



Low-Temperature Predicted Structures of Ag2S (Silver Sulfide)

 ,
,  and
and 
Abstract
:1. Introduction
2. Computational Simulations
3. Results and Discussion
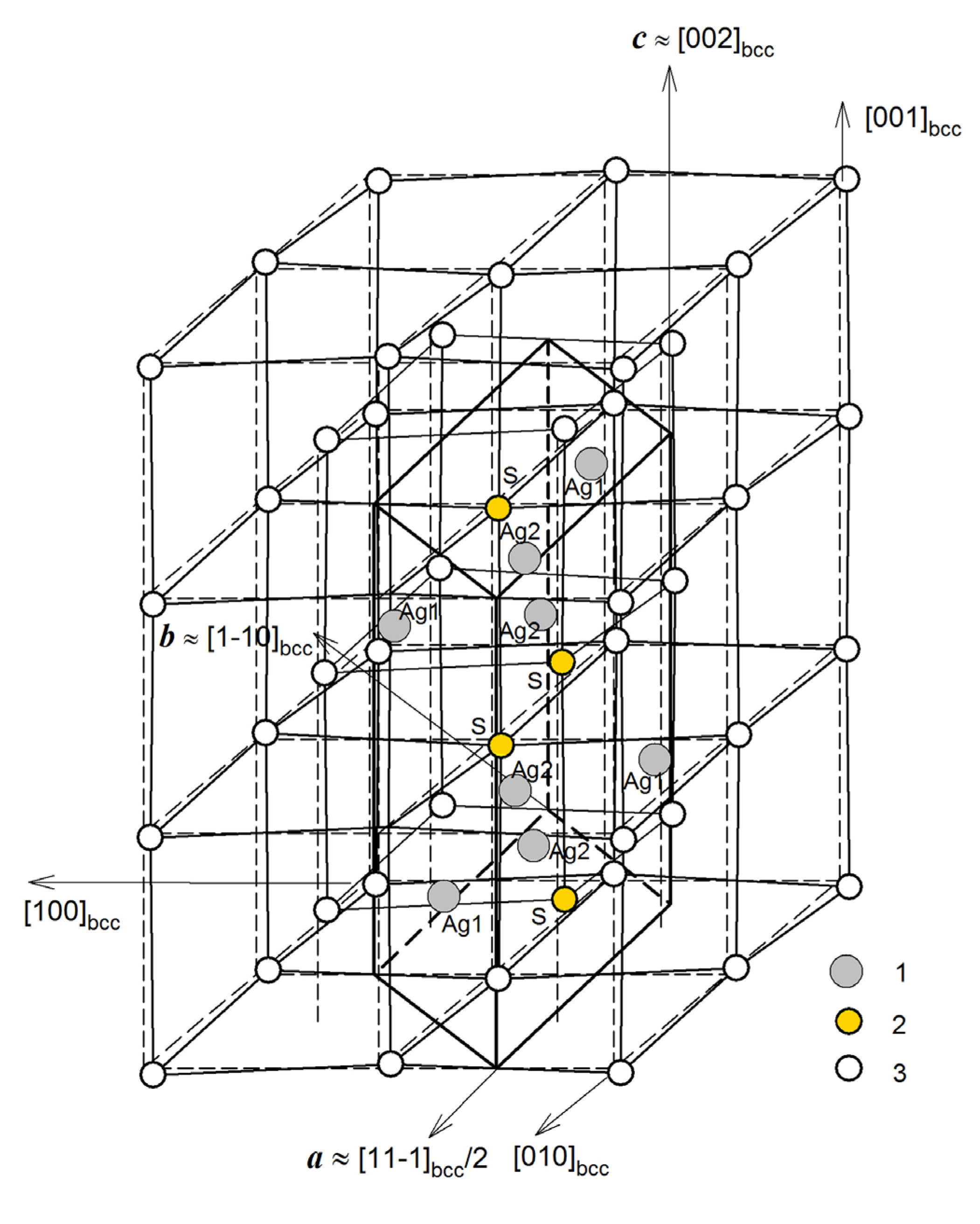
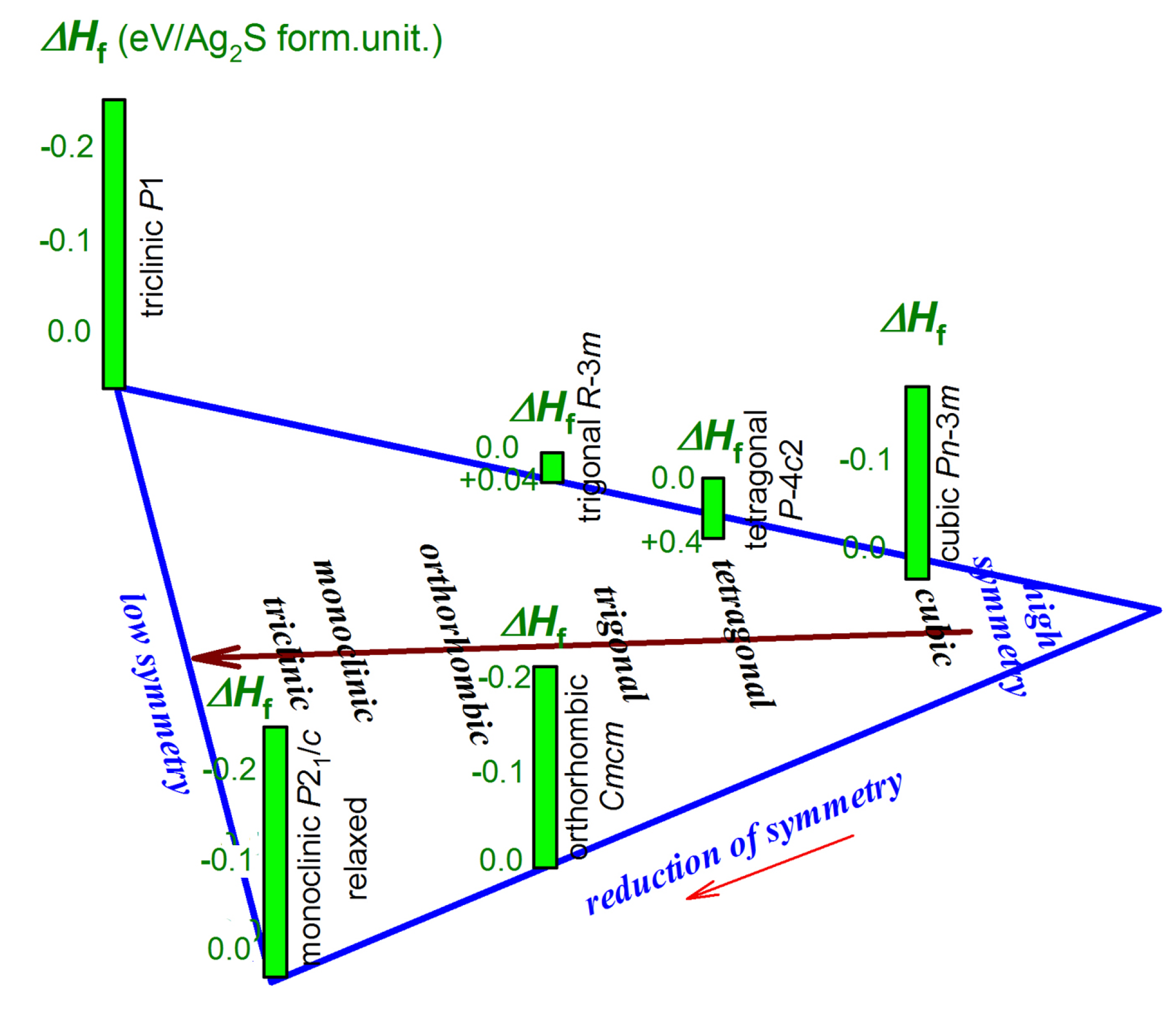
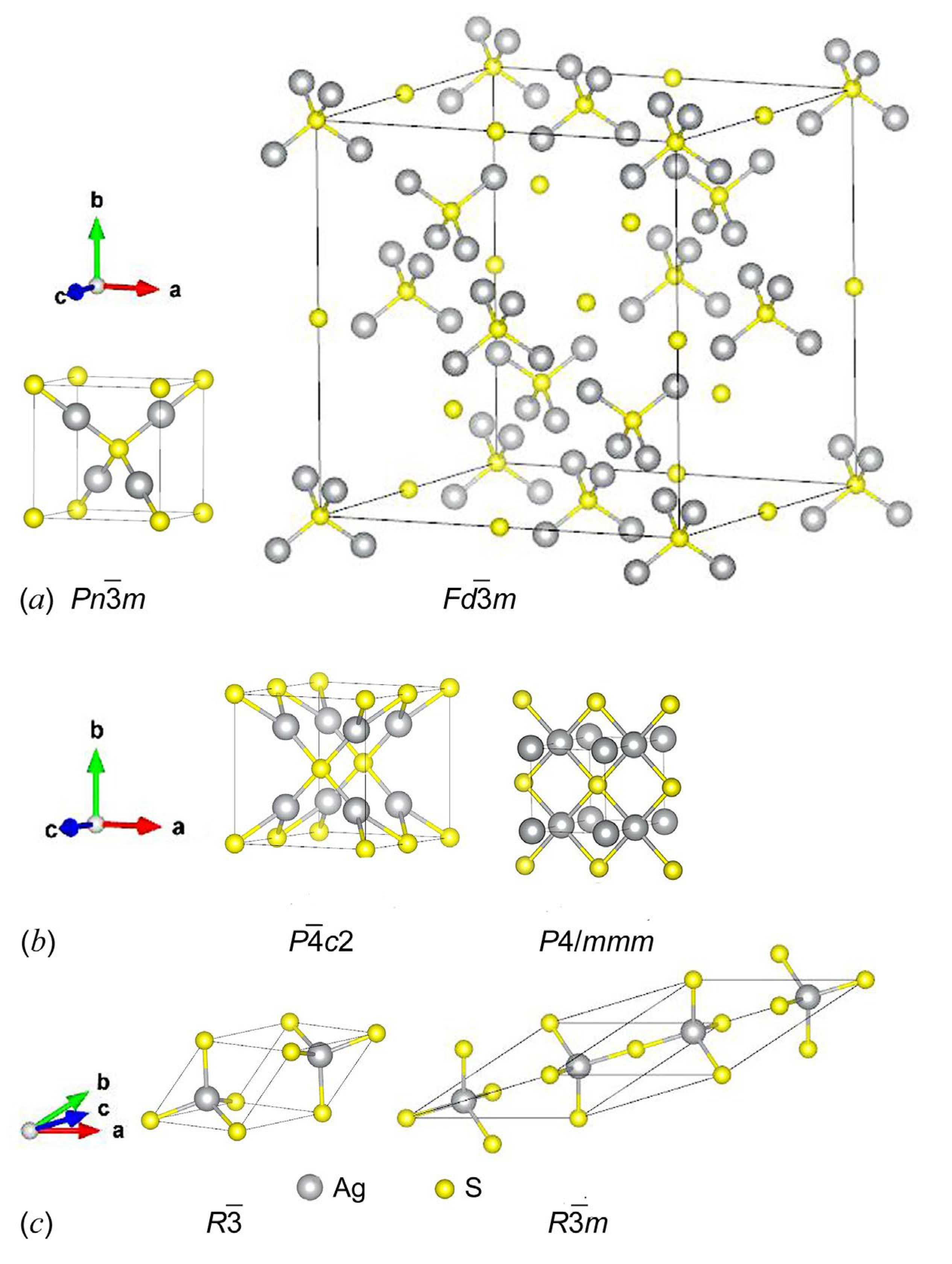
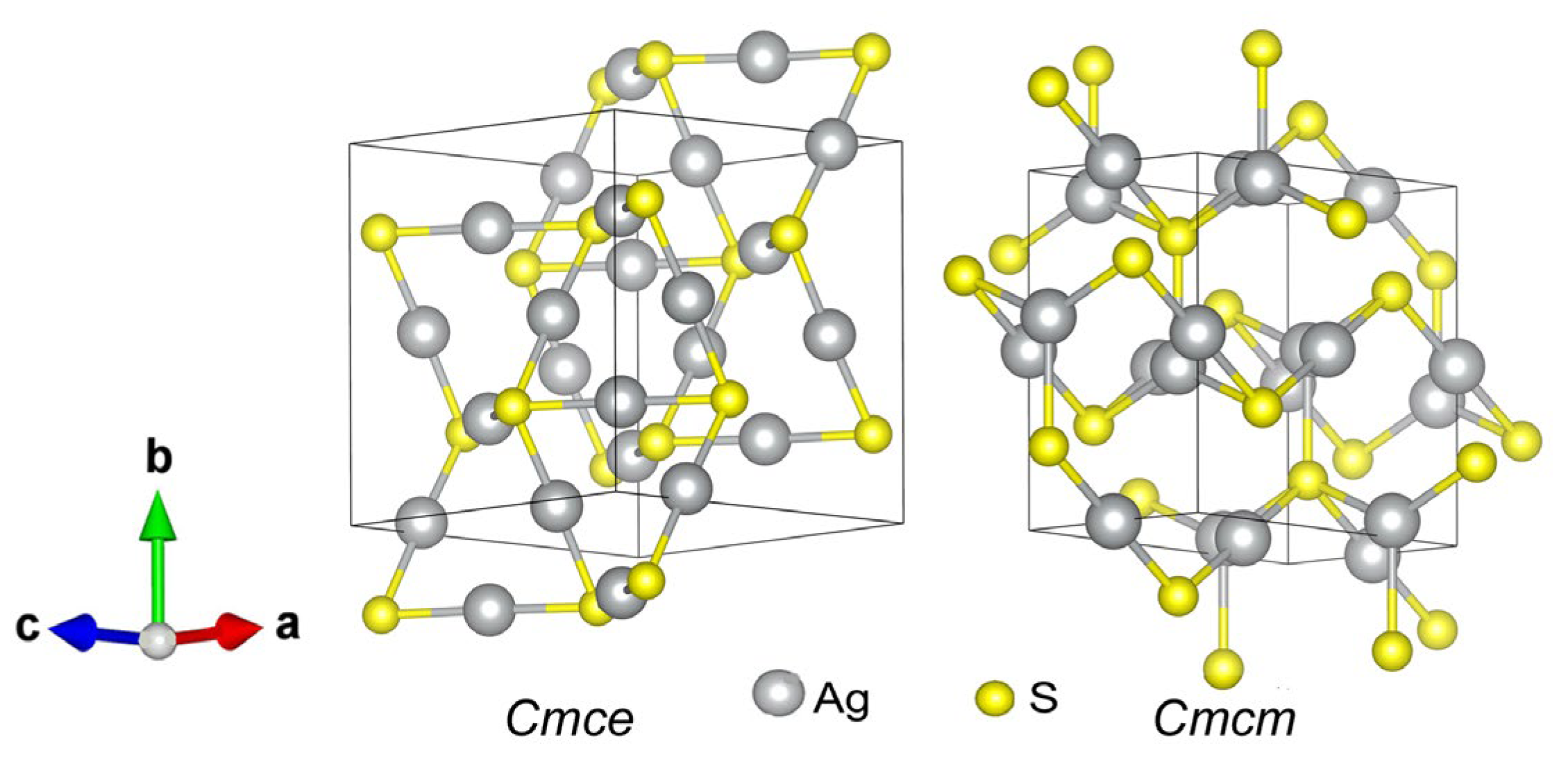
3.1. Structures and Energies of the Predicted Ag2S (Silver Sulfide) Phases
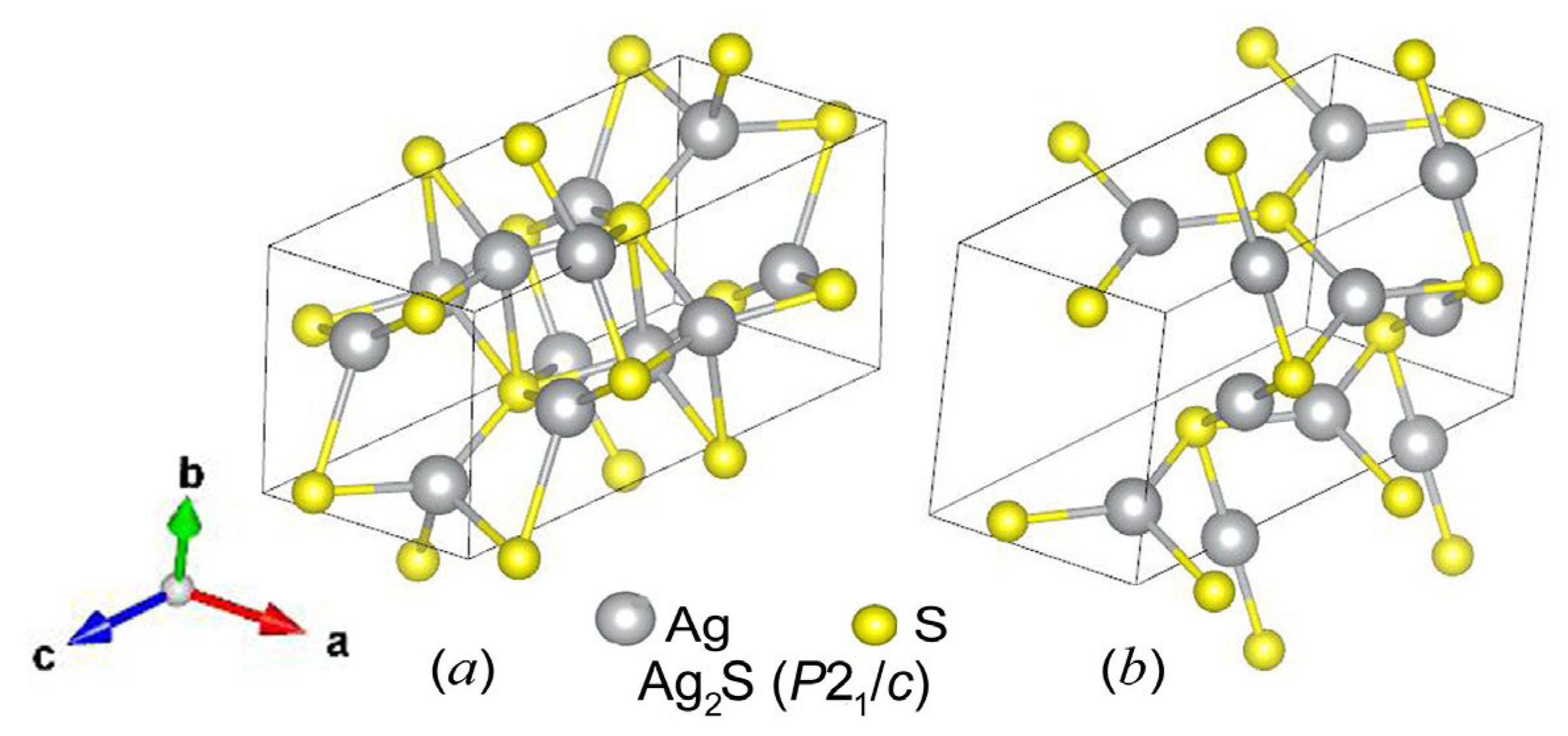
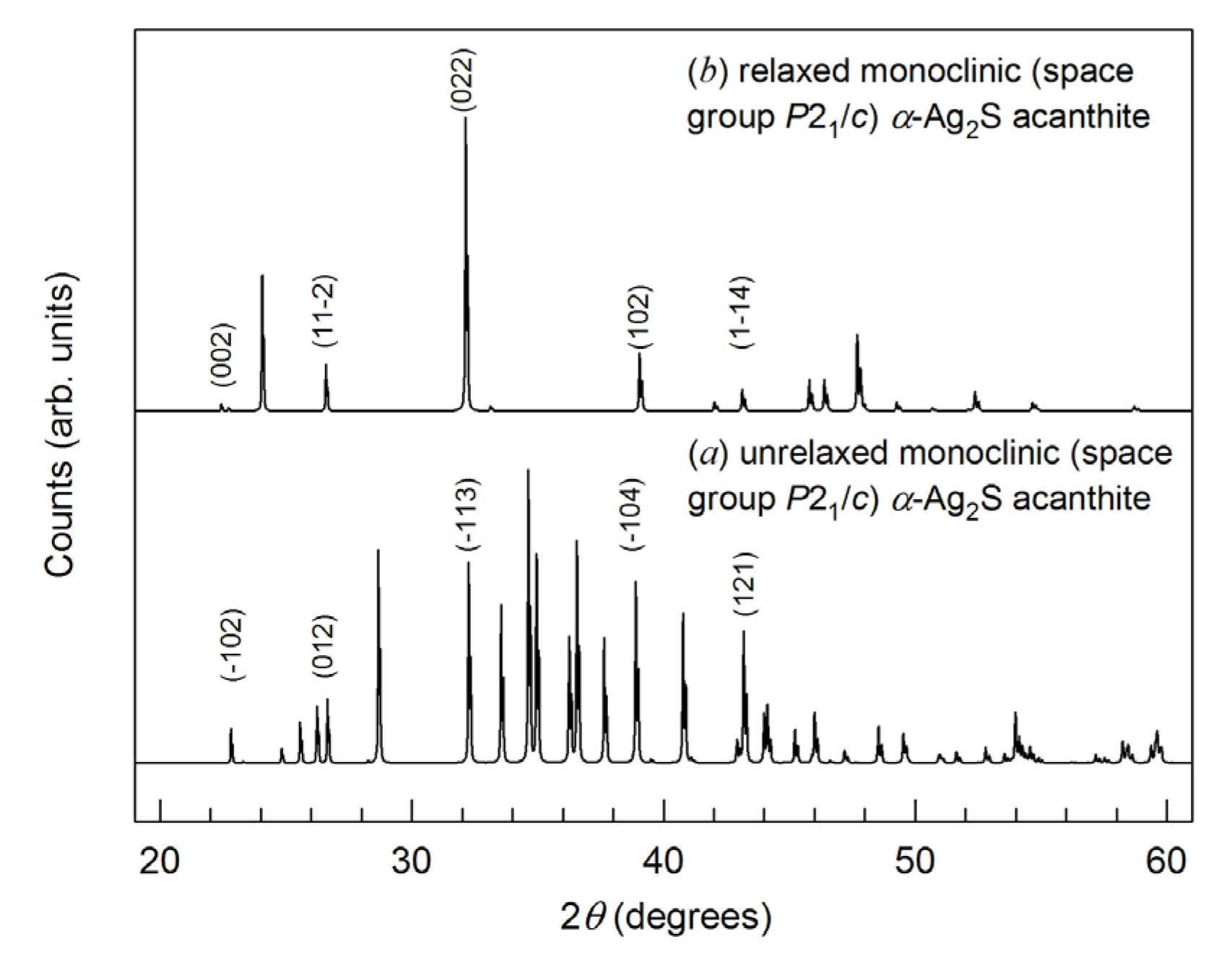
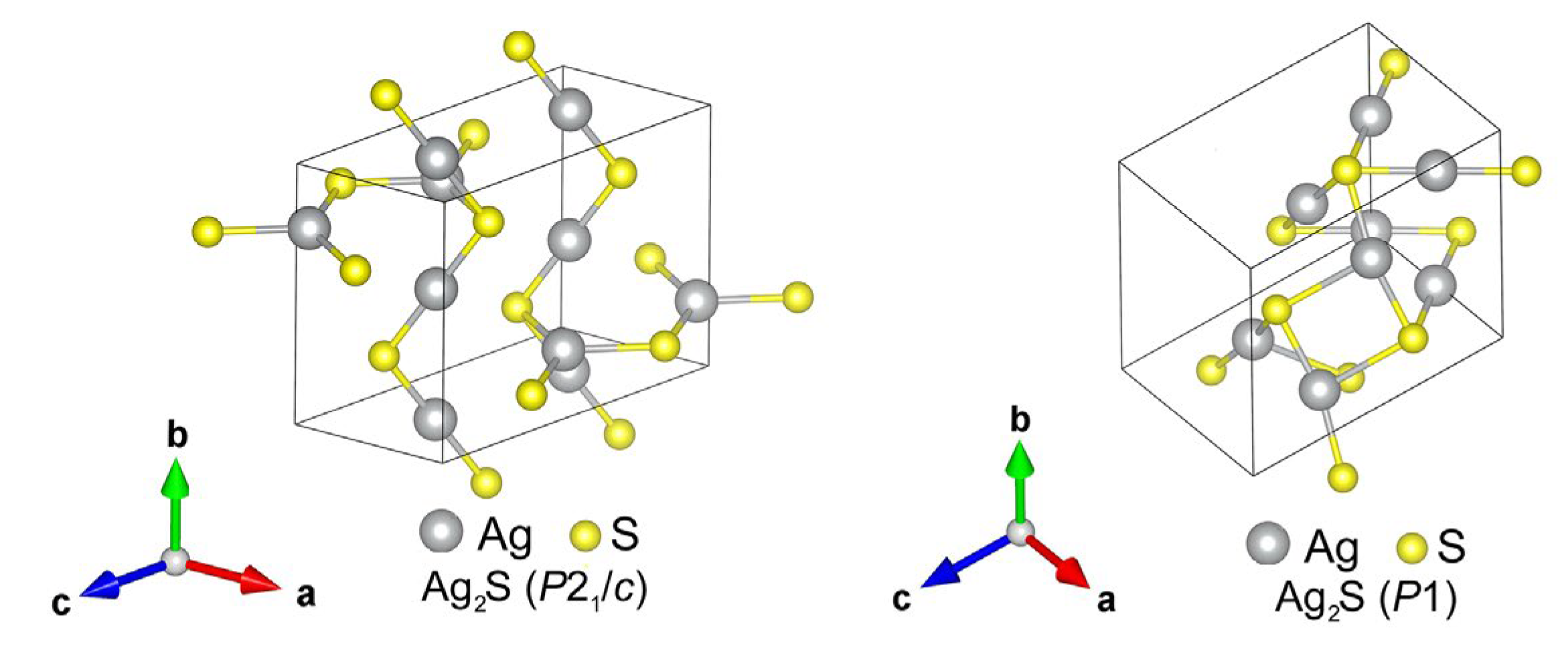
3.2. Symmetry Analysis of the Predicted Structures
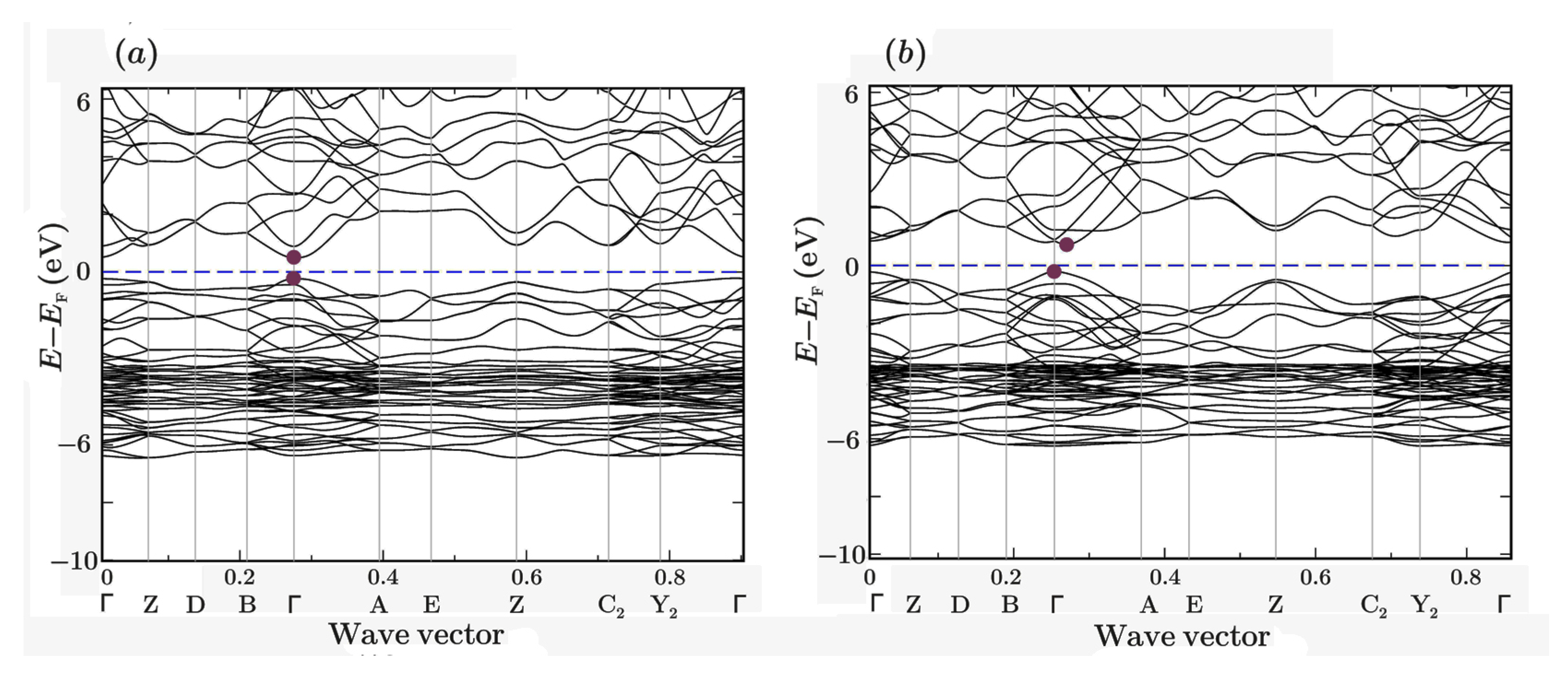
3.3. Electronic Structure
3.4. Elastic Properties and Hardness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sharma, R.C.; Chang, Y.A. The Ag-S (Silver-Sulfur) system. Bull. Alloy Phase Diagr. 1986, 7, 263–269. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Rempel, A.A.; Gusev, A.I. Nanostructured Lead, Cadmium and Silver Sulfides: Structure, Nonstoichiometry and Properties; Springer International Publishing AG: Cham/Heidelberg, Germany, 2018; 331p. [Google Scholar]
- Thompson, W.T.; Flengas, S.N. Drop calorimetric measurements on some chlorides, sulfides, and binary melts. Can. J. Chem. 1971, 49, 1550–1563. [Google Scholar] [CrossRef]
- Grønvold, F.; Westrum, E.F. Silver(I) sulfide: Ag2S heat capacity from 5 to 1000 K, thermodynamic properties, and transitions. J. Chem. Therm. 1986, 18, 381–401. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. Nonstoichiometry of nanocrystalline monoclinic silver sulfide. Phys. Chem. Chem. Phys. 2015, 17, 12466–12471. [Google Scholar] [CrossRef] [PubMed]
- Sadanaga, R.; Sueno, S. X-ray study on the α-β transition of Ag2S. Mineralog. J. Jpn. 1967, 5, 124–148. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I. Recent progress in nanostructured silver sulfide Ag2S: From synthesis and nonstoichiometry to properties. J. Mater. Chem. A 2017, 5, 17676–17704. [Google Scholar] [CrossRef]
- Van Doorselaer, M.K. Solid state properties and photographic activiti of crystalline Ag2S and (Ag, Au)2S-specks at the surface of silver halide crystalls. J. Photograph. Sci. 1987, 35, 42–52. [Google Scholar] [CrossRef]
- Simonnin, P.; Sassi, M.; Gilbert, B.; Charlet, L.; Rosso, K.M. Phase transition and liquid-like superionic conduction in Ag2S. J. Phys. Chem. C 2020, 124, 10150–10158. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gerasimov, E.Y. Direct TEM observation of the “acanthite α-Ag2S–argentite β-Ag2S” phase transition in a silver sulfide nanoparticle. Nanoscale Adv. 2019, 1, 1581–1588. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. High-temperature scanning electron microscopy study of acanthite—Argentite phase transformation in nanocrystalline silver sulfide powder. Phys. Chem. Chem. Phys. 2015, 17, 20495–20501. [Google Scholar] [CrossRef]
- Alekperov, O.; Jahangirli, Z.; Paucar, R. First-principles lattice dynamics and Raman scattering in ionic conductor β-Ag2S. Phys. stat. Sol. 2016, 253, 2049–2055. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I. Acanthite-argentite transformation in silver sulfide as a disorder-order transition. JETP Lett. 2019, 109, 584–588. [Google Scholar] [CrossRef]
- Blanton, T.; Misture, S.; Dontula, N.; Zdzieszynski, S. In situ high-temperature X-ray diffraction characterization of silver sulfide, Ag2S. Powder Diff. 2011, 26, 110–118. [Google Scholar] [CrossRef]
- Kashida, S.; Watanabe, N.; Hasegawa, T.; Iida, H.; Mori, M.; Savrasov, S. Electronic structure of Ag2S, band calculation and photoelectron spectroscopy. Sol. State Ion. 2003, 158, 167–175. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Available online: https://next-gen.materialsproject.org (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32884 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-556225 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-1095694 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32669 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-36216 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-31053 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32791 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-610517 (accessed on 20 August 2023).
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Cond. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Murugadoss, G.; Jayavel, R.; Rajesh Kumar, M.; Thangamuthu, R. Synthesis, optical, photocatalytic, and electrochemical studies on Ag2S/ZnS and ZnS/Ag2S nanocomposites. Appl. Nanosci. 2016, 6, 503–510. [Google Scholar] [CrossRef]
- Tang, A.; Wang, Y.; Ye, H.; Zhou, C.; Yang, C.; Li, X.; Peng, H.; Zhang, F.; Hou, Y.; Teng, F. Controllable synthesis of silver and silver sulfide nanocrystals via selective cleavage of chemical bonds. Nanotechnology 2013, 24, 355602. [Google Scholar] [CrossRef]
- Du, N.; Zhang, H.; Sun, H.Z.; Yang, D.R. Sonochemical synthesis of amorphous long silver sulfide nanowires. Mater. Lett. 2007, 61, 235–238. [Google Scholar] [CrossRef]
- Liu, L.; Hu, S.; Dou, Y.-P.; Liu, T.; Lin, J.; Wang, Y. Nonlinear optical properties of near-infrared region Ag2S quantum dots pumped by nanosecond laser pulses. Beilst. J. Nanotechnol. 2015, 6, 1781–1787. [Google Scholar] [CrossRef]
- Terabe, K.; Hasegawa, T.; Nakayama, T.; Aono, M. Quantized conductance atomic switch. Nature 2005, 433, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.H.; Terabe, K.; Hasegawa, T.; Aono, M. Resistance switching of an individual Ag2S/Ag nanowire heterostructure. Nanotechnology 2007, 18, 485202. [Google Scholar] [CrossRef]
- Xu, Z.; Bando, Y.; Wang, W.; Bai, X.; Golberg, D. Real-time in situ HRTEM-resolved resistance switching of Ag2S Nanoscale ionic conductor. ACS Nano 2010, 4, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.N.; Pyatilova, O.V.; Vorobiev, M.I. Synthesis of Ag/Ag2S nanoclusters resistive switches for memory cells. Adv. Nanopart. 2014, 3, 1–4. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Liu, Y.; Peng, L.; Chen, X.; Wang, Q. Manganese-doped Ag2S-ZnS heteronanostructures. Chem. Mater. 2012, 24, 2407–2413. [Google Scholar] [CrossRef]
- Nasrallah, T.B.; Dlala, H.; Amlouk, M.; Belgacem, S.; Bernede, J.C. Some physical investigations on Ag2S thin films prepared by sequential thermal evaporation. Synth. Met. 2005, 151, 225–230. [Google Scholar] [CrossRef]
- Karashanova, D.; Nihtianova, D.; Starbova, K.; Starbov, N. Crystalline structure and phase composition of epitaxially grown Ag2S thin films. Sol. State Ion. 2004, 171, 269–275. [Google Scholar] [CrossRef]
- Jadhav, U.M.; Patel, S.N.; Patil, R.S. Synthesis of silver sulphide nanoparticles by modified chemical route for solar cell applications. Res. J. Chem. Sci. 2013, 3, 69–74. [Google Scholar]
- Yang, J.; Ying, J.Y. Nanocomposites of Ag2S and noble metals. Angew. Chem. Int. Ed. 2011, 50, 4637–4643. [Google Scholar] [CrossRef]
- Hasegawa, T.; Terabe, K.; Tsuruoka, T.; Aono, M. Atomic switch: Atom/ion movement controlled devices for beyond Von-Neumann computers. Adv. Mater. 2012, 24, 252–267. [Google Scholar] [CrossRef]
- Wang, D.; Liu, L.; Kim, Y.; Huang, Z.; Pantel, D.; Hesse, D.; Alexe, M. Fabrication and characterization of extended arrays of Ag2S/Ag nanodot resistive switches. Appl. Phys. Lett. 2011, 98, 243109. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Wang, M.; Zhang, Y.; Chen, G.; Li, L.; Wu, D.; Wang, Q. In vivo real-time visualization of tissue blood flow and angiogenesis using Ag2S quantum dots in the NIR-II window. Biomaterials 2014, 35, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.; Hasegawa, T.; Chikyow, T. Ag2S atomic switch-based ‘tug of war’ for decision making. Nanoscale 2016, 8, 14031–14035. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Nie, S. Quantum dot-encoded mesoporous beads with hgh brightness and uniformity: Rapid readout using flow cytometry. Anal. Chem. 2004, 76, 2406–2410. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, Y.; Jiang, X.; Liu, W.; Liu, S.; Tang, Z. Facile preparation of Ag2S/Ag semiconductor/metal heteronanostructures with remarkable antibacterial properties. ChemPhysChem 2012, 13, 2531–2535. [Google Scholar] [CrossRef]
- Chang, P.; Cheng, H.; Lin, W.; Li, X.; Zhao, F. A stable and active AgxS crystal preparation and its performance as photocatalyst. Chin. J. Catal. 2015, 36, 564–571. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, G.; Chen, F. Hollow spheric Ag–Ag2S/TiO2 composite and its applicationfor photocatalytic reduction of Cr(VI). Appl. Surf. Sci. 2015, 351, 962–968. [Google Scholar] [CrossRef]
- Xiao, C.; Xu, J.; Li, K.; Feng, J.; Yang, J.; Xie, Y. Superionic phase transition in silver chalcogenide nanocrystals realizing optimized thermoelectric performance. J. Amer. Chem. Soc. 2012, 134, 4287–4293. [Google Scholar] [CrossRef]
- Universal Structure Predictor: Evolutionary Xtallography. Manual. Version 9.4.4. Available online: http://uspex-team.org (accessed on 20 August 2023).
- Etris, S.F. Silver and Silver Alloys. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons (Wiley): New York, NY, USA, 2001; Volume 4, pp. 761–803. [Google Scholar]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How evolutionary crystal structure prediction works—And why. Accounts Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comp. Phys. Comm. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Vienna Ab-initio Simulation Package. VASP the GUIDE. 20 April 2016. Available online: http://cms.mpi.univie.ac.at/VASP/ (accessed on 20 August 2023).
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Hinuma, Y.; Pizzi, G.; Kumagai, Y.; Oba, F.; Tanaka, I. Band structure diagram paths based on crystallography. Comp. Mater. Sci. 2017, 128, 140–184. [Google Scholar] [CrossRef]
- Mouhat, P.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Gusev, A.I.; Sadovnikov, S.I. Conditions of mechanical stability and elastic properties of crystal structures with different symmetry. Fiz. Tverd. Tela 2022, 64, 671–675. (In Russian) [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. Artificial silver sulfide Ag2S: Crystal structure and particle size in deposited powders. Superlat. Microstr. 2015, 83, 35–47. [Google Scholar] [CrossRef]
- International Tables for X-ray Crystallography; Volume A1: Symmetry Relations between Space Groups; Wondratschek, H.; Müller, U. (Eds.) Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, NY, USA; London, UK, 2004; 731p. [Google Scholar]
- Kovalev, O.V. Representations of the Crystallographic Space Groups: Irreducible Representations, Induced Representations and Corepresentation, 2nd ed.; Gordon & Breach Science Publications: Yverdon, Switzerland; Paris, France; Berlin, Germany; London, UK; Tokyo, Japan; Amsterdam, The Netherlands, 1993; 390p. [Google Scholar]
- Flurry, R.L. Symmetry Groups: Theory and Chemical Applications; Prentice-Hall Inc.: Englewood Cliffs, NJ, USA, 1980; p. 356. [Google Scholar]
- Gusev, A.I.; Rempel, A.A.; Magerl, A.J. Disorder and Order in Strongly Nonstoichiometric Compounds: Transition Metal Carbides, Nitrides and Oxides; Springer: Berlin, Germany, 2001; 608p. [Google Scholar]
- Kryukov, A.I.; Stroyuk, O.L.; Zin’chuk, N.N.; Korzhak, A.V.; Kuchmii, S.Y. Optical and catalytical properties of Ag2S nanoparticles. J. Mol. Catal. A 2004, 221, 209–221. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Kuznetsova, Y.V.; Rempel, A.A. Ag2S silver sulfide nanoparticles and colloidal solutions: Synthesis and properties. Nanostr. Nano-Object. 2016, 7, 81–91. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Kozlova, E.A.; Gerasimov, E.Y.; Rempel, A.A.; Gusev, A.I. Enhanced photocatalytic hydrogen evolution from aqueous solutions on Ag2S/Ag heteronanostructure. Int. J. Hydrog. Energy 2017, 42, 25258–25266. [Google Scholar] [CrossRef]
- Liu, Q.; Pu, Y.; Zhao, Z.; Wang, J.; Wang, D. Synthesis of silver sulfide qantum dots via the liquid–liquid interface reaction in a rotating packed bed reactor. Transact. Tianjin Univ. 2020, 26, 273–282. [Google Scholar] [CrossRef]
- Available online: https://matrix.reshish.ru (accessed on 20 August 2023).
- Born, M. On the stability of crystal lattices. Math. Proc. Camb. Phil. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: New York, NY, USA, 1998; 432p. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Sadovnikov, S.I. Bulk modulus of coarse-crystalline and nanocrystalline silver sulfides. Phys. Solid State 2018, 69, 2437–2441. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermet 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refr. Metal Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Mazhnik, E.; Oganov, A.R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 2019, 126, 125109. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symmetry and Space Group | Number of Atoms | 1 Z | 2 Ephase, eV | E coh | 3 ΔHf | N rot | 4 Ntr | N tot | |
|---|---|---|---|---|---|---|---|---|---|
| N Ag | N S | eV/(Ag2S form.unit) | |||||||
| Cubic (No.224-) | 4 | 2 | 2 | −19.498 | −8.272 | −0.191 | 1 | 1.5 | 1.5 |
| Cubic (No.227-) | 32 | 16 | 16 | −95.749 | −4.507 | 3.573 | 1 | 8 | 8 |
| Tetragonal (No.116-) | 8 | 4 | 4 | −36.655 | −7.687 | 0.394 | 6 | 2 | 12 |
| Tetragonal (No.123-P4/mmm) | 2 | 1 | 1 | −8.2445 | −6.767 | 1.313 | 3 | 0.5 | 1.5 |
| Trigonal (No.148 -) | 2 | 1 | 1 | −9.516 | −8.039 | 0.042 | 8 | 0.5 | 4 |
| Trigonal (No.166 -) | 4 | 2 | 2 | −19.033 | −8.039 | 0.041 | 4 | 1 | 4 |
| Orthorhombic (No.64-Cmce) | 16 | 8 | 8 | −78.216 | −8.300 | −0.219 | 6 | 5 | 30 |
| Orthorhombic (No.63-Cmcm) | 8 | 4 | 4 | −39.027 | −8.280 | −0.199 | 6 | 2 | 12 |
| Orthorhombic (No.36-Cmc21) | 8 | 4 | 4 | −39.027 | −8.280 | −0.199 | 12 | 2 | 24 |
| Orthorhombic (No.19-P212121) | 8 | 4 | 4 | −38.384 | −8.119 | −0.038 | 12 | 2 | 24 |
| Monoclinic (No.11-P21/m) | 4 | 2 | 2 | −19.514 | −8.280 | −0.199 | 12 | 1 | 12 |
| Monoclinic (No.4-P21) | 4 | 2 | 2 | −19.532 | −8.289 | −0.208 | 24 | 1.5 | 36 |
| Monoclinic (No.14-P21/c) unrelaxed acanthite | 8 | 4 | 4 | −38.361 | −8.113 | −0.033 | 12 | 2 | 24 |
| Monoclinic (No.14-P21/c) relaxed acanthite | 8 | 4 | 4 | −39.028 | −8.280 | −0.199 | 12 | 2.5 | 30 |
| Monoclinic (No.14-P21/c) | 8 | 4 | 4 | −39.107 | −8.300 | −0.219 | 12 | 3 | 36 |
| Triclinic (No.1-P1) | 8 | 4 | 4 | −39.125 | −8.304 | −0.223 | 48 | 2 | 96 |
| Space Group | Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|---|
| x/a ≡ x/aorthorh | y/b ≡ y/borthorh | z/c ≡ z/corthorh | |||
| * No.63-Cmcm | Ag1 | 4(b) | 0 | 0.5 | 0 |
| Ag2 | 4(c) | 0 | 0.04657 | 0.25 | |
| S | 4(c) | 0 | 0.68496 | 0.25 | |
| ** No. 64-Cmce | Ag1 | 8(e) | 0.25 | 0.22162 | 0.25 |
| Ag2 | 8(f) | 0 | 0.02841 | 0.75496 | |
| S | 8(f) | 0 | 0.72452 | 0.39559 | |
| *** No.36-Cmc21 | Ag | 8(b) | 0 | 0.04705 | 0.06242 |
| S | 4(a) | 0 | 0.31480 | 0.56242 | |
| **** No.19-P212121 | Ag1 | 4(a) | 0.00985 | 0.77359 | 0.64205 |
| Ag2 | 4(a) | 0.15219 | 0.38829 | 0.45552 | |
| S | 4(a) | 0.12297 | 0.00356 | 0.34389 | |
| Monoclinic (Space Group P21/c) α-Ag2S | Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|---|
| x/a ≡ x/amon | y/b ≡ y/bmon | z/c ≡ z/cmon | |||
| * Unrelaxed unit cell [46] | Ag1 | 4(e) | 0.07157 | 0.48487 | 0.80943 |
| Ag2 | 4(e) | 0.27353 | 0.67586 | 0.56247 | |
| S | 4(e) | 0.4922 | 0.2341 | 0.13217 | |
| ** Relaxed unit cell | Ag1 | 4(e) | 0.04498 | 0.74996 | 0.47750 |
| Ag2 | 4(e) | 0.50004 | 0.00001 | 0.25002 | |
| S | 4(e) | 0.31581 | 0.25002 | 0.34210 | |
| Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|
| x/a ≡ x/amon | y/b ≡ y/bmon | z/c ≡ z/cmon | ||
| Ag1 | 4(e) | 0.52641 | 0.24248 | 0.47333 |
| Ag2 | 4(e) | 0.02658 | 0.25188 | 0.47346 |
| S | 4(e) | 0.52441 | 0.39461 | 0.22983 |
| Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|
| x/a ≡ x/atricl | y/b ≡ y/btricl | z/c ≡ z/ctricl | ||
| Ag1 | 1(a) | 0.86160 | 0.11768 | 0.65392 |
| Ag2 | 1(a) | 0.18251 | 0.45397 | 0.34767 |
| Ag3 | 1(a) | 0.68702 | 0.45957 | 0.35685 |
| Ag4 | 1(a) | 0.02431 | 0.56922 | 0.00840 |
| Ag5 | 1(a) | 0.52371 | 0.56411 | 0.00802 |
| Ag6 | 1(a) | 0.36410 | 0.10713 | 0.66484 |
| Ag7 | 1(a) | 0.00327 | 0.00177 | 0.00153 |
| Ag8 | 1(a) | 0.50720 | 0.00312 | 0.01042 |
| S1 | 1(a) | 0.62789 | 0.41309 | 0.70515 |
| S2 | 1(a) | 0.90766 | 0.15612 | 0.30911 |
| S3 | 1(a) | 0.11466 | 0.85369 | 0.70273 |
| S4 | 1(a) | 0.42086 | 0.71554 | 0.31158 |
| Symmetry, Space Group | G V | G R | G H | B V | B R | B H | k | μ | E H | H V | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Equation (5) | Equation (6) | ||||||||||
| Orthorhombic (Cmce) | 13.0 | 3.6 | 8.3 | 18.0 | 10.3 | 14.1 | 0.588 | 0.277 | 20.0 | 2.0 | 1.0 |
| Orthorhombic (Cmcm) | 11.8 | 8.6 | 10.2 | 34.9 | 21.9 | 28.4 | 0.360 | 0.359 | 25.9 | 1.2 | 1.5 |
| Monoclinic (P21) | 13.4 | −2.3 | 5.6 | 13.7 | −2.9 | 5.4 | 1.032 | 0.133 | 12.2 | 3.2 | 1.0 |
| Monoclinic (P21/c) unrelaxed acanthite | 11.6 | 8.6 | 10.1 | 34.3 | 27.9 | 31.1 | 0.326 | 0.371 | 25.9 | 1.0 | 1.5 |
| Monoclinic (P21/c) relaxed acanthite | 11.3 | 8.3 | 9.8 | 35.1 | 21.5 | 28.3 | 0.348 | 0.363 | 25.0 | 1.1 | 1.4 |
| Monoclinic (P21/c) | −0.8 | 12.4 | 5.8 | 25.3 | 61.3 | 43.3 | 0.134 | 0.445 | 15.5 | 0.2 | 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadovnikov, S.I.; Kostenko, M.G.; Gusev, A.I.; Lukoyanov, A.V. Low-Temperature Predicted Structures of Ag2S (Silver Sulfide). Nanomaterials 2023, 13, 2638. https://doi.org/10.3390/nano13192638
Sadovnikov SI, Kostenko MG, Gusev AI, Lukoyanov AV. Low-Temperature Predicted Structures of Ag2S (Silver Sulfide). Nanomaterials. 2023; 13(19):2638. https://doi.org/10.3390/nano13192638
Chicago/Turabian StyleSadovnikov, Stanislav I., Maksim G. Kostenko, Aleksandr I. Gusev, and Aleksey V. Lukoyanov. 2023. "Low-Temperature Predicted Structures of Ag2S (Silver Sulfide)" Nanomaterials 13, no. 19: 2638. https://doi.org/10.3390/nano13192638
APA StyleSadovnikov, S. I., Kostenko, M. G., Gusev, A. I., & Lukoyanov, A. V. (2023). Low-Temperature Predicted Structures of Ag2S (Silver Sulfide). Nanomaterials, 13(19), 2638. https://doi.org/10.3390/nano13192638