
Theoretical Study of the Defects and Doping in Tuning the Electrocatalytic Activity of Graphene for CO2 Reduction

Abstract
:1. Introduction
2. Methods
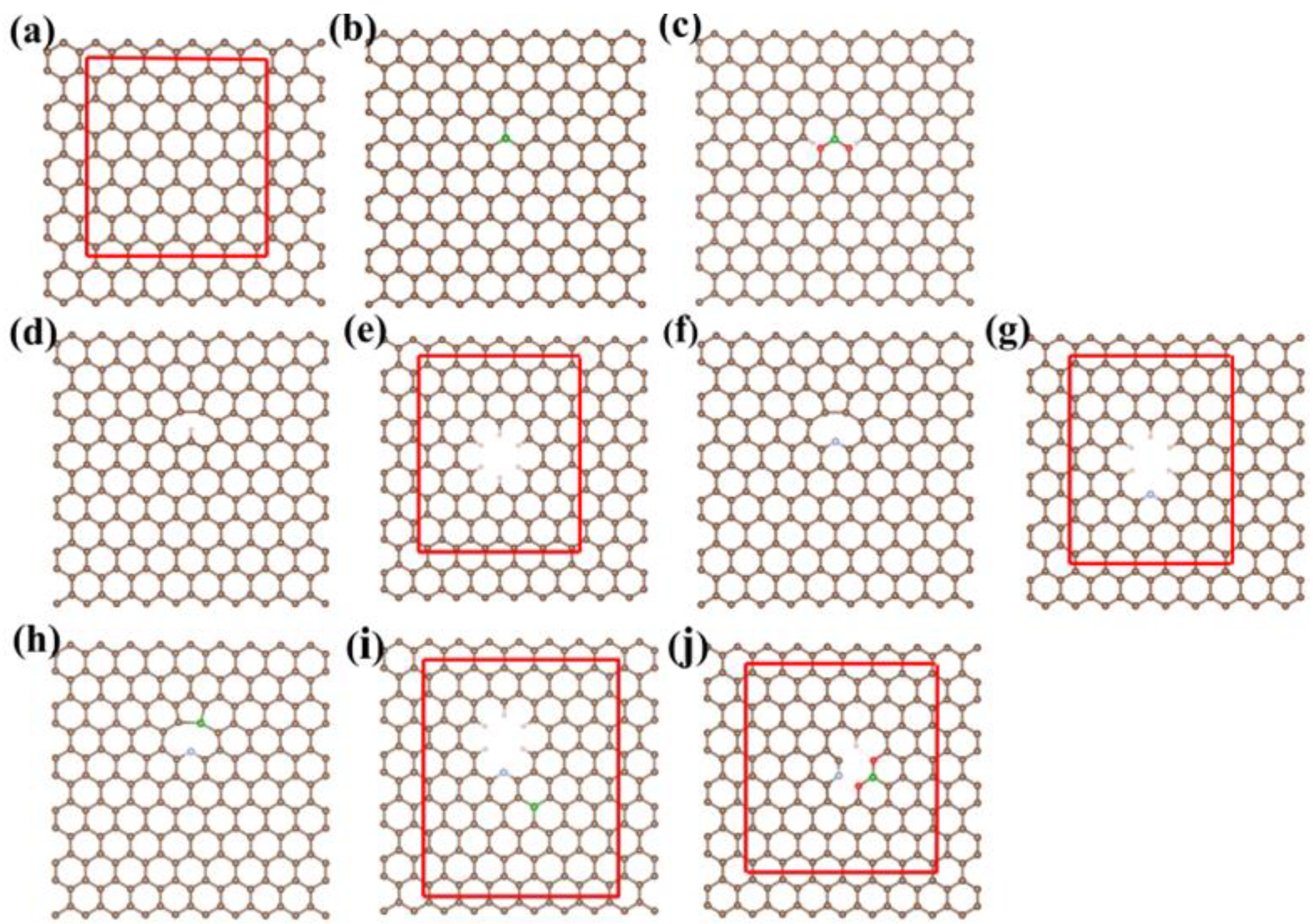
2.1. Computational Models
2.2. Computational Details
3. Results and Discussion
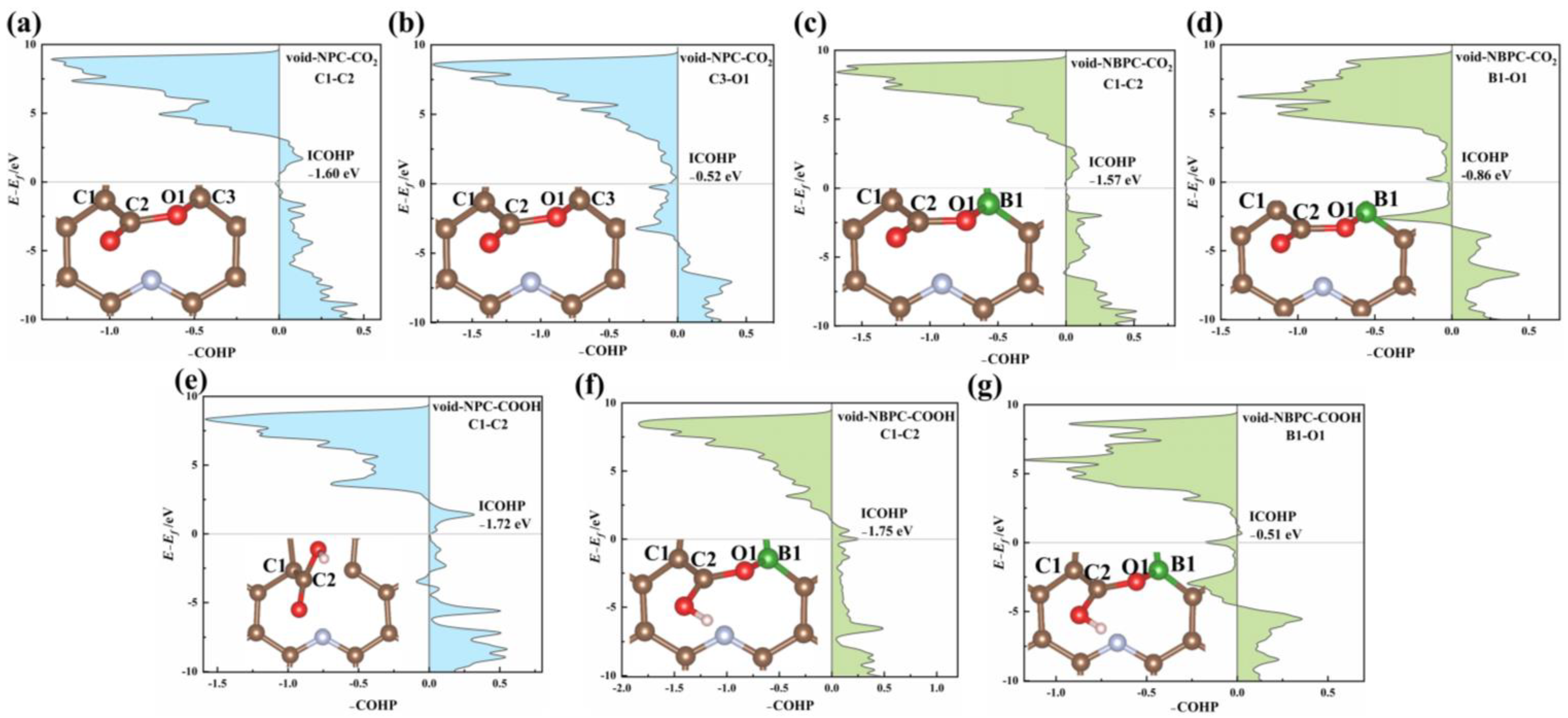
3.1. Reaction Pathways for ECO2RR
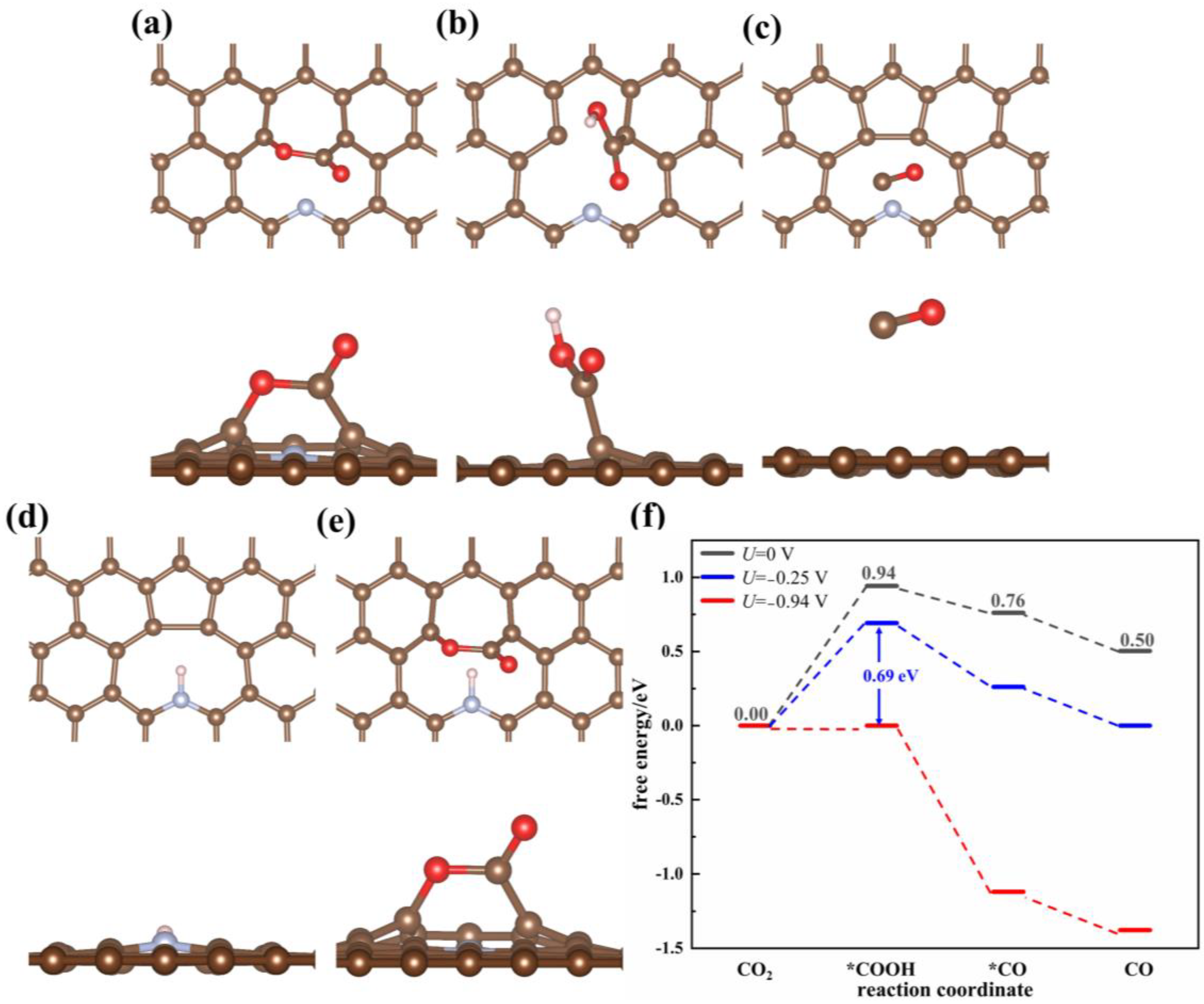
3.1.1. ECO2RR on Pristine Graphene and the Effect of the Defects
3.1.2. Monoatomic-Doped Graphene
N Doping
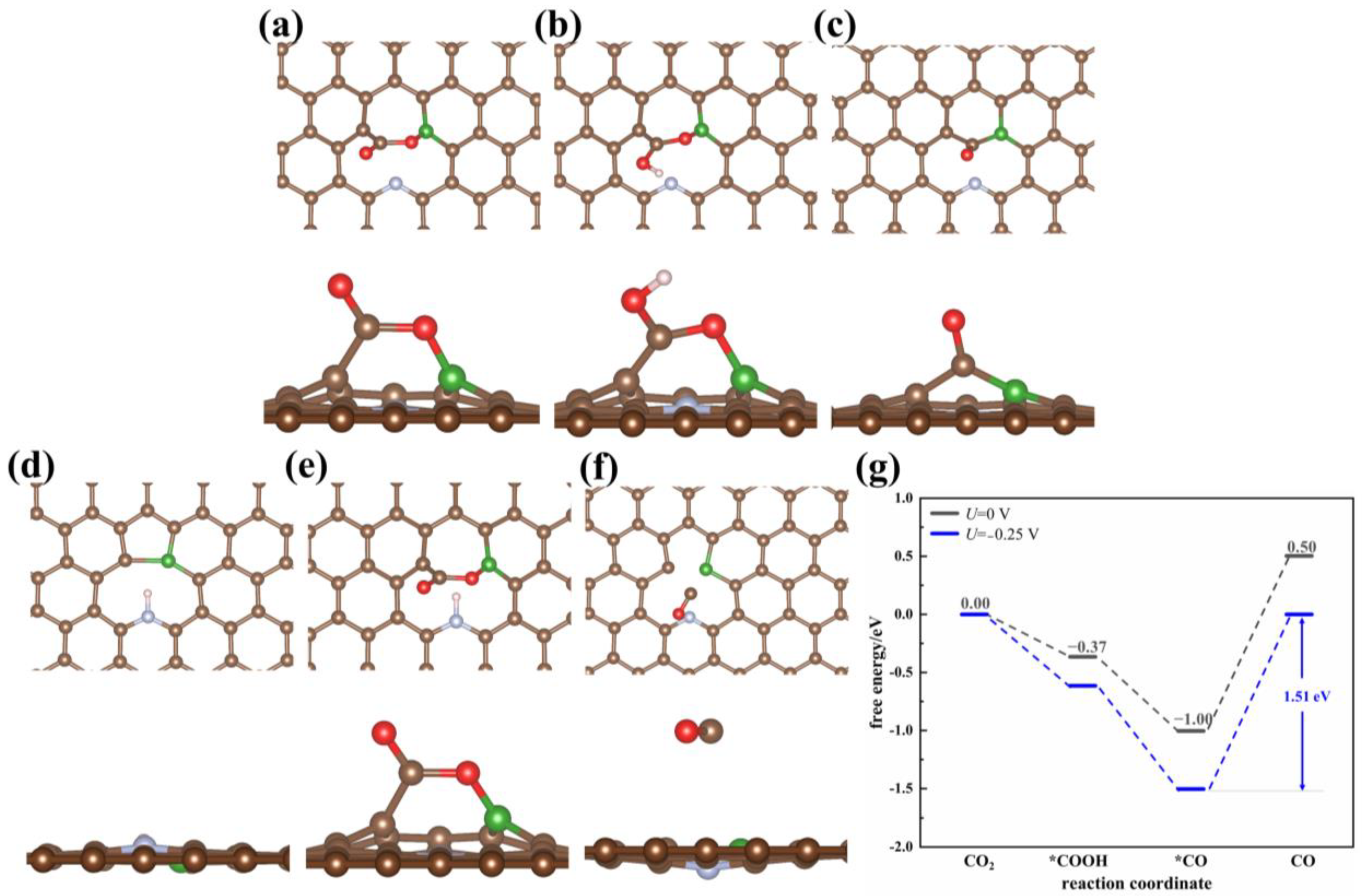
B-Doping
3.1.3. Diatomic-Doped Graphene
3.1.4. Triatomic-Doped Graphene
3.2. Discussion
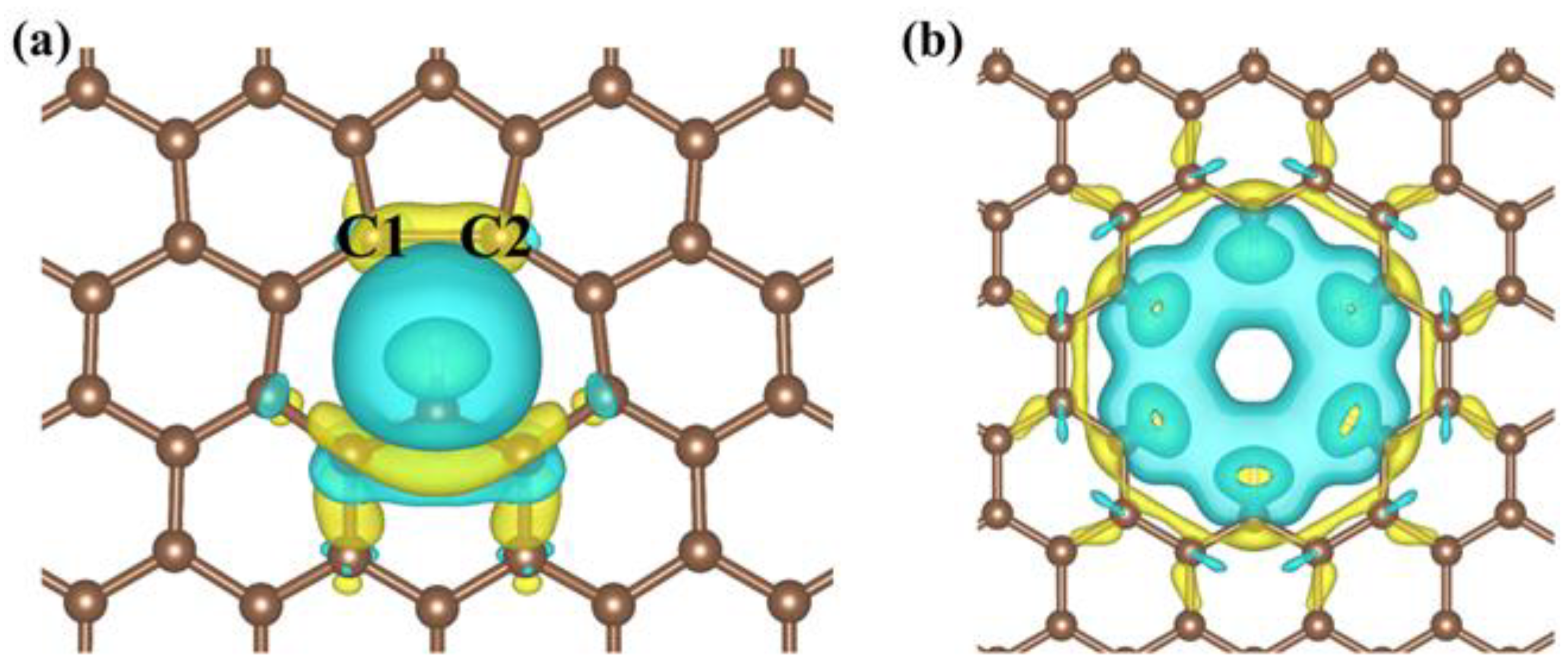
3.2.1. Defect Effect
3.2.2. Single-Atom Doping
3.2.3. Synergistic Effect between Doped Heteroatoms
3.2.4. Doping Effects of the O Atom
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, L.; Zhao, N.; Wei, W.; Sun, Y. A review of research progress on CO2 capture, storage, and utilization in Chinese Academy of Sciences. Fuel 2013, 108, 112–130. [Google Scholar] [CrossRef]
- Yaashikaa, P.R.; Senthil Kumar, P.; Varjani, S.J.; Saravanan, A. A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Osman, A.I.; Hefny, M.; Abdel Maksoud, M.I.A.; Elgarahy, A.M.; Rooney, D.W. Recent advances in carbon capture storage and utilisation technologies: A review. Environ. Chem. Lett. 2021, 19, 797–849. [Google Scholar] [CrossRef]
- Wang, L.; Chen, W.; Zhang, D.; Du, Y.; Amal, R.; Qiao, S.; Wu, J.; Yin, Z. Surface strategies for catalytic CO2 reduction: From two-dimensional materials to nanoclusters to single atoms. Chem. Soc. Rev. 2019, 48, 5310–5349. [Google Scholar] [CrossRef]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, S.-X.; Gandionco, K.A.; Bond, A.M.; Zhang, J. Electrocatalytic carbon dioxide reduction: From fundamental principles to catalyst design. Mater. Today Adv. 2020, 7, 100074. [Google Scholar] [CrossRef]
- Karapinar, D.; Creissen, C.E.; Rivera de la Cruz, J.G.; Schreiber, M.W.; Fontecave, M. Electrochemical CO2 Reduction to Ethanol with Copper-Based Catalysts. ACS Energy Lett. 2021, 6, 694–706. [Google Scholar] [CrossRef]
- Johnson, D.; Qiao, Z.; Djire, A. Progress and Challenges of Carbon Dioxide Reduction Reaction on Transition Metal Based Electrocatalysts. ACS Appl. Energy Mater. 2021, 4, 8661–8684. [Google Scholar] [CrossRef]
- Gao, F.-Y.; Wu, Z.-Z.; Gao, M.-R. Electrochemical CO2 Reduction on Transition-Metal Chalcogenide Catalysts: Recent Advances and Future Perspectives. Energy Fuels 2021, 35, 12869–12883. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Liu, J.; Huang, K.; Chen, Y.; Li, Y.; Zhu, J.-J. Low Overpotential for Electrochemically Reducing CO2 to CO on Nitrogen-Doped Graphene Quantum Dots-Wrapped Single-Crystalline Gold Nanoparticles. ACS Energy Lett. 2018, 3, 946–951. [Google Scholar] [CrossRef]
- Ma, S.; Luo, R.; Gold, J.I.; Yu, A.Z.; Kim, B.; Kenis, P.J.A. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 2016, 4, 8573–8578. [Google Scholar] [CrossRef]
- Liang, C.; Kim, B.; Yang, S.; Yang, L.; Francisco Woellner, C.; Li, Z.; Vajtai, R.; Yang, W.; Wu, J.; Kenis, P.J.A.; et al. High efficiency electrochemical reduction of CO2 beyond the two-electron transfer pathway on grain boundary rich ultra-small SnO2 nanoparticles. J. Mater. Chem. A 2018, 6, 10313–10319. [Google Scholar] [CrossRef]
- Shi, Y.; Ji, Y.; Long, J.; Liang, Y.; Liu, Y.; Yu, Y.; Xiao, J.; Zhang, B. Unveiling hydrocerussite as an electrochemically stable active phase for efficient carbon dioxide electroreduction to formate. Nat. Commun. 2020, 11, 3415. [Google Scholar] [CrossRef]
- O’Mara, P.B.; Wilde, P.; Benedetti, T.M.; Andronescu, C.; Cheong, S.; Gooding, J.J.; Tilley, R.D.; Schuhmann, W. Cascade Reactions in Nanozymes: Spatially Separated Active Sites inside Ag-Core–Porous-Cu-Shell Nanoparticles for Multistep Carbon Dioxide Reduction to Higher Organic Molecules. J. Am. Chem. Soc. 2019, 141, 14093–14097. [Google Scholar] [CrossRef]
- Ren, D.; Ang, B.S.-H.; Yeo, B.S. Tuning the Selectivity of Carbon Dioxide Electroreduction toward Ethanol on Oxide-Derived CuxZn Catalysts. ACS Catal. 2016, 6, 8239–8247. [Google Scholar] [CrossRef]
- Hu, C.; Xiao, Y.; Zou, Y.; Dai, L. Carbon-Based Metal-Free Electrocatalysis for Energy Conversion, Energy Storage, and Environmental Protection. Electrochem. Energy Rev. 2018, 1, 84–112. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Quan, X. Carbon-Based Materials for Electrochemical Reduction of CO2 to C2+ Oxygenates: Recent Progress and Remaining Challenges. ACS Catal. 2021, 11, 2076–2097. [Google Scholar] [CrossRef]
- Hu, C.; Paul, R.; Dai, Q.; Dai, L. Carbon-based metal-free electrocatalysts: From oxygen reduction to multifunctional electrocatalysis. Chem. Soc. Rev. 2021, 50, 11785–11843. [Google Scholar] [CrossRef]
- Jeon, I.-Y.; Zhang, S.; Zhang, L.; Choi, H.-J.; Seo, J.-M.; Xia, Z.; Dai, L.; Baek, J.-B. Edge-Selectively Sulfurized Graphene Nanoplatelets as Efficient Metal-Free Electrocatalysts for Oxygen Reduction Reaction: The Electron Spin Effect. Adv. Mater. 2013, 25, 6138–6145. [Google Scholar] [CrossRef]
- Yang, S.; Feng, X.; Wang, X.; Müllen, K. Graphene-Based Carbon Nitride Nanosheets as Efficient Metal-Free Electrocatalysts for Oxygen Reduction Reactions. Angew. Chem. Int. Ed. 2011, 50, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, L.; Xia, Z.; Roy, A.; Chang, D.W.; Baek, J.-B.; Dai, L. BCN Graphene as Efficient Metal-Free Electrocatalyst for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2012, 51, 4209–4212. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Mao, Y.; Wang, H.-F.; Hu, P. Theoretical Study of Heteroatom Doping in Tuning the Catalytic Activity of Graphene for Triiodide Reduction. ACS Catal. 2016, 6, 6804–6813. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bui, V.Q.; Lee, J.; Wang, L.; Jadhav, A.R.; Liu, X.; Shao, X.; Liu, Y.; Yu, J.; Hwang, Y.; et al. Moving beyond bimetallic-alloy to single-atom dimer atomic-interface for all-pH hydrogen evolution. Nat. Commun. 2021, 12, 6766. [Google Scholar] [CrossRef]
- Jia, C.; Ren, W.; Chen, X.; Yang, W.; Zhao, C. (N, B) Dual Heteroatom-Doped Hierarchical Porous Carbon Framework for Efficient Electroreduction of Carbon Dioxide. ACS Sustain. Chem. Eng. 2020, 8, 6003–6010. [Google Scholar] [CrossRef]
- Chen, X.; Ge, F.; Lai, N. N, O Co-Doped Graphene as a Potential Catalyst for the Oxygen Reduction Reaction. J. Electrochem. Soc. 2019, 166, F847. [Google Scholar] [CrossRef]
- Feng, Z.; Tang, Y.; Chen, W.; Wei, D.; Ma, Y.; Dai, X. O-doped graphdiyne as metal-free catalysts for nitrogen reduction reaction. Mol. Catal. 2020, 483, 110705. [Google Scholar] [CrossRef]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Zhang, L.; Du, A.; Gao, G.; Chen, J.; Yan, X.; Brown, C.L.; Yao, X. Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions. Adv. Mater. 2016, 28, 9532–9538. [Google Scholar] [CrossRef]
- Qiu, Y.; Ali, S.; Lan, G.; Tong, H.; Fan, J.; Liu, H.; Li, B.; Han, W.; Tang, H.; Liu, H.; et al. Defect-rich activated carbons as active and stable metal-free catalyst for acetylene hydrochlorination. Carbon 2019, 146, 406–412. [Google Scholar] [CrossRef]
- Tian, W.; Li, W.; Yu, W.; Liu, X. A Review on Lattice Defects in Graphene: Types, Generation, Effects and Regulation. Micromachines 2017, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Kleis, J.; Greeley, J.; Romero, N.A.; Morozov, V.A.; Falsig, H.; Larsen, A.H.; Lu, J.; Mortensen, J.J.; Dułak, M.; Thygesen, K.S.; et al. Finite Size Effects in Chemical Bonding: From Small Clusters to Solids. Catal. Lett. 2011, 141, 1067–1071. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G. Ab initio molecular dynamics for liquid metals. J. Non-Cryst. Solids 1995, 192–193, 222–229. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396–1396. [Google Scholar] [CrossRef] [Green Version]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef]
- Mathew, K.; Kolluru, V.S.C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Valdés, Á.; Qu, Z.W.; Kroes, G.J.; Rossmeisl, J.; Nørskov, J.K. Oxidation and Photo-Oxidation of Water on TiO2 Surface. J. Phys. Chem. C 2008, 112, 9872–9879. [Google Scholar] [CrossRef]
- Choi, J.I.; Kim, H.S.; Sohn, Y.-J.; Yim, S.-D.; Alamgir, F.M.; Jang, S.S. Density Functional Theory Study of Oxygen Reduction on Graphene and Platinum Surfaces of Pt–Graphene Hybrids. ACS Appl. Nano Mater. 2021, 4, 1067–1075. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef]
- Dronskowski, R.; Bloechl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO2 | CO | H | |
|---|---|---|---|
| graphene | −0.15 | −0.13 | 1.31 |
| void-graphene | −0.64 | −3.00 | −0.34 |
| hole-graphene | −0.14 | −0.13 | 0.26 |
| BPC | −0.15 | −0.14 | 0.56 |
| BPC-O | −0.17 | −0.14 | −0.46 |
| void-NPC | −0.68 | −0.14 | −0.75 |
| hole-NPC | −0.19 | −0.15 | −0.42 |
| void-NBPC | −1.48 | −2.16 | −1.00 |
| hole-NBPC | −0.19 | −0.15 | −0.78 |
| void-NBPC-O | −0.19 | −0.15 | −0.59 |
| CO2→*COOH | *COOH→*CO | *CO→CO | Overpotential | |
|---|---|---|---|---|
| graphene | 2.45 | −1.64 | −0.31 | 2.20 |
| void-graphene | 0.97 | −2.76 | 2.29 | 0.72 |
| hole-graphene | 1.30 | −0.45 | −0.34 | 1.05 |
| BPC | 1.52 | −0.76 | −0.26 | 1.27 |
| BPC-O | 1.53 | −0.72 | −0.31 | 1.28 |
| void-NPC | 0.94 | −0.18 | −0.26 | 0.69 |
| hole-NPC | 1.30 | −0.59 | −0.21 | 1.05 |
| void-NBPC | −0.37 | −0.64 | 1.51 | - |
| hole-NBPC | 1.24 | −0.52 | −0.21 | 0.99 |
| void-NBPC-O | 0.89 | −0.12 | −0.27 | 0.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.; Meng, F.; Li, X.; Liu, Y.; Tan, H.; Chen, G. Theoretical Study of the Defects and Doping in Tuning the Electrocatalytic Activity of Graphene for CO2 Reduction. Nanomaterials 2023, 13, 2273. https://doi.org/10.3390/nano13152273
Su X, Meng F, Li X, Liu Y, Tan H, Chen G. Theoretical Study of the Defects and Doping in Tuning the Electrocatalytic Activity of Graphene for CO2 Reduction. Nanomaterials. 2023; 13(15):2273. https://doi.org/10.3390/nano13152273
Chicago/Turabian StyleSu, Xiao, Fanqi Meng, Xiang Li, Yueying Liu, Hongwei Tan, and Guangju Chen. 2023. "Theoretical Study of the Defects and Doping in Tuning the Electrocatalytic Activity of Graphene for CO2 Reduction" Nanomaterials 13, no. 15: 2273. https://doi.org/10.3390/nano13152273