef]
- Aguilar-Armenta, G.; Hernandez-Ramirez, G.; Flores-Loyola, E.; Ugarte-Castaneda, A.; Silva-Gonzalez, R.; Tabares-Munoz, C.; Jimenez-Lopez, A.; Rodriguez-Castellon, E. Adsorption kinetics of CO2, O2, N2, and CH4 in cation-exchanged clinoptilolite. J. Phys. Chem. B 2001, 105, 1313–1319. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; Munir, T.; Jia, B.; Gul, A. Various morphologies of clinoptilolites synthesized in alcohol-solvent hydrothermal system and their selective adsorption of CH4 and N2. Microporous Mesoporous Mater. 2021, 323, 111235. [Google Scholar] [CrossRef]
- Jia, B.; Sun, J.; Bing, L.; Bai, S. Nonionic surfactant-assisted strategy towards disorderly layered UiO-66-on-clinoptilolites heterostructure as efficient adsorbent for selective adsorption of CO2 and CH4. Mater. Lett. 2022, 328, 133147. [Google Scholar] [CrossRef]
- Jia, B.; Xu, B.; Bing, L.; Sun, J.; Bai, S. Cationic surfactant-assisted delamination of disorderly layered clinoptilolites for selective adsorption of CO2 and CH4. J. Environ. Chem. Eng. 2022, 10, 108033. [Google Scholar] [CrossRef]
- Iwakai, K.; Tago, T.; Konno, H.; Nakasaka, Y.; Masuda, T. Preparation of nano-crystalline MFI zeolite via hydrothermal synthesis in water/surfactant/organic solvent using fumed silica as the Si source. Microporous Mesoporous Mater. 2011, 141, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.F.; Ferracine, E.D.; Cardoso, D. Effects of different variables on the formation of mesopores in Y zeolite by the action of CTA+ surfactant. Appl. Sci. 2018, 8, 1299. [Google Scholar] [CrossRef] [Green Version]
- Koohsaryan, E.; Anbia, M. Facile and rapid synthesis of highly crystalline mesoporous zeolite FAU. Mater. Lett. 2019, 236, 390–393. [Google Scholar] [CrossRef]
- Corma, A.; Fornés, V.; Pergher, S.B.; Maesen, T.L.M.; Bugless, J.G. Delaminated zeolite precursors as selective acidic catalysts. Nature 1998, 396, 353–356. [Google Scholar] [CrossRef]
- Gorgojo, P.; Galve, A.; Uriel, S.; Téllez, C.; Coronas, J. Direct exfoliation of layered zeolite Nu-6 (1). Microporous Mesoporous Mater. 2011, 142, 122–129. [Google Scholar] [CrossRef]
- Okrut, A.; Grosso-Giordano, N.A.; Schttle, C.; Zones, S.; Katz, A. Understanding the role of Zn2+ in surfactant-free layered silicate delamination: Exfoliation of magadiite. Microporous Mesoporous Mater. 2019, 283, 55–63. [Google Scholar] [CrossRef]
- Roth, W.J. MCM-22 zeolite family and the delaminated zeolite MCM-56 obtained in one-step synthesis. Stud. Surf. Sci. Catal. 2005, 158, 19–26. [Google Scholar]
- Zhang, Z.; Zhao, Y.; Gong, Q.; Zhong, L.; Li, J. MOFs for CO2 capture and separation from flue gas mixtures: The effect of multifunctional sites on their adsorption capacity and selectivity. Chem. Commun. 2013, 49, 653–661. [Google Scholar] [CrossRef]
- Chui, S.S.Y.; Lo, S.M.F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Chen, Y.; Mu, X.; Lester, E.; Wu, T. High efficiency synthesis of HKUST–1 under mild conditions with high BET surface area and CO2 uptake capacity. Prog. Nat. Sci. Mater. Int. 2018, 28, 584–589. [Google Scholar] [CrossRef]
- Al-Naddaf, Q.; Thakkar, H.; Rezaei, F. Novel zeolite-5A@MOF–74 composite adsorbents with core-shell structure for H2 purification. ACS Appl. Mater. Interfaces 2018, 10, 29656–29666. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, F.; Amini, M.M.; Sillanpää, M. Hydrothermal synthesis of novel MIL–100(Fe)@SBA–15 composite material with high adsorption efficiency towards dye pollutants for wastewater remediation. J. Taiwan Inst. Chem. Eng. 2020, 116, 303–313. [Google Scholar] [CrossRef]
- Tari, N.E.; Tadjarodi, A.; Tamnanloo, J.; Fatemi, S. Synthesis and property modification of MCM-41 composited with Cu(BDC) MOF for improvement of CO2 adsorption Selectivity. J. CO2 Util. 2016, 14, 126–134. [Google Scholar] [CrossRef]
- Mu, X.; Chen, Y.; Lester, E.; Wu, T. Optimized synthesis of nano-scale high quality HKUST-1 under mild conditions and its application in CO2 capture. Microporous Mesoporous Mater. 2018, 270, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Karimzadeha, R.; Moosavib, E.S. Modified and ion exchanged clinoptilolite for the adsorptive removal of sulfur compounds in a model fuel: New adsorbents for desulfurization. Fuel 2018, 217, 467–477. [Google Scholar] [CrossRef]
- Li, Z. A program for SAXS data processing and analysis. Chin. Phys. C 2013, 37, 108002. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Lach-hab, M.; Blaisten-Barojas, E.; Li, X.; Karen, V.L. Machine learning study of the heulandite family of zeolites. Microporous Mesoporous Mater. 2010, 130, 309–313. [Google Scholar] [CrossRef]
- Uzcátegui, D.; González, G. Study of the kinetics of crystallization of zeolite MEL. Catal. Today 2005, 107, 901–905. [Google Scholar] [CrossRef]
- Corma, A.; Fornés, V.; Martínez-Triguero, J.; Pergher, S.B. Delaminated zeolites: Combining the benefits of zeolites and mesoporous materials for catalytic uses. J. Catal. 1999, 186, 57–63. [Google Scholar] [CrossRef]
- Alvarez-Puebla, R.A.; Aisa, C.; Blasco, J.; Echeverría, J.C.; Mosquera, B.; Garrido, J.J. Copper heterogeneous nucleation on a palygorskitic clay: An XRD, EXAFS and molecular modeling study. Appl. Clay Sci. 2004, 25, 103–110. [Google Scholar] [CrossRef]
- Zheng, X.G.; Xu, C.N. Antiferromagnetic transition in botallackite Cu2Cl(OH)3. Solid State Commun. 2004, 131, 509–511. [Google Scholar] [CrossRef]
- Lin, K.S.; Adhikari, A.K.; Ku, C.N.; Chiang, C.L.; Kuo, H. Synthesis and characterization of porous HKUST-1 metal organic frameworks for hydrogen storage. Int. J. Hydrogen Energy 2012, 37, 13865–13871. [Google Scholar] [CrossRef]
- Moor, P.E.A.D.; Beelen, T.P.M.; Santen, R.A.V.; Beck, L.W.; Davis, M.E. Si-MFI crystallization using a “Dimer” and “Trimer” of TPA studied with small-angle X-ray scattering. J. Phys. Chem. B 2000, 104, 7600–7611. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Crystallogr. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Boukari, H.; Lin, J.S.; Harris, M.T. Small-angle X-ray scattering study of the formation of colloidal silica particles from alkoxides: Primary particles or not? J. Colloid Interf. Sci. 1997, 194, 311–318. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, R.; Zhang, H.; Shang, Y.; Song, Y.; Liu, C.; Wang, T.; Gong, Y.; Li, Z. Structure evolution of aluminosilicate sol and its structure-directing effect on the synthesis of NaY zeolite. J. Appl. Crystallogr. 2017, 50, 231–239. [Google Scholar] [CrossRef]
- Radlinski, A.P.; Mastalerz, M.; Hinde, A.L.; Hainbuchner, M.; Rauch, H.; Baron, M.; Lin, J.S.; Fan, L.; Thiyagarajan, P. Application of SAXS and SANS in evaluation of porosity, pore size distribution and surface area of coal. Int. J. Coal Geol. 2004, 59, 245–271. [Google Scholar] [CrossRef]
- Svergun, D.I.; Koch, M.H.J. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 2003, 66, 1735. [Google Scholar] [CrossRef]
- Xu, Y.; Koga, Y.; Strausz, O.P. Characterization of Athabasca asphaltenes by small-angle X-ray scattering. Fuel 1995, 74, 960–964. [Google Scholar] [CrossRef]
- Corral-Capulin, N.G.; Vilchis-Nestor, A.R.; Gutiérrez-Segura, E.; Solache Ríos, M. The influence of chemical and thermal treatments on the fluoride removal from water by three mineral structures and their characterization. J. Fluor. Chem. 2018, 213, 42–50. [Google Scholar] [CrossRef]
- Rakitskaya, T.L.; Kiose, T.A.; Golubchik, K.O.; Ennan, A.A.; Volkova, V.Y. Acid-modified clinoptilolite as a support for palladium-copper complexes catalyzing carbon monoxide oxidation with air oxygen. Chem. Cent. J. 2017, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Basabe, Y.; Rodriguez-Iznaga, I.; de Menorval, L.; Llewellyn, P.; Maurin, G.; Lewis, D.W.; Binions, R.; Autie, M.; Ruiz-Salvador, A.R. Step-wise dealumination of natural clinoptilolite: Structural and physicochemical characterization. Microporous Mesoporous Mater. 2010, 135, 187–196. [Google Scholar] [CrossRef]
- Maia, R.A.; Louis, B.; Baudron, S.A. HKUST-1 MOF in reline deep eutectic solvent: Synthesis and phase transformation. Dalton T. 2021, 50, 4145–4151. [Google Scholar] [CrossRef]
- Lin, S.; Song, Z.; Che, G.; Ren, A.; Li, P.; Liu, C.; Zhang, J. Adsorption behavior of metal-organic frameworks for methylene blue from aqueous solution. Microporous Mesoporous Mater. 2014, 193, 27–34. [Google Scholar] [CrossRef]
- Hunger, B.; Miessner, H.; Szombathely, M.; Geidel, E. Heterogeneity of Si-OH-Al groups in HNaY zeolites. J. Chem. Soc. Faraday Trans. 1996, 92, 499–504. [Google Scholar] [CrossRef]
- Kaneko, K. Determination of pore size and pore size distribution: 1. Adsorbents and catalysts. J. Membr. Sci. 1994, 96, 59–89. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Majchrzak-Kucęba, I.; Bukalak-Gaik, D. Regeneration performance of metal-organic frameworks. J. Therm. Anal. Calorim. 2016, 125, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, J.; Ahn, W. Synthesis of metal-organic frameworks: A mini review. Korean J. Chem. Eng. 2013, 30, 1667–1680. [Google Scholar] [CrossRef]
- Klimakow, M.; Klobes, P.; Rademann, K.; Emmerling, F. Characterization of mechanochemically synthesized MOFs. Microporous Mesoporous Mater. 2012, 154, 113–118. [Google Scholar] [CrossRef]
- Omri, A.; Lamber, S.D.; Geen, J.; Bennour, F.; Benzina, M. Synthesis, surface characterization and photocatalytic activity of TiO2 supported on almond shell activated carbon. J. Mater. Sci. Technol. 2014, 30, 894–902. [Google Scholar] [CrossRef]
- Mendoza, H.R.; Jordens, J.; Pereira, M.V.L.; Lutz, C.; Gerven, T.V. Effects of ultrasonic irradiation on crystallization kinetics, morphological and structural properties of zeolite FAU. Ultrason. Sonochem. 2020, 64, 105010. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.L.G.; Renzo, F.D.; Fajula, F.; Bousquet, J. Crystallization kinetics of zeolite omega, the synthetic analog of mazzite. Microporous Mesoporous Mater. 2006, 90, 221–228. [Google Scholar] [CrossRef]
- Jiao, J.; Sun, J.; Ullah, R.; Bai, S.; Zhai, C. One-step synthesis of hydrophobic clinoptilolite modified by silanization for the degradation of crystal violet dye in aqueous solution. RSC Adv. 2020, 10, 22809–22818. [Google Scholar] [CrossRef]
- Shalmani, F.M.; Halladj, R.; Askari, S. An investigation of the crystallization kinetics of zeotype SAPO-34 crystals synthesized by hydrothermal and sonochemical methods. Ultrason. Sonochem. 2016, 29, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Amin, G.M.; Esmail, A. Application of nano silica to improve self-healing of asphalt mixes. J. Cent. South Univ. 2017, 24, 1019–1026. [Google Scholar] [CrossRef]
- Majano, G.; Pérez-Ramírez, J. Room Temperature Synthesis and Size Control of HKUST-1. Helv. Chim. Acta 2012, 95, 2278–2286. [Google Scholar] [CrossRef]
- Wu, Y.; Yuan, D.; He, D.; Xing, J.; Liu, Z. Decorated traditional zeolites with subunits of metal-organic frameworks for CH4/N2 separation. Angew. Chem. Int. Edit. 2019, 58, 10241–10244. [Google Scholar] [CrossRef]
- Lestari, W.W.; Adreane, M.; Purnawan, C.; Fansuri, H.; Widiastuti, N.; Rahardjo, S.B. Solvothermal and electrochemical synthetic method of HKUST-1 and its methane storage capacity. IOP Conf. Ser. Mater. Sci. Eng. 2016, 107, 012030. [Google Scholar] [CrossRef] [Green Version]
- Schlichte, K.; Kratzke, T.; Kaskel, S. Improved synthesis, thermal stability and catalytic properties of the metal-organic framework compound Cu3(BTC)2. Microporous Mesoporous Mater. 2004, 73, 81–88. [Google Scholar] [CrossRef]
- Yörükoğulları, E.; Yılmaz, G.; Dikmen, S. Thermal treatment of zeolitic tuff. J. Therm. Anal. Calorim. 2010, 100, 925–928. [Google Scholar] [CrossRef]
- Hao, X.; Hu, H.; Li, Z.; Wu, L.; Liu, X.; Zhang, Y. Adsorption Properties of Modified Clinoptilolite for Methane and Nitrogen. Materials 2018, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Xin, C.; Zhan, H.; Huang, X.; Li, H.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. Effect of various alkaline agents on the size and morphology of nano-sized HKUST-1 for CO2 adsorption. RSC Adv. 2015, 5, 27901–27911. [Google Scholar] [CrossRef]
- Spanopoulos, I.; Tsangarakis, C.; Klontzas, E.; Tylianakis, E.; Froudakis, G.; Adil, K.; Belmabkhout, Y.; Eddaoudi, M.; Trikalitis, P.N. Reticular synthesis of HKUST-like tbo MOFs with enhanced CH4 storage. J. Am. Chem. Soc. 2016, 138, 1568–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, K.; Takéuchi, Y. Clinoptilolite: The distribution of potassium atoms and its role in thermal stability. Z. Krist-Cryst. Mater. 1977, 145, 216–239. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Shelyapina, M.G.; Petranovskii, V. Ion Exchange in Natural Clinoptilolite: Aspects Related to Its Structure and Applications. Minerals 2022, 12, 1628. [Google Scholar] [CrossRef]
- Iznaga, I.R.; Petranovskii, V.; Fuentes, G.R.; Mendoza, C.; Aguilar, A.B. Exchange and reduction of Cu2+ ions in clinoptilolitem. J. Colloid Interf. Sci. 2007, 316, 877–886. [Google Scholar] [CrossRef]
- Garcia-Basabe, Y.; Ruiz-Salvador, A.R.; Maurin, G.; Menorval, L.; Rodriguez-Iznaga, I.; Gomez, A. Location of extra-framework Co2+, Ni2+, Cu2+ and Zn2+ cations in natural and dealuminated clinoptilolite. Microporous Mesoporous Mater. 2012, 155, 233–239. [Google Scholar] [CrossRef]
- Park, J.M.; Woo, H.C.; Jhung, S.H. Effective CO2 adsorption at low pressure over nitrogen-enriched porous carbons, derived from melamine-loaded polyaniline. Chem. Eng. J. 2021, 412, 128641. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Mujčin, M.; Abou-Zied, C.; Tezel, F.H. Cation exchange modification of clinoptilolite-thermodynamic effects on adsorption separations of carbon dioxide, methane, and nitrogen. Microporous Mesoporous Mater. 2019, 274, 327–341. [Google Scholar] [CrossRef]
- Zhang, K.; Meng, Z.; Liu, S. Comparisons of methane adsorption/desorption, diffusion behaviors on intact coals and deformed coals: Based on experimental analysis and isosteric heat of adsorption. Energy Fuels 2021, 35, 5975–5987. [Google Scholar] [CrossRef]
- Yan, X.; Komarneni, S.; Zhang, Z.; Yan, Z. Extremely enhanced CO2 uptake by HKUST-1 metal-organic framework via a simple chemical treatment. Microporous Mesoporous Mater. 2014, 183, 69–73. [Google Scholar] [CrossRef]
- Wu, X.; Liu, J.; He, T.; Zhang, P.; Yu, J.; Li, J. Understanding how pore surface fluorination influences light hydrocarbon separation in metal-organic frameworks. Chem. Eng. J. 2021, 407, 127183. [Google Scholar] [CrossRef]
- Álvarez-Gutiérrez, N.; Gil, M.V.; Rubiera, F.; Pevida, C. Kinetics of CO2 adsorption on cherry stone-based carbons in CO2/CH4 separations. Chem. Eng. J. 2017, 307, 249–257. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Temperature | CO2/CH4 Selectivity |
|---|---|---|
| HKUST−1@CP−0−E−10.5 | 273 K 298 K | 13.51 13.65 |
| HKUST−1@CP−24−E−10.1 | 273 K 298 K | 7.23 9.48 |
| HKUST−1@CP−72−E−3.4 | 273 K 298 K | 3.58 3.57 |
| HKUST−1@CP−72−E−7.3 | 273 K 298 K | 4.89 5.87 |
| HKUST−1 | 273 K 298 K | 3.13 3.12 |
| HKUST−1 1 cited in reference [72] | 273 K | 7.19 |
| HKUST−1 2 cited in references [24,59] | 273 K | 4.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Zhou, J.; Wan, C.; Liu, M.; Wu, X.; Sun, J. Constructing Randomly Lamellar HKUST–1@Clinoptilolite through Polyethylene Glycol—Assisted Hydrothermal Method and Coordinated Complexation for Enhanced Adsorptive Separation for CO2 and CH4. Nanomaterials 2023, 13, 1860. https://doi.org/10.3390/nano13121860
Zhang M, Zhou J, Wan C, Liu M, Wu X, Sun J. Constructing Randomly Lamellar HKUST–1@Clinoptilolite through Polyethylene Glycol—Assisted Hydrothermal Method and Coordinated Complexation for Enhanced Adsorptive Separation for CO2 and CH4. Nanomaterials. 2023; 13(12):1860. https://doi.org/10.3390/nano13121860
Chicago/Turabian StyleZhang, Mingxuan, Jiawei Zhou, Chunlei Wan, Ming Liu, Xia Wu, and Jihong Sun. 2023. "Constructing Randomly Lamellar HKUST–1@Clinoptilolite through Polyethylene Glycol—Assisted Hydrothermal Method and Coordinated Complexation for Enhanced Adsorptive Separation for CO2 and CH4" Nanomaterials 13, no. 12: 1860. https://doi.org/10.3390/nano13121860