
First-Principles Study of χ3-Borophene as a Candidate for Gas Sensing and the Removal of Harmful Gases

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
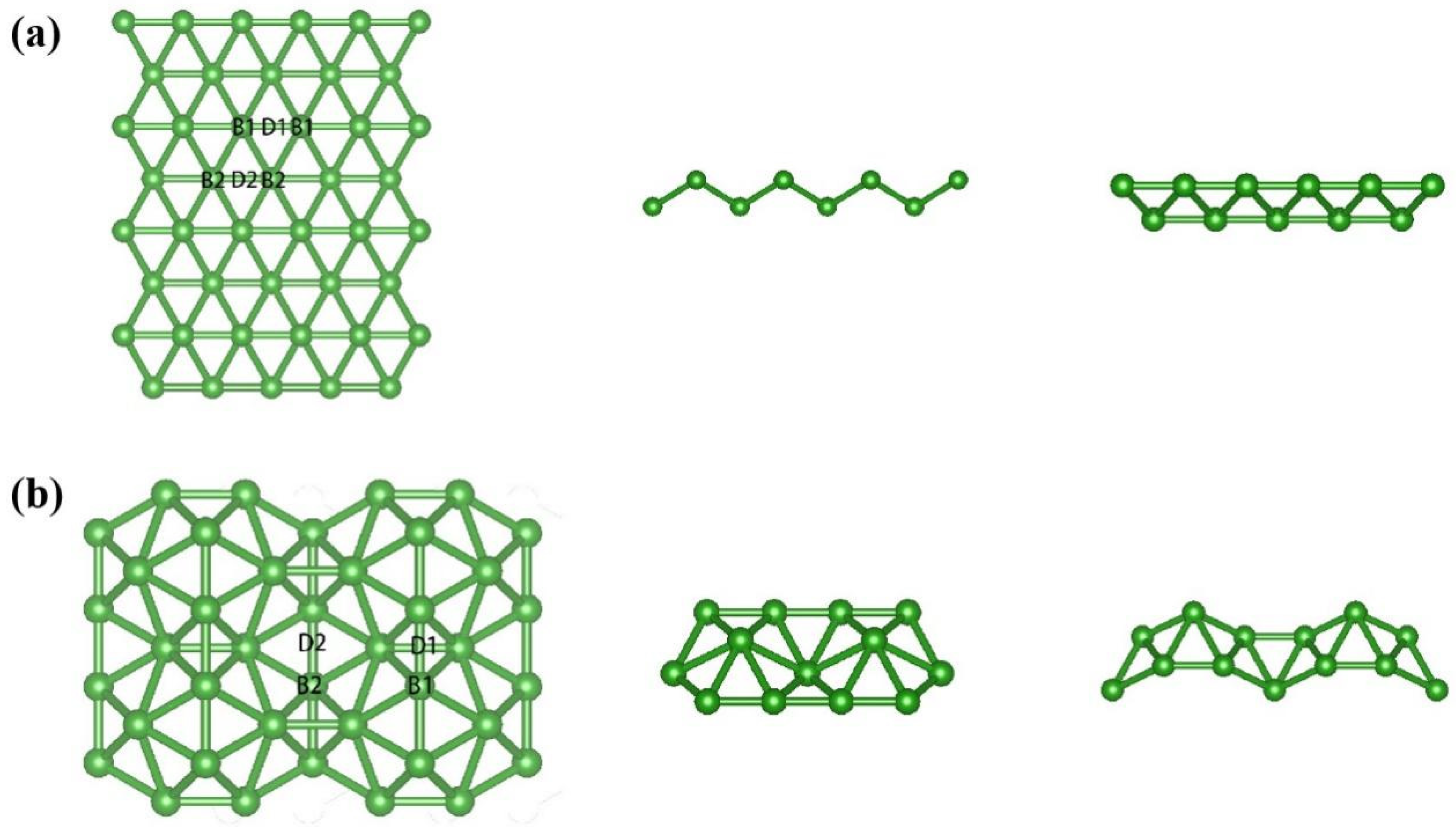
3.1. Geometric and Electronic Structures of Pristine χ3-Borophene
3.2. Adsorption of Gases on Pristine χ3-Borophene
3.2.1. Analysis of the Overall Trend of Gas Adsorption for Five Gases
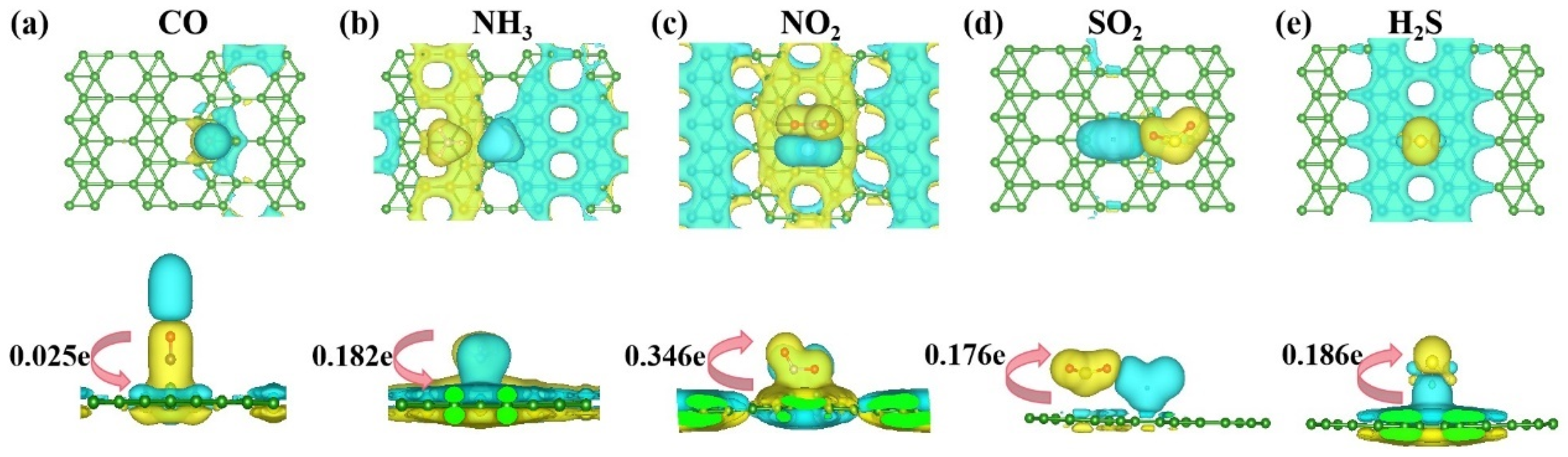
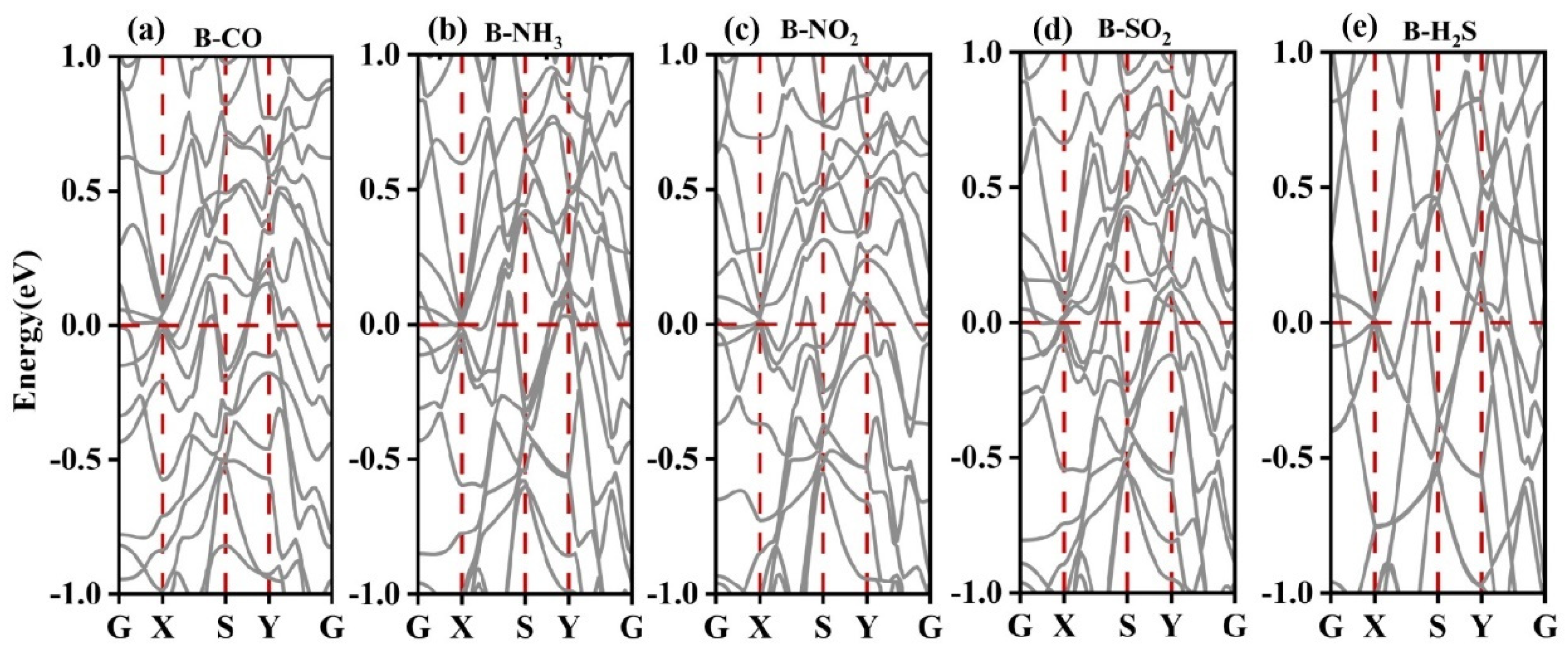
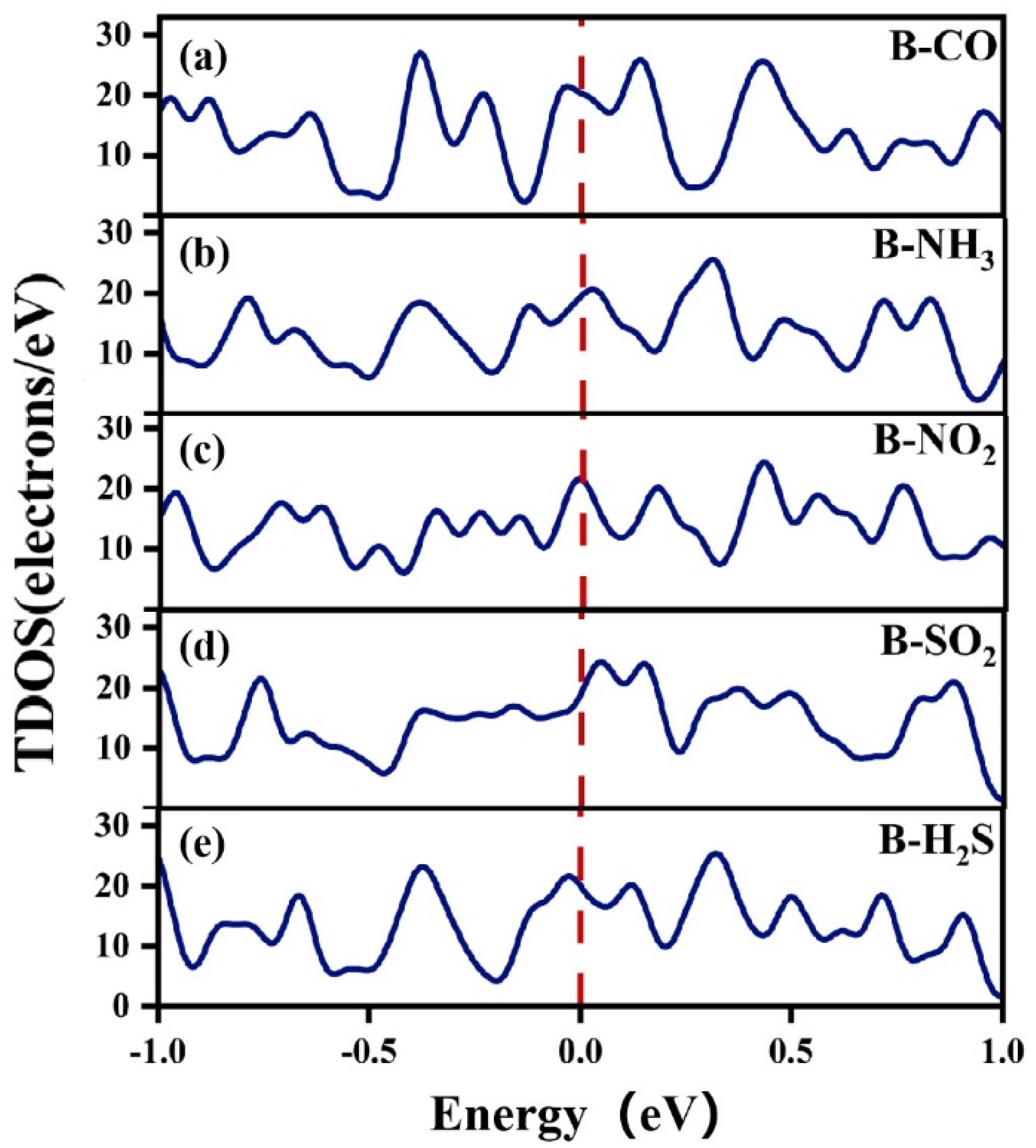
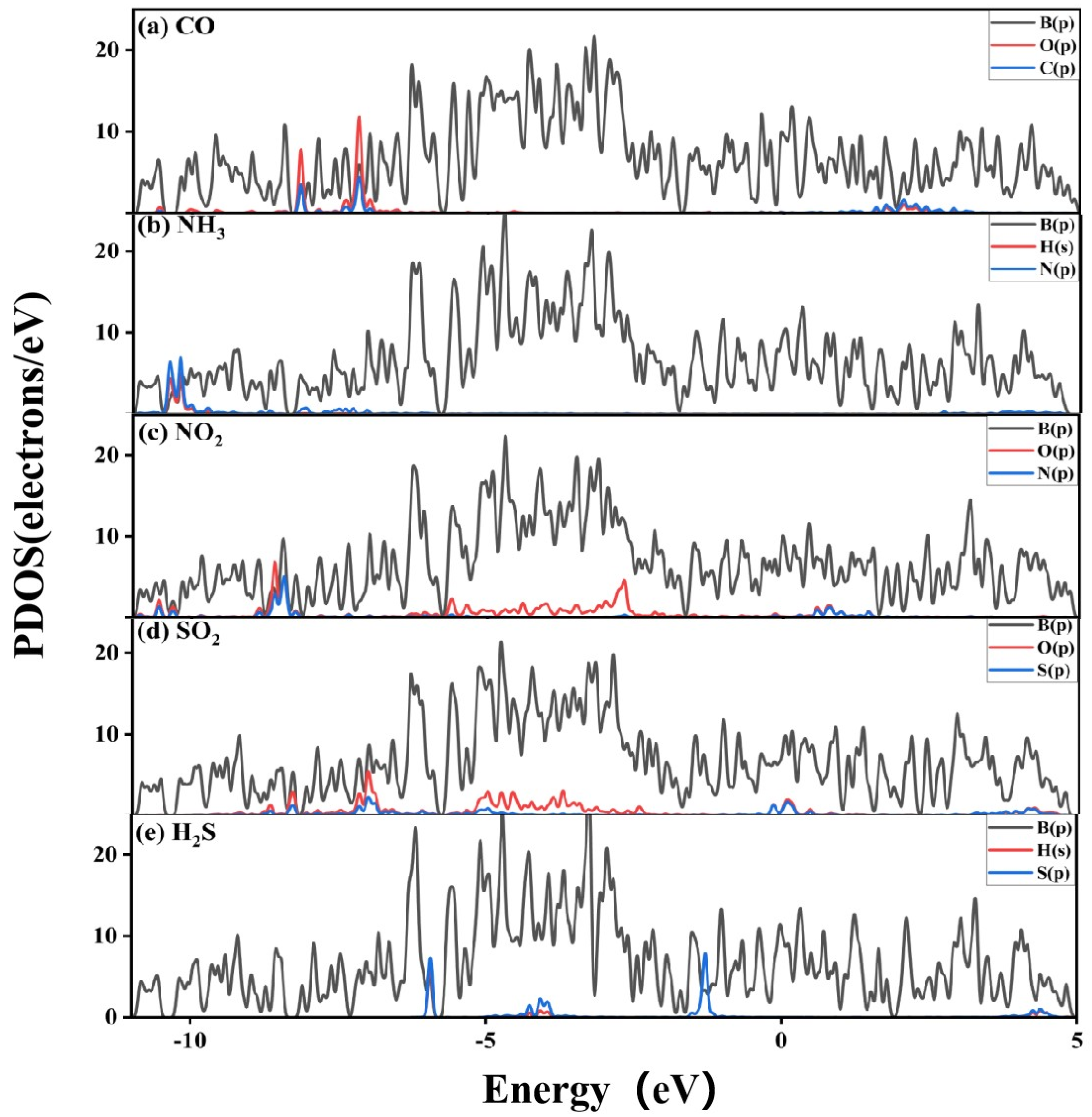
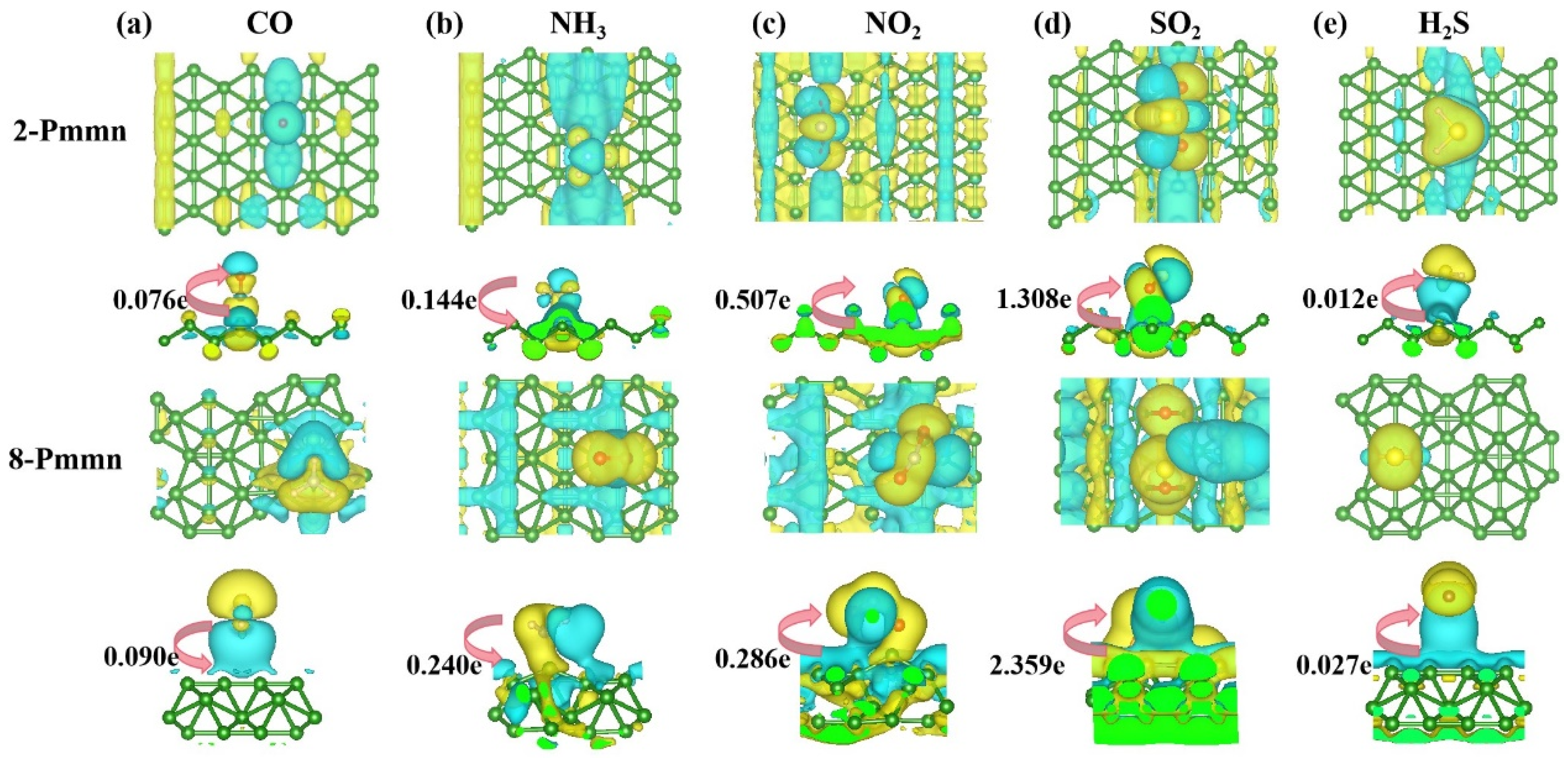
3.2.2. Electronic Structure of a System of a Gas Molecule Adsorbed on χ3-Borophene for Five Different Gases
3.3. 2-Pmmn Borophene and 8 Pmmn Borophene
4. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nicoletti, G.; Arcuri, N.; Nicoletti, G.; Bruno, R. A technical and environmental comparison between hydrogen and some fossil fuels. Energy Convers. Manag. 2015, 89, 205–213. [Google Scholar] [CrossRef]
- Ritchie, H.; Roser, M.; Rosado, P. CO2, and Greenhouse Gas Emissions. In Our World in Data; 2020; Available online: https://ourworldindata.org/co2-and-greenhouse-gas-emissions (accessed on 18 June 2023).
- Zhao, B.; Wang, S.X.; Xu, J.Y.; Fu, K.; Amann, M. NOx emissions in China: Historical trends and future perspectives. Atmospheric Chem. Phys. 2013, 13, 16047–16112. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Cao, X.; Wu, X.J.; He, Q.; Yang, J.; Zhang, X.; Chen, J.; Zhao, W.; Han, S.; Nam, G.H. Recent Advances in Ultrathin Two-Dimensional Nanomaterials. Chem. Rev. 2017, 117, 6225–6331. [Google Scholar]
- Zhang, H. Ultrathin Two-Dimensional Nanomaterials. ACS Nano 2015, 9, 9451–9469. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chen, W.; Chen, Y.; Chen, Y.; Chen, Y.; Ding, F.; Fan, C.; Fan, H.J.; Fan, Z.; Gong, C.; et al. Recent Progress on Two-Dimensional Materials. Acta Phys.-Chim. Sin. 2021, 37, 2108010–2108017. [Google Scholar] [CrossRef]
- Shen, J.; Yang, Z.; Wang, Y.; Xu, L.C.; Liu, X. The gas sensing performance of borophene/MoS2 heterostructure. Appl. Surf. Sci. 2020, 504, 14412. [Google Scholar] [CrossRef]
- Khan, K.; Tareen, A.K.; Aslam, M.; Khan, M.F.; Shi, Z.; Ma, C.; Shams, S.S.; Khatoon, R.; Zhang, H.; Guo, Z. Synthesis, Properties and novel electrocatalytic applications of the 2D-borophene Xenes. Prog. Solid State Chem. 2020, 59, 100283. [Google Scholar]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Lebon, A.; Aguilera-del-Toro, R.H.; Gallego, L.J.; Vega, A. Li-decorated Pmmn8 phase of borophene for hydrogen storage. A van der Waals corrected density-functional theory study. Int. J. Hydrogen Energy 2019, 44, 1021–1033. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Agrawal, M.; Becerril, H.A.; Bao, Z.; Liu, Z.; Chen, Y.; Peumans, P. Organic Light-Emitting Diodes on Solution-Processed Graphene Transparent Electrodes. ACS Nano 2010, 4, 43–48. [Google Scholar] [CrossRef]
- Khan, K.; Tareen, A.K.; Aslam, M.; Mahmood, A.; Zhang, Y.; Ouyang, Z.; Guo, Z.; Zhang, H. Going green with batteries and supercapacitor: Two-dimensional materials and their nanocomposites based energy storage applications. Prog. Solid State Chem. 2020, 58, 10025. [Google Scholar]
- KKhan; Tareen, A.K.; Aslam, M.; Wang, R.; Zhang, Y.; Mahmood, A.; Ouyang, Z.; Zhang, H.; Guo, Z. Recent developments in emerging two-dimensional materials and their applications. J. Mater. Chem. C 2010, 8, 387. [Google Scholar]
- Tang, H.; Ismail-Beigi, S. Novel precursors for boron nanotubes: The competition of two-center and three-center bonding in boron sheets. Phys. Rev. Lett. 2007, 99, 115501. [Google Scholar] [CrossRef] [Green Version]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.J.; Wang, H.T. Semimetallic Two-Dimensional Boron Allotrope with Massless Dirac Fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bezanilla, A.; Littlewood, P.B. Electronic properties of 8 -Pmmn borophene. Phys. Rev. B 2016, 93, 241405. [Google Scholar] [CrossRef] [Green Version]
- Carrete, J.; Li, W.; Lindsay, L.; Broido, D.A.; Gallego, L.J.; Mingo, N. Physically founded phonon dispersions of few-layer materials and the case of borophene. Mater. Res. Lett. 2016, 4, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fuente, A.; Carrete, J.; Vega, A.; Gallego, L.J. What will freestanding borophene nanoribbons look like? An analysis of their possible structures, magnetism, and transport properties. Phys. Chem. Chem. Phys 2017, 19, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, P.; Sahu, T.K.; Bhushan, R.; Yamijala, S.S.; Late, D.J.; Kumar, P.; Vinu, A. Borophene: Freestanding borophene and its hybrids. Adv. Mater. 2019, 31, 201900353. [Google Scholar]
- Zhong, Q.; Zhang, J.; Cheng, P.; Feng, B.; Wu, K. Metastable phases of 2D boron sheets on Ag(111). J. Phys. Condens. Matter. 2017, 29, 095002. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Song, L.; Liu, Z. Adsorption of gas molecules on the defective stanene nanosheets with single vacancy: A DFT study. Appl. Surf. Sci. 2020, 512, 14572. [Google Scholar] [CrossRef]
- Yue, Q.; Shao, Z.; Chang, S.; Li, J. Adsorption of gas molecules on monolayer MoS2 and effect of applied electric field. Nanoscale Res. Lett. 2013, 8, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Kang, W.; Xue, J. The potential application of phosphorene as an anode material in Li-ion batteries. J. Mater. Chem. A 2014, 2, 19046–19052. [Google Scholar] [CrossRef]
- Kou, L.; Frauenheim, T.; Chen, C. Phosphorene as a Superior Gas Sensor: Selective Adsorption and Distinct I-V Response. J. Phys. Chem. Lett. 2014, 5, 2675–2681. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Ke, Q.; Zhang, G.; Zhang, Y.W. Nergetics, Charge Transfer, and Magnetism of Small Molecules Physisorbed on Phosphorene. J. Phys. Chem. C 2015, 119, 3102–3110. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Hu, W.; Li, Z.; Yang, J. A first-principles study of gas adsorption on germanene. Phys. Chem. Chem. Phys. 2014, 16, 22495–22498. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, X.; Chen, D. Borophene: A promising adsorbent material with strong ability and capacity for SO2 adsorption. Appl. Phys. A 2018, 124, 1. [Google Scholar] [CrossRef]
- Allal, H.; Belhocine, Y.; Rahali, S.; Damous, M.; Ammouchi, N. Structural, electronic, and energetic investigations of acrolein adsorption on B36 borophene nanosheet: A dispersion-corrected DFT insight. J. Mol. Model. 2020, 26, 1. [Google Scholar]
- Omidvar, A. Borophene: A novel boron sheet with a hexagonal vacancy offering high sensitivity for hydrogen cyanide detection. Comput. Theor. Chem. 2017, 1115, 179. [Google Scholar] [CrossRef]
- MFazilaty; Pourahmadi, M.; Shayesteh, M.R.; Hashemian, S. χ3-borophene-based detection of hydrogen sulfide via gas nanosensors. Chem. Phys. Lett. 2020, 741, 137066. [Google Scholar] [CrossRef]
- Kootenaei, A.S.; Ansari, G. B36 borophene as an electronic sensor for formaldehyde: Quantum chemical analysis. Phys. Lett. A 2016, 380, 2664. [Google Scholar] [CrossRef]
- Sabokdast, S.; Horri, A.; Azar, Y.T.; Momeni, M.; Tavakoli, M.B. Adsorption of adenine molecule on χ3 borophene nanosheets: A density functional theory study. Phys. E Lowdimens Syst. Nanostruct. 2020, 119, 11402. [Google Scholar] [CrossRef]
- Bhuvaneswari, R.; Chandiramouli, R. DFT investigation on the adsorption behavior of dimethyl and trimethyl amine molecules on borophene nanotube. Chem. Phys. Lett. 2018, 701, 34. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, H.; Li, J.; Hu, S.; Cao, S.; Ren, W.; Wang, Y. A comprehensive first-principle study of borophene-based nano gas sensor with gold electrodes. Front. Phys. 2022, 17, 13501. [Google Scholar] [CrossRef]
- Yu, X.; Chen, F.; Yu, Z.; Li, Y. Computational study of borophene with line defects as sensors for nitrogen-containing gas molecules. ACS Appl. Nano Mater. 2020, 3, 9961. [Google Scholar] [CrossRef]
- Abhervé, A.; Manas-Valero, S.; Clemente-León, M.; Coronado, E. Graphene related magnetic materials: Micromechanical exfoliation of 2D layered magnets based on bimetallic anilate complexes with inserted Fe-III(acac(2)-trien) (+) and Fe-III(sal(2)-trien) (+) molecules. Chem. Sci. 2015, 6, 4665–4673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Zhang, J.M.; Xu, K.W.; Ji, V. A first-principles study on gas sensing properties of graphene and Pd-doped graphene. Appl. Surf. Sci. 2015, 343, 121–127. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 031758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1977, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, H.; Wang, Z.; Liu, G.; Liu, S.; Ouyang, C. First-principles study of chi(3)-borophene for charge-modulated switchable CO2 capture. Phys. Chem. Chem. Phys. 2010, 22, 8864–8869. [Google Scholar] [CrossRef]
- He, J.; Ouyang, Y.; Yu, C.; Jiang, P.; Chen, J. Lattice thermal conductivity of beta(12) and chi(3) borophene. Chin. Phys. B 2020, 29, 126503. [Google Scholar] [CrossRef]
- Xiao, H.; Cao, W.; Ouyang, T.; Guo, S.; He, C.; Zhong, J. Lattice thermal conductivity of borophene from first principle calculation. Sci. Rep. 2017, 7, 45986. [Google Scholar] [CrossRef] [Green Version]
- Meshginqalam, B.; Barvestani, J. Vacancy defected blue and black phosphorene nanoribbons as gas sensor of NOx and SOx molecules. Appl. Surf. Sci. 2020, 526, 146692. [Google Scholar] [CrossRef]
- Thi, L.T.; Hamada, I.; Morikawa, Y.; Dinh, V.A. Adsorption of toxic gases on borophene: Surface deformation links to chemisorptions. RSC Adv. 2021, 11, 18279. [Google Scholar]
- Zaremba, E.; Kohn, W. Theory of helium adsorption on simple and noble-metal surfaces. Phys. Rev. B 1977, 15, 1769–1781. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a/Å | b/Å | |
|---|---|---|
| This work | 8.48 | 2.94 |
| Ref. [47] | 8.42 | 2.90 |
| Position | B1-1 | B1-2 | B2-1 | B2-2 | D1-1 | D1-2 | D2-1 | D2-2 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | −0.444 | −0.087 | −0.685 | −0.104 | −0.091 | −0.083 | −0.498 | −0.103 | −0.532 | −0.121 | |
| Δq | 0.025e | 0.043e | 0.034e | −0.055e | −0.066e | 0.053e | 0.058e | 0.008e | 0.052e | 0.050e | |
| 1.51 | 3.48 | 1.50 | 3.31 | 3.15 | 3.05 | 1.55 | 3.50 | 1.53 | 3.42 | ||
| NH3 | −0.538 | −0.124 | −0.764 | −0.138 | −0.134 | −0.135 | −0.758 | −0.118 | −0.148 | −0.129 | |
| Δq | 0.191e | 0.148e | 0.182e | −0.147e | −0.011e | 0.167e | 0.197e | 0.142e | 0.023e | 0.158e | |
| 1.65 | 2.97 | 1.64 | 2.73 | 2.86 | 2.82 | 1.64 | 2.94 | 2.72 | 2.82 | ||
| NO2 | −1.245 | −1.559 | −2.071 | −2.221 | −2.073 | −1.436 | −2.072 | −1.555 | −2.067 | −2.175 | |
| Δq | 0.450e | 0.438e | 0.348e | −0.450e | −0.346e | 0.481e | 0.352e | 0.439e | 0.356e | 0.447e | |
| 1.61 | 1.48 | 1.55 | 1.54 | 1.54 | 1.47 | 1.56 | 1.48 | 1.55 | 1.52 | ||
| SO2 | −0.183 | −0.058 | −0.186 | −0.145 | −0.112 | −0.037 | −0.185 | −0.193 | −0.178 | −0.176 | |
| Δq | 0.168e | 0.315e | 0.176e | −0.339e | −0.108e | 0.117e | 0.176e | 0.187e | 0.164e | 0.158e | |
| 2.95 | 2.27 | 2.96 | 2.34 | 2.88 | 3.00 | 2.98 | 2.98 | 2.91 | 2.95 | ||
| H2S | −0.080 | −0.039 | −0.081 | −0.038 | −0.081 | −0.040 | −0.080 | −0.032 | −0.074 | −0.035 | |
| Δq | 0.180e | 0.004e | 0.184e | −0.001e | −0.186e | 0.006e | 0.176e | 0.001e | 0.195e | 0.003e | |
| 2.49 | 3.43 | 2.40 | 3.34 | 2.58 | 3.24 | 2.70 | 3.43 | 2.71 | 3.34 |
| CO | NH3 | NO2 | SO2 | H2S | |
|---|---|---|---|---|---|
| DFT-D3 | −0.444 | −0.764 | −2.073 | −0.186 | −0.081 |
| DFT | −0.323 | −0.577 | −1.933 | −0.003 | 0.788 |
| Position | B1-1 | B1-2 | B2-1 | B2-2 | D1-1 | D1-2 | D2-1 | D2-2 | |
|---|---|---|---|---|---|---|---|---|---|
| CO | −0.762 | −0.113 | −0.163 | −0.120 | −0.113 | −0.114 | −0.967 | −0.121 | |
| Δq | 0.142e | −0.046e | 0.053e | −0.062e | 0.034e | 0.045e | 0.076e | −0.061e | |
| NH3 | −1.332 | −1.333 | −0.230 | −0.230 | −1.334 | −0.224 | −0.230 | −0.229 | |
| Δq | 0.152e | 0.144e | 0.159e | −0.152e | 0.151e | 0.130e | 0.153e | −0.155e | |
| NO2 | −1.712 | −2.275 | −1.712 | −2.156 | −1.711 | −2.178 | −1.713 | −2.158 | |
| Δq | 0.349e | −0.507e | 0.409e | −0.356e | 0.367e | 0.438e | 0.424e | −0.357e | |
| SO2 | −0.132 | −2.058 | −0.100 | −0.111 | −0.121 | −1.095 | −0.168 | −0.423 | |
| Δq | 0.106e | −1.308e | 0.097e | −0.067e | 0.109e | 1.250e | 0.481e | −1.672e | |
| H2S | −0.105 | −0.226 | −0.231 | −0.231 | −0.106 | −0.210 | −0.243 | −0.245 | |
| 0.093e | 0.049e | 0.047e | −0.026e | 0.102e | 0.012e | 0.027e | 0.034e |
| Position | B1-1 | B1-2 | B2-1 | B2-2 | D1-1 | D1-2 | D2-1 | D2-2 | |
|---|---|---|---|---|---|---|---|---|---|
| CO | −0.540 | −0.070 | −0.138 | −0.108 | −0.051 | −0.054 | −0.110 | −0.109 | |
| Δq | 0.090e | −0.030e | 0.054e | −0.066e | 0.011e | 0.030e | 0.063e | −0.063e | |
| NH3 | −0.707 | −0.108 | 0.193 | −0.160 | −0.086 | −0.084 | −0.168 | −0.164 | |
| Δq | 0.240e | −0.099e | 0.205e | −0.211e | 0.810e | 0.871e | 0.197e | −0.715e | |
| NO2 | −1.253 | −1.251 | −0.706 | −0;813 | −1.255 | −1.660 | −0.601 | −0.503 | |
| Δq | 0.333e | −0.346e | 0.475e | −0.463e | 0.334e | 0.286e | 0.968e | −0.798e | |
| SO2 | −0.071 | −0.094 | −0.467 | −0.110 | 0.868 | −0.090 | −0.489 | −0.489 | |
| Δq | 0.079e | −0.093e | 1.282e | −0.095e | 0.763e | 0.090e | 2.383e | −2.359e | |
| H2S | −0.049 | −0.023 | −0.127 | −0.119 | −0.048 | −0.016 | −0.119 | −0.074 | |
| 0.021 | 0.006 | 0.025e | −0.082e | 0.027e | 0.001e | 0.026e | 0.001e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, J.-X.; Tian, Y.-P.; Wang, C.-B.; Zhang, L.-L. First-Principles Study of χ3-Borophene as a Candidate for Gas Sensing and the Removal of Harmful Gases. Nanomaterials 2023, 13, 2117. https://doi.org/10.3390/nano13142117
Duan J-X, Tian Y-P, Wang C-B, Zhang L-L. First-Principles Study of χ3-Borophene as a Candidate for Gas Sensing and the Removal of Harmful Gases. Nanomaterials. 2023; 13(14):2117. https://doi.org/10.3390/nano13142117
Chicago/Turabian StyleDuan, Jia-Xing, Yu-Ping Tian, Chao-Bo Wang, and Lian-Lian Zhang. 2023. "First-Principles Study of χ3-Borophene as a Candidate for Gas Sensing and the Removal of Harmful Gases" Nanomaterials 13, no. 14: 2117. https://doi.org/10.3390/nano13142117