
Hierarchical Self-Assembly and Conformation of Tb Double-Decker Molecular Magnets: Experiment and Molecular Dynamics

 ,
,
Abstract
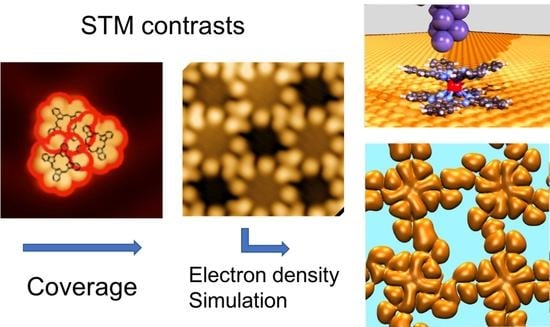
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
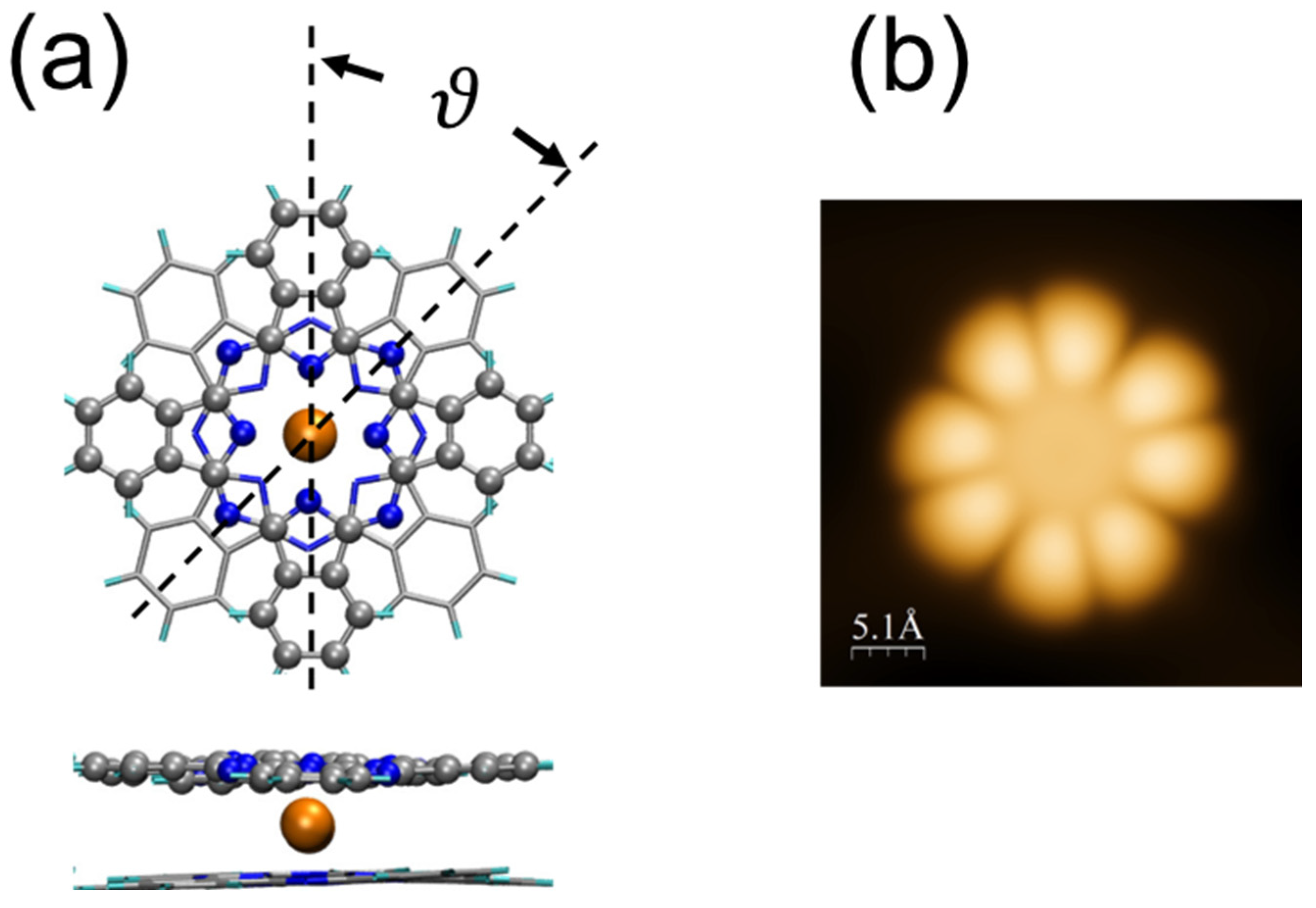
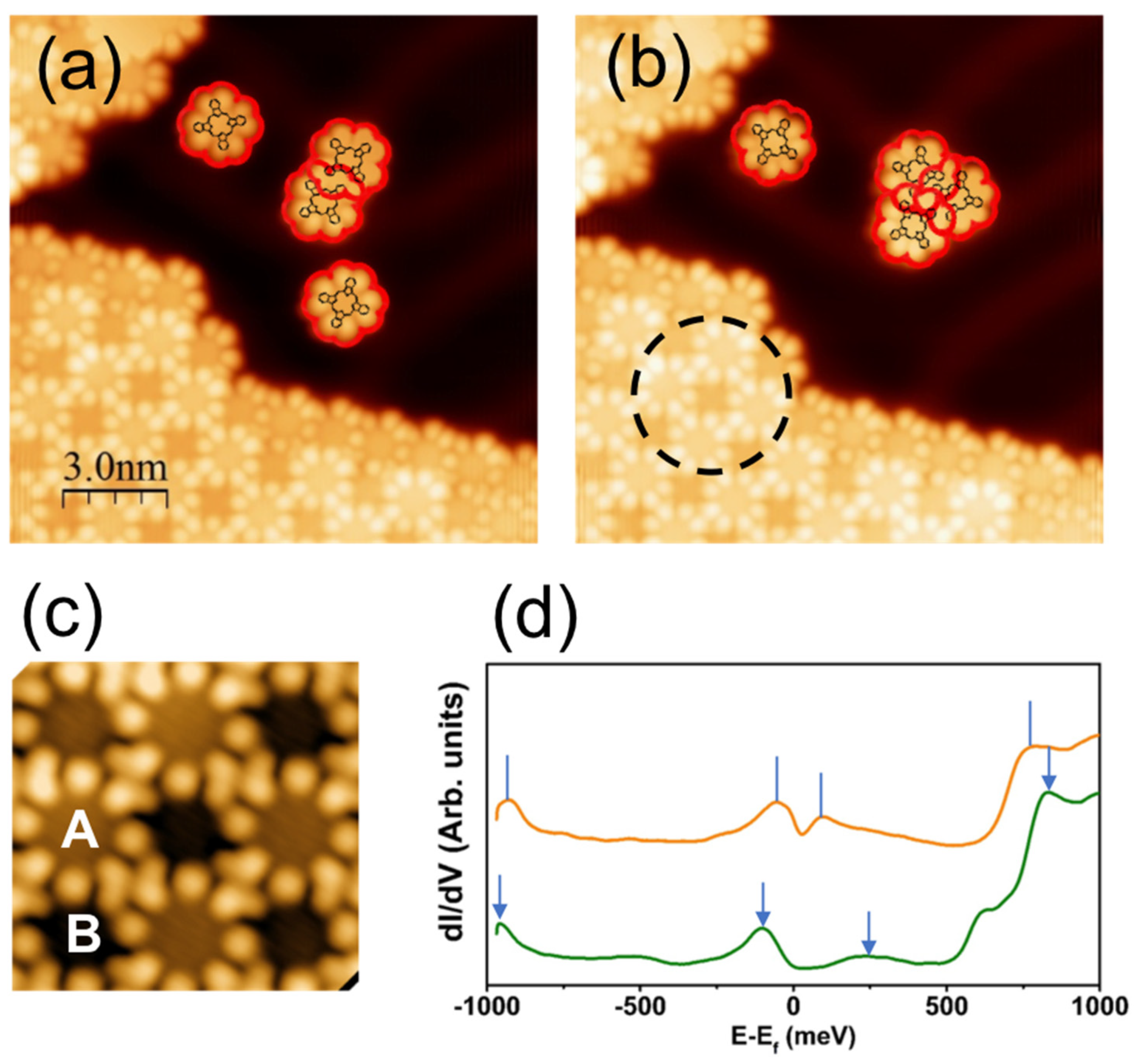
2.1. Scanning Tunneling Microscopy and Spectroscopy
2.2. Computational Method
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gabarro-Riera, G.; Aromi, G.; Sanudo, E.C. Magnetic molecules on surfaces: SMMs and beyond. Coord. Chem. Rev. 2023, 475, 214858. [Google Scholar]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef]
- Koike, N.; Uekusa, H.; Ohashi, Y.; Harnoode, C.; Kitamura, F.; Ohsaka, T.; Tokuda, K. Relationship between the Skew Angle and Interplanar Distance in Four Bis(phthalocyaninato)lanthanide(III) Tetrabutylammonium Salts ([NBun4][LnIIIPc2]; Ln = Nd, Gd, Ho, Lu). Inorg. Chem. 1996, 35, 5798. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.-Y.; Kaizu, Y. Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef] [PubMed]
- Komeda, T.; Isshiki, H.; Liu, J.; Zhang, Y.F.; Lorente, N.; Katoh, K.; Breedlove, B.K.; Yamashita, M. Observation of Electric Current Control of a Local Spin in a Single-Molecule Magnet. Nat. Commun. 2011, 2, 217. [Google Scholar] [CrossRef] [Green Version]
- Amokrane, A.; Klyatskaya, S.; Boero, M.; Ruben, M.; Bucher, J.P. The Role of π-Radicals in the Spin Connectivity of Clusters and Networks of Tb Double-Decker Single Molecule Magnets. ACS Nano 2017, 11, 10750–10760. [Google Scholar] [CrossRef] [PubMed]
- Ara, F.; Qi, Z.K.; Hou, J.; Komeda, T.; Katoha, K.; Yamashita, M. A Scanning Tunneling Microscopy Study of the Electronic and Spin States of Bis(phthalocyaninato) terbium(III) (TbPc2) Molecules on Ag(111). Dalton Trans. 2016, 45, 16644–16652. [Google Scholar]
- Katoh, K.; Komeda, T.; Yamashita, M. Surface Morphologies, Electronic Structures, and Kondo Effect of Lanthanide(III)-Phthalocyanine Molecules on Au(111) by Using STM, STS and FET Properties for Next Generation Devices. Dalton Trans. 2010, 39, 4708–4723. [Google Scholar] [PubMed]
- Deng, Z.; Rauschenbach, S.; Stepanov, S.; Klyatskaya, S.; Ruben, M.; Kern, K. Self-assembly of bis(phthalocyaninato)terbium on metal surfaces. Phys. Scr. 2015, 90, 098003. [Google Scholar] [CrossRef]
- Gonidec, M.; Biagi, R.; Corradini, V.; Moro, F.; De Renzi, V.; del Pennino, U.; Summa, D.; Muccioli, L.; Zannoni, C.; Amabilino, D.B.; et al. Surface supramolecular organization of a terbium (III) double-decker complex on graphite and its single molecule magnet behavior. Am. Chem. Soc. 2011, 133, 6603–6612. [Google Scholar] [CrossRef]
- Rogez, G.; Donnio, B.; Terazzi, E.; Gallani, J.-L.; Kappler, J.P.; Bucher, J.-P.; Drillon, M. The Quest for Nanoscale Magnets: The example of [Mn12] Single Molecule Magnets. Adv. Mater. 2009, 21, 4323–4333. [Google Scholar]
- Biard, H.; Moreno-Pineda, E.; Ruben, M.; Bonet, E.; Wernsdorfer, W.; Balestro, F. Increasing the Hilbert space dimension using a single coupled molecular spin. Nat. Commun. 2021, 12, 4443. [Google Scholar] [CrossRef] [PubMed]
- Wernsdorfer, W.; Ruben, M. Synthetic Hilbert Space Engineering of Molecular Qudits: Isotopologue Chemistry. Adv. Mater. 2019, 31, 1806687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaita-Ariño, A.; Luis, F.; Hill, S.; Coronado, E. Molecular spins for quantum computation. Nat. Chem. 2019, 11, 301–309. [Google Scholar] [PubMed]
- Vincent, R.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W.; Balestro, F. Electronic Read-Out of a Single Nuclear Spin Using a Molecular Spin Transistor. Nature 2012, 488, 357–359. [Google Scholar] [PubMed]
- Urdampilleta, M.; Klayatskaya, S.; Ruben, M.; Wernsdorfer, W. Magnetic Interaction Between a Radical Spin and a Single-Molecule Magnet in a Spin Valve. ACS Nano 2015, 9, 4458–4464. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, R.; Amokrane, A.; Klyatskaya, S.; Boero, M.; Ruben, M.; Bucher, J.P. Screening the 4f-electron spin of TbPc2 single-molecule magnets on metal substrates by ligand channeling. Nanoscale 2019, 11, 21167. [Google Scholar] [CrossRef]
- Tuerhong, R.; Ngassam, F.; Watanabe, S.; Onoe, J.; Alouani, M.; Bucher, J.P. Two-Dimensional Organometallic Kondo Lattice with Long-Range Antiferromagnetic Order. J. Phys. Chem. 2018, 122, 20046. [Google Scholar]
- Tuerhong, R.; Ngassam, F.; Alouani, M.; Bucher, J.P. When Molecular Dimerization Induces Magnetic Bi-Stability at the Metal–Organic Interface. Adv. Phys. Res. 2023, 2, 2200005. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- CPMD, Copyright IBM Corp. 1990–2023 and Max Planck Institute, Stuttgart, 1994–2001. Available online: http://www.cpmd.org (accessed on 30 July 2023).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electronic density. Phys. Rev. B 1988, 37, 785. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Field. J. Phys. Chem. 1994, 98, 11623. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787. [Google Scholar]
- Martin, E.; Essomba, I.B.A.; Ishisone, K.; Boero, M.; Ori, G.; Massobrio, C. Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases. Molecules 2022, 27, 9034. [Google Scholar] [CrossRef] [PubMed]
- Silvestrelli, P.L.; Martin, E.; Boero, M.; Bouzid, A.; Ori, G.; Massobrio, C. Atomic Structure of Glassy GeTe4 as a Playground to Assess the Performances of Density Functional Schemes Accounting for Dispersion Forces. Phys. Chem. B 2020, 124, 11273–11279. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probert, M.I.J. Improved algorithm for geometry optimization using damped molecular dynamics. J. Comput. Phys. 2003, 191, 130–146. [Google Scholar] [CrossRef]
- Briganti, M.; Serrano, G.; Poggini, L.; Cortigiani, B.; Carol de Camargo, L.; Fernandes Soares, J.; Motta, A.; Caneschi, A.; Totti, F.; Sessoli, R. Mixed-Sandwich Titanium(III) Qubits on Au(111): Electron Delocalization Ruled by Molecular Packing. Nano Lett. 2022, 22, 8626–8632. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawes, P.; Boero, M.; Barhoumi, R.; Klyatskaya, S.; Ruben, M.; Bucher, J.-P. Hierarchical Self-Assembly and Conformation of Tb Double-Decker Molecular Magnets: Experiment and Molecular Dynamics. Nanomaterials 2023, 13, 2232. https://doi.org/10.3390/nano13152232
Lawes P, Boero M, Barhoumi R, Klyatskaya S, Ruben M, Bucher J-P. Hierarchical Self-Assembly and Conformation of Tb Double-Decker Molecular Magnets: Experiment and Molecular Dynamics. Nanomaterials. 2023; 13(15):2232. https://doi.org/10.3390/nano13152232
Chicago/Turabian StyleLawes, Patrick, Mauro Boero, Rabei Barhoumi, Svetlana Klyatskaya, Mario Ruben, and Jean-Pierre Bucher. 2023. "Hierarchical Self-Assembly and Conformation of Tb Double-Decker Molecular Magnets: Experiment and Molecular Dynamics" Nanomaterials 13, no. 15: 2232. https://doi.org/10.3390/nano13152232