
Magnetoelastic and Magnetoelectric Coupling in Two-Dimensional Nitride MXenes: A Density Functional Theory Study



Abstract
:1. Introduction
2. Methods (Computational Approach)
2.1. Density Functional Theory
2.2. LKAG Formalism and Monte Carlo Simulations
3. Results and Discussion
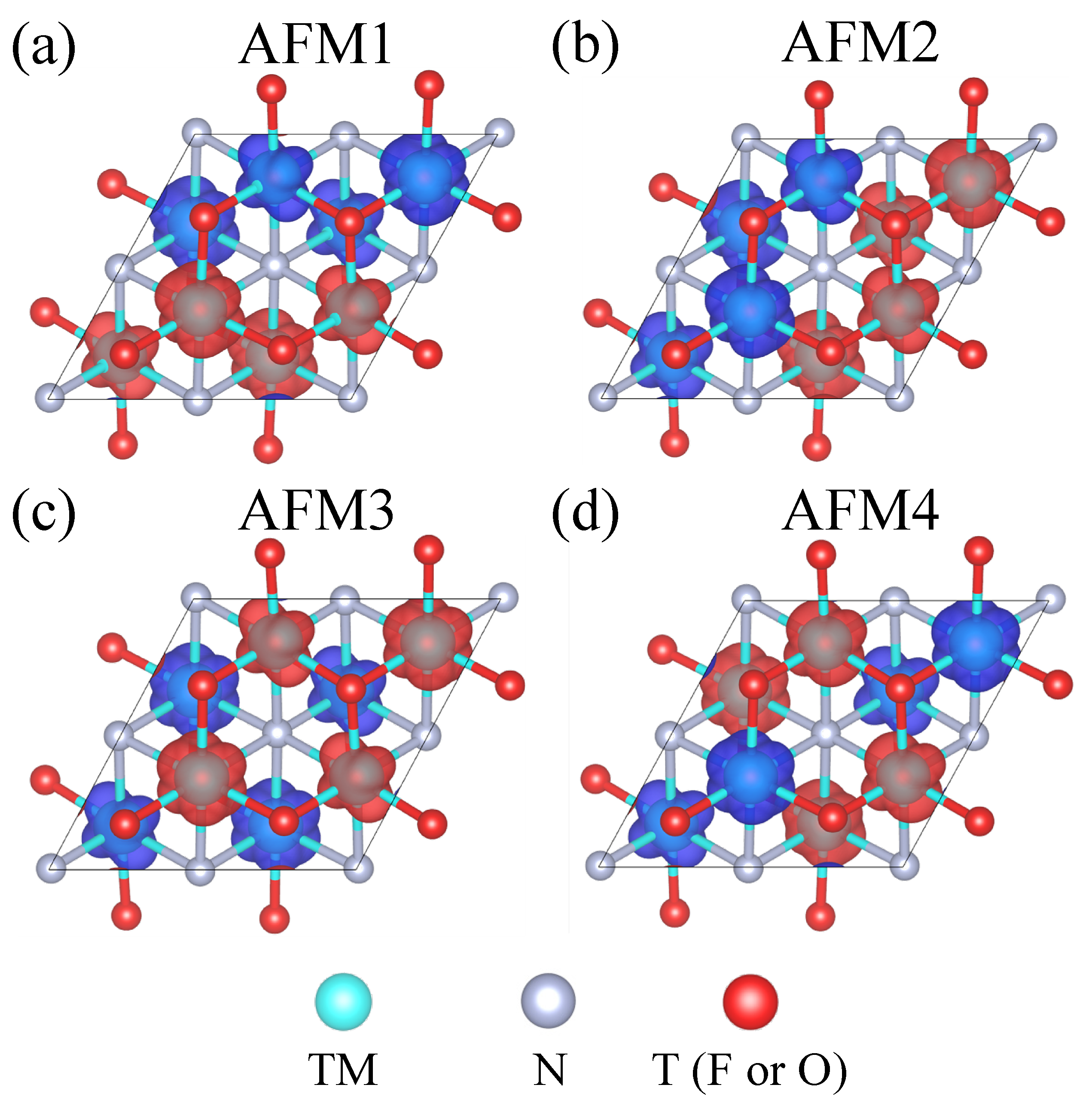
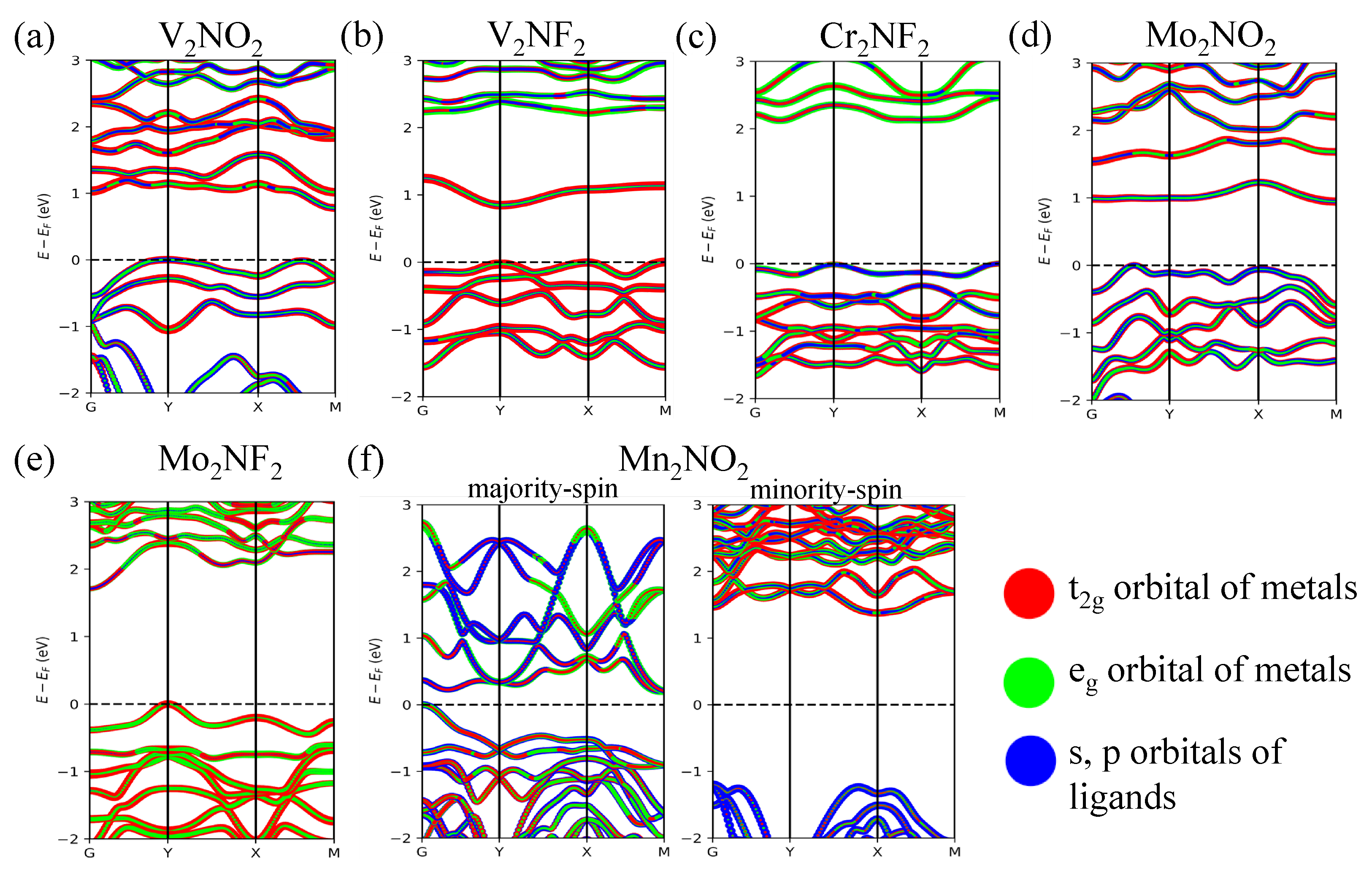
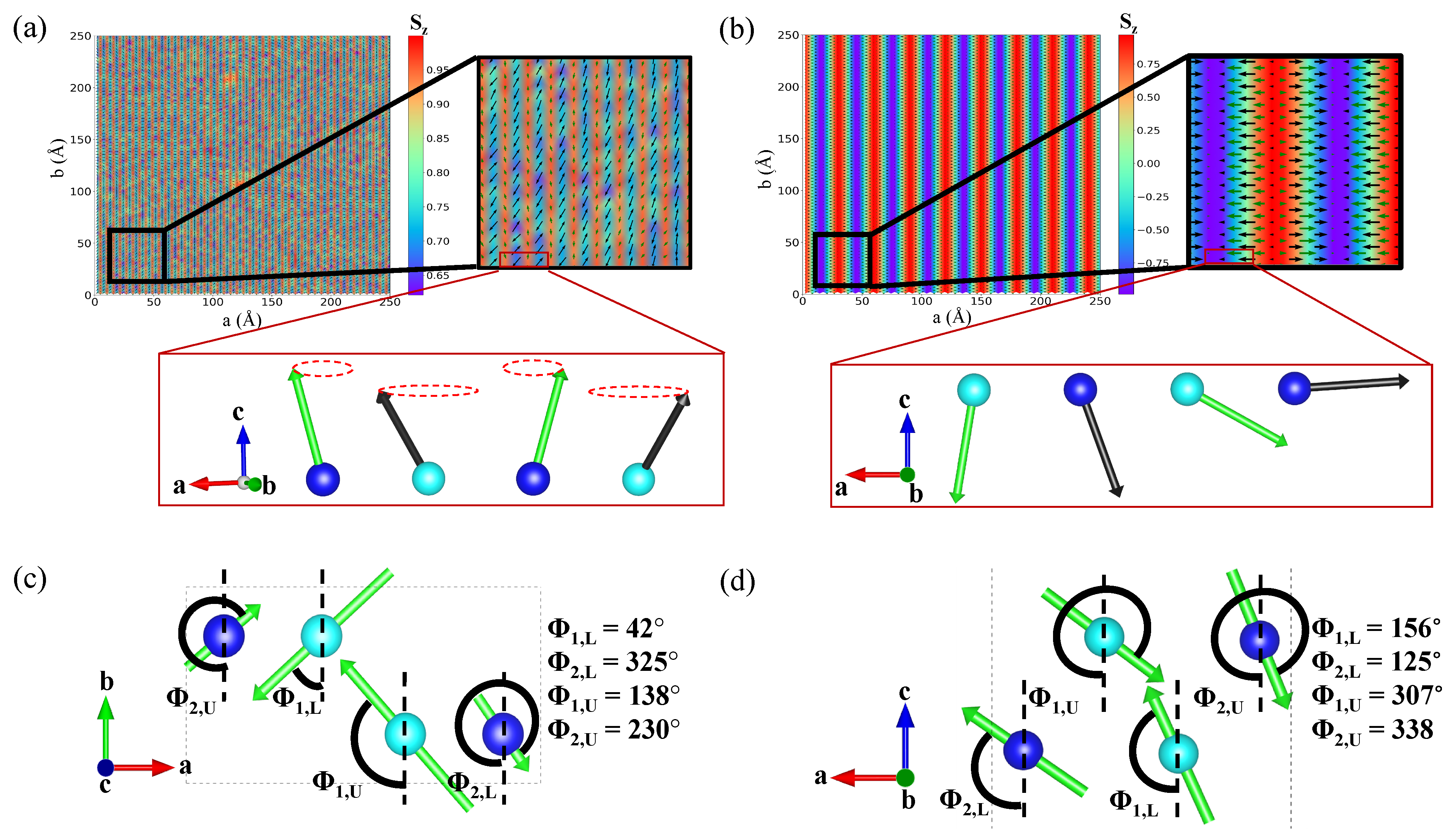
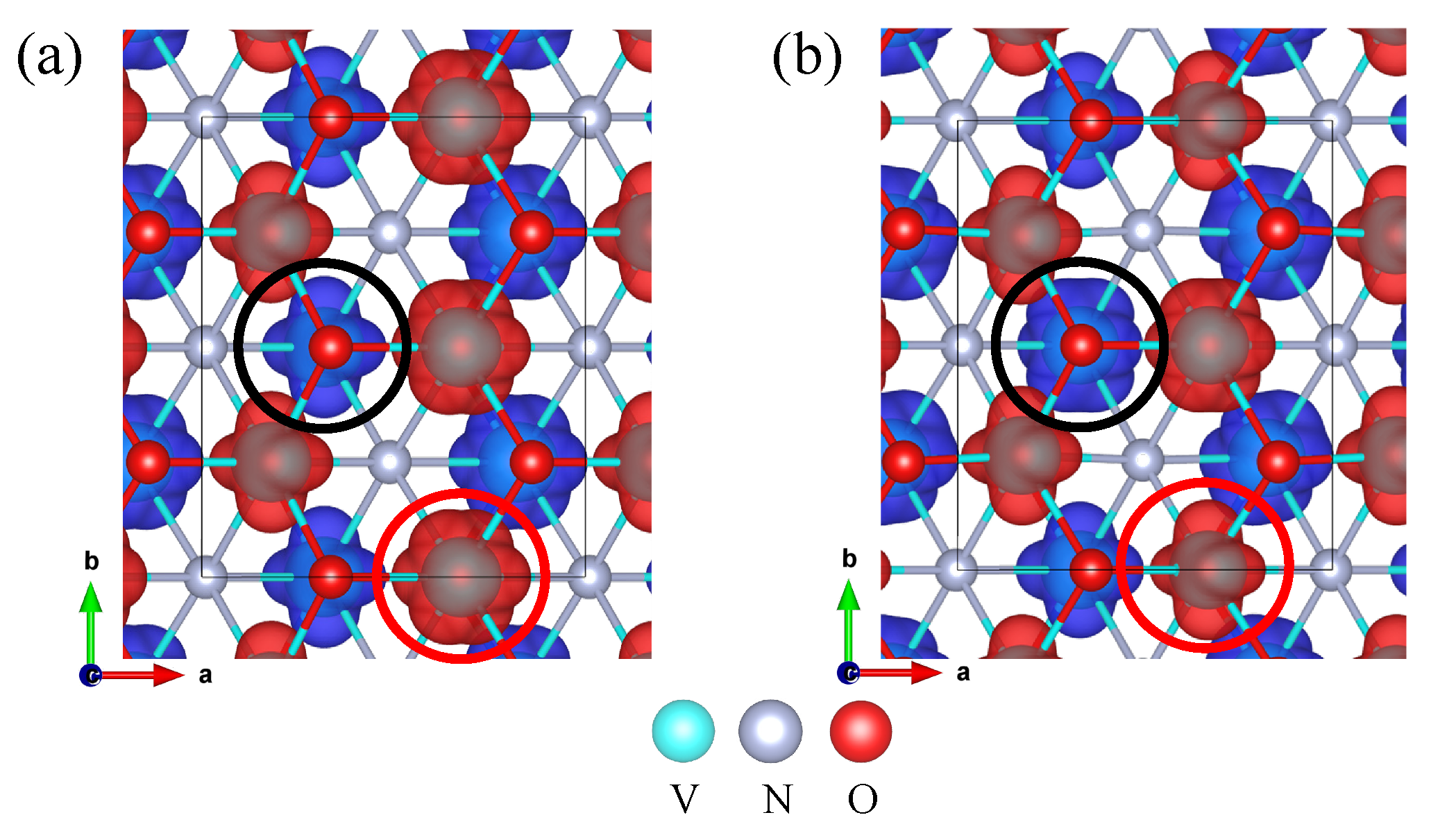
3.1. Orbital Ordering, Electronic, and Magnetic Properties
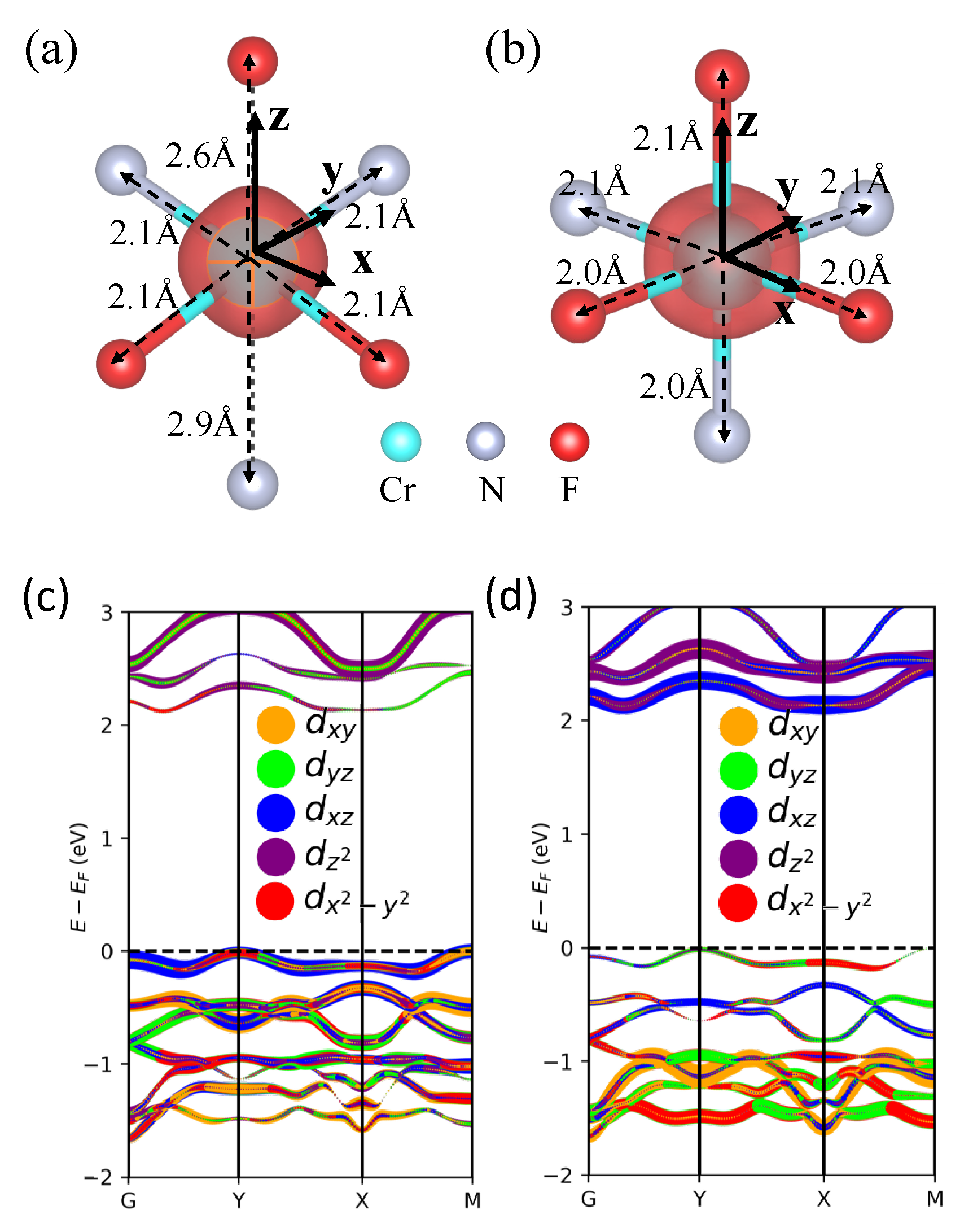
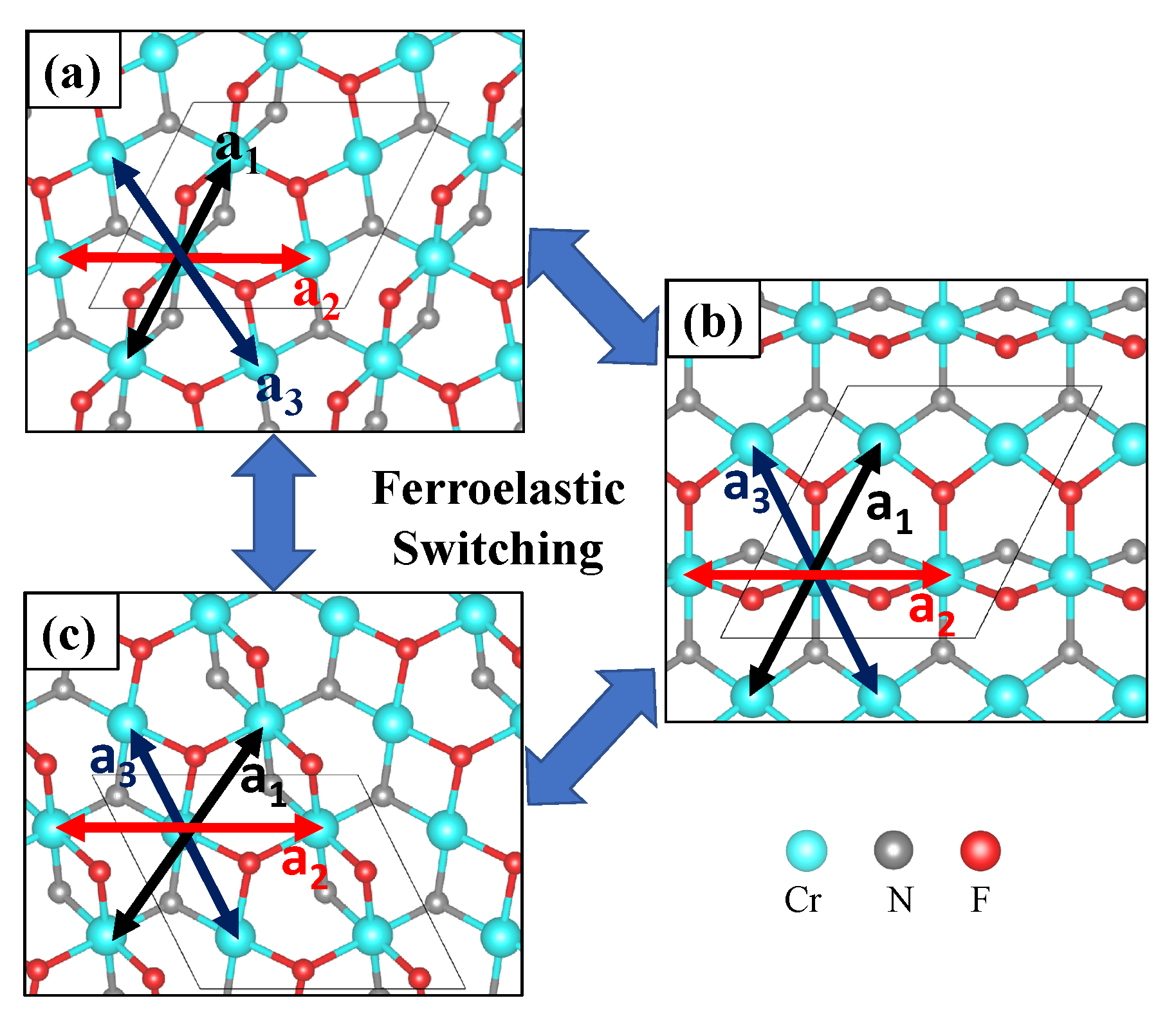
3.2. Magnetoelastic Coupling in Cr2NF2
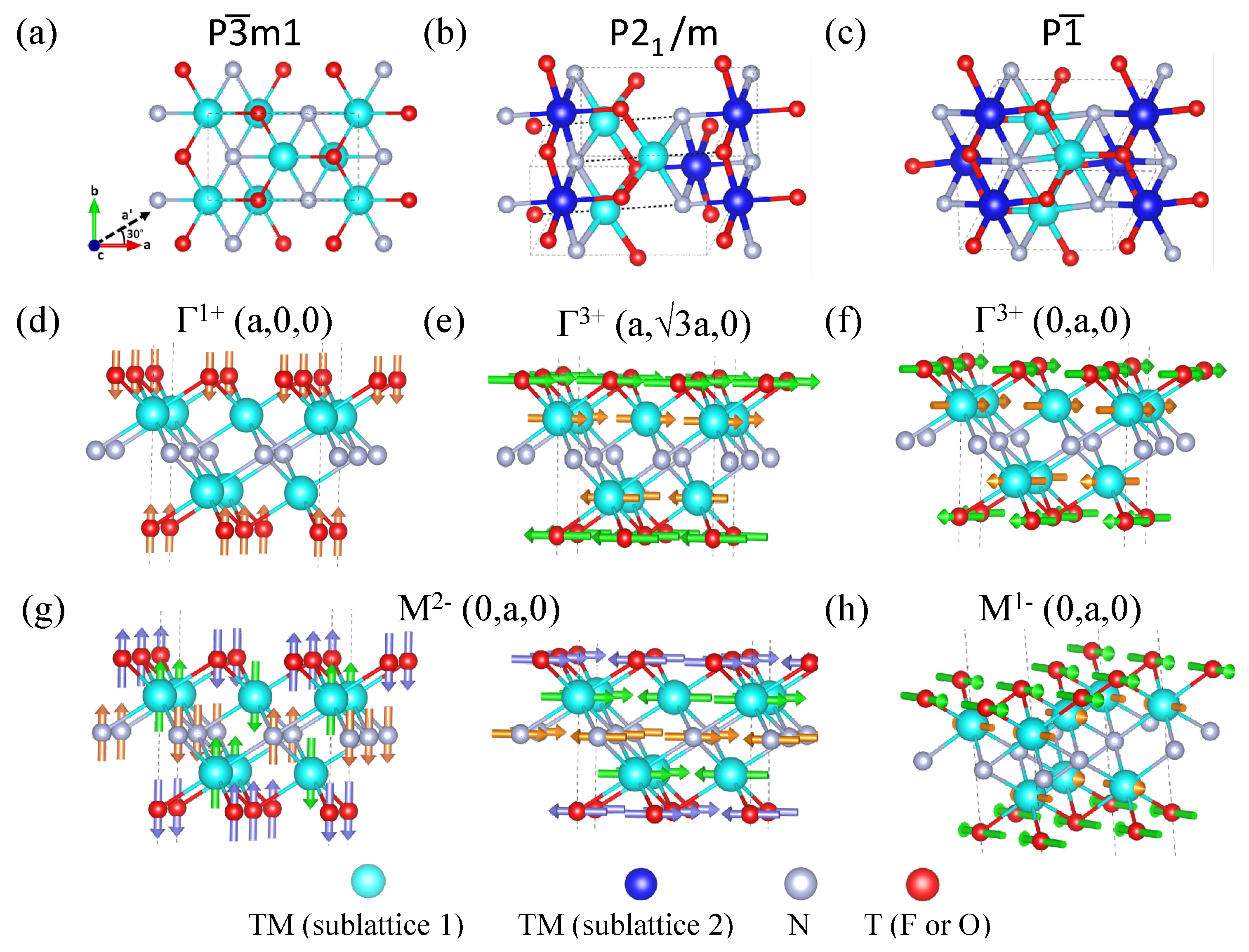
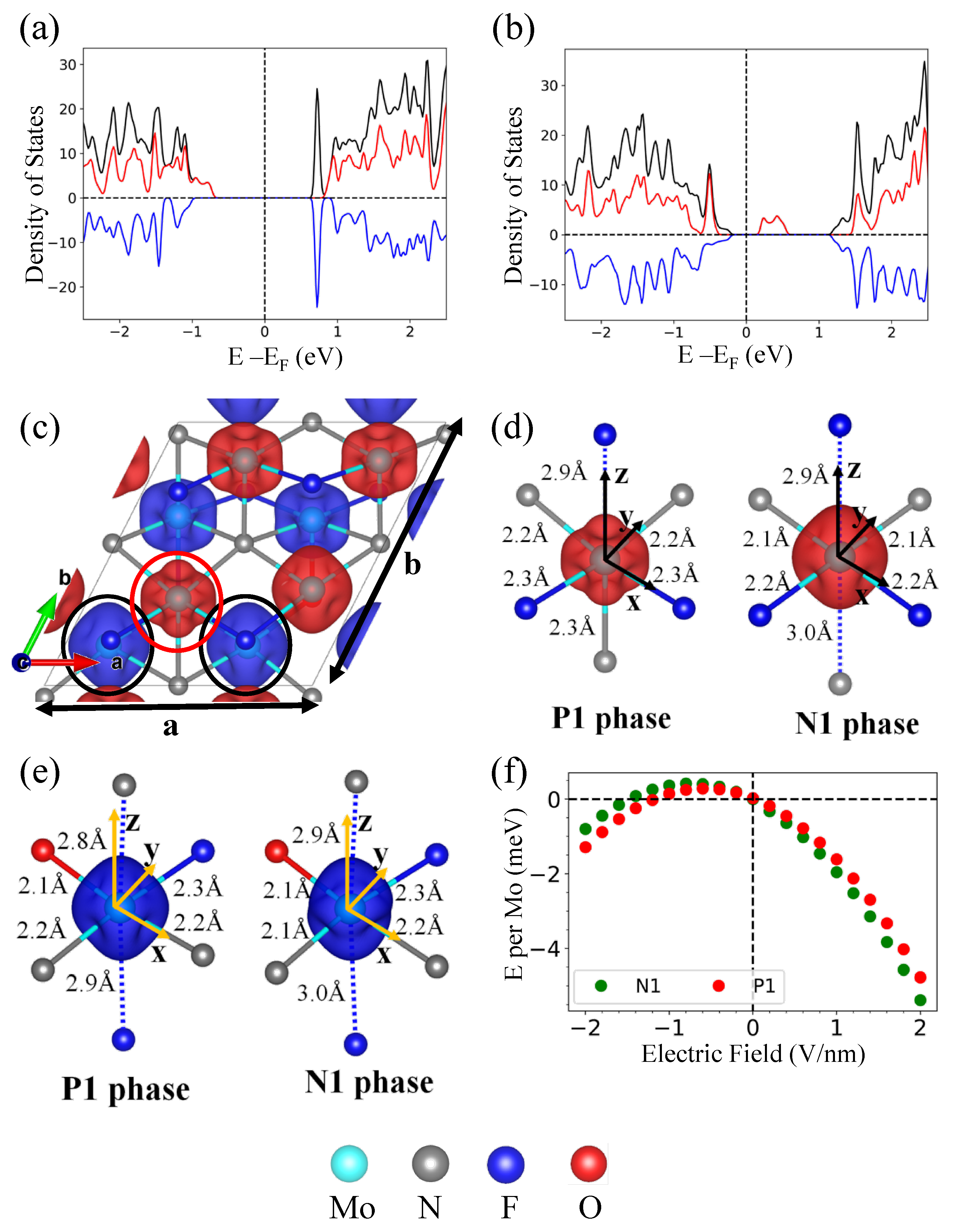
3.3. Orbital-Ordering-Induced Magnetoelectric Coupling
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, Y.; Gao, M.; Lu, Y. Two-dimensional multiferroics. Nanoscale 2021, 13, 19324–19340. [Google Scholar] [CrossRef]
- Tian, S.; Zhang, J.F.; Li, C.; Ying, T.; Li, S.; Zhang, X.; Liu, K.; Lei, H. Ferromagnetic van der Waals Crystal VI3. J. Am. Chem. Soc. 2019, 141, 5326–5333. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, J.; Jiang, C.; Rasmita, A.; Gao, W.; Yu, T. Direct Photoluminescence Probing of Ferromagnetism in Monolayer Two-Dimensional CrBr3. Nano Lett. 2019, 19, 3138–3142. [Google Scholar] [CrossRef]
- Bonilla, M.; Kolekar, S.; Ma, Y.; Diaz, H.C.; Kalappattil, V.; Das, R.; Eggers, T.; Gutierrez, H.R.; Phan, M.H.; Batzill, M. Strong room-temperature ferromagnetism in VSe2 monolayers on van der Waals substrates. Nat. Nanotechnol. 2018, 13, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, X.; Dai, Y.; Huang, B.; Ma, Y. Intrinsic ferromagnetic triferroicity in bilayer T’-VTe2. Appl. Phys. Lett. 2022, 120, 192903. [Google Scholar] [CrossRef]
- Li, J.; Zhao, B.; Chen, P.; Wu, R.; Li, B.; Xia, Q.; Guo, G.; Luo, J.; Zang, K.; Zhang, Z.; et al. Synthesis of Ultrathin Metallic MTe2 (M = V, Nb, Ta) Single-Crystalline Nanoplates. Adv. Mater. 2018, 30, 1801043. [Google Scholar] [CrossRef] [PubMed]
- OHara, D.J.; Zhu, T.; Trout, A.H.; Ahmed, A.S.; Luo, Y.K.; Lee, C.H.; Brenner, M.R.; Rajan, S.; Gupta, J.A.; McComb, D.W.; et al. Room Temperature Intrinsic Ferromagnetism in Epitaxial Manganese Selenide Films in the Monolayer Limit. Nano Lett. 2018, 18, 3125–3131. [Google Scholar] [CrossRef]
- O’Neill, A.; Rahman, S.; Zhang, Z.; Schoenherr, P.; Yildirim, T.; Gu, B.; Su, G.; Lu, Y.; Seidel, J. Enhanced Room Temperature Ferromagnetism in Highly Strained 2D Semiconductor Cr2Ge2Te6. ACS Nano 2023, 17, 735–742. [Google Scholar] [CrossRef]
- Ostwal, V.; Shen, T.; Appenzeller, J. Efficient Spin-Orbit Torque Switching of the Semiconducting Van Der Waals Ferromagnet Cr2Ge2Te6. Adv. Mater. 2020, 32, 1906021. [Google Scholar] [CrossRef]
- Cai, X.; Song, T.; Wilson, N.P.; Clark, G.; He, M.; Zhang, X.; Taniguchi, T.; Watanabe, K.; Yao, W.; Xiao, D.; et al. Atomically Thin CrCl3: An In-Plane Layered Antiferromagnetic Insulator. Nano Lett. 2019, 19, 3993–3998. [Google Scholar] [CrossRef]
- Lee, Y.; Son, S.; Kim, C.; Kang, S.; Shen, J.; Kenzelmann, M.; Delley, B.; Savchenko, T.; Parchenko, S.; Na, W.; et al. Giant Magnetic Anisotropy in the Atomically Thin van der Waals Antiferromagnet FePS3. Adv. Electron. Mater. 2023, 9, 2200650. [Google Scholar] [CrossRef]
- Wang, X.; Du, K.; Liu, Y.Y.F.; Hu, P.; Zhang, J.; Zhang, Q.; Owen, M.H.S.; Lu, X.; Gan, C.K.; Sengupta, P.; et al. Raman spectroscopy of atomically thin two-dimensional magnetic iron phosphorus trisulfide (FePS3) crystals. 2D Mater. 2016, 3, 031009. [Google Scholar] [CrossRef]
- Suzuki, H.; Liu, H.; Bertinshaw, J.; Ueda, K.; Kim, H.; Laha, S.; Weber, D.; Yang, Z.; Wang, L.; Takahashi, H.; et al. Proximate ferromagnetic state in the Kitaev model material α-RuCl3. Nat. Commun. 2021, 12, 4512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Li, L.; Weber, D.; Goldberger, J.; Mak, K.F.; Shan, J. Gate-tunable spin waves in antiferromagnetic atomic bilayers. Nat. Mater. 2020, 19, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Clark, G.; Klein, D.R.; MacNeill, D.; Navarro-Moratalla, E.; Seyler, K.L.; Wilson, N.; McGuire, M.A.; Cobden, D.H.; Xiao, D.; et al. Electrical control of 2D magnetism in bilayer CrI3. Nat. Nanotechnol. 2018, 13, 544–548. [Google Scholar] [CrossRef]
- Jiang, S.; Li, L.; Wang, Z.; Mak, K.F.; Shan, J. Controlling magnetism in 2D CrI3 by electrostatic doping. Nat. Nanotechnol. 2018, 13, 549–553. [Google Scholar] [CrossRef]
- Verzhbitskiy, I.A.; Kurebayashi, H.; Cheng, H.; Zhou, J.; Khan, S.; Feng, Y.P.; Eda, G. Controlling the magnetic anisotropy in Cr2Ge2Te6 by electrostatic gating. Nat. Electron. 2020, 3, 460–465. [Google Scholar] [CrossRef]
- Telford, E.J.; Dismukes, A.H.; Dudley, R.L.; Wiscons, R.A.; Lee, K.; Chica, D.G.; Ziebel, M.E.; Han, M.G.; Yu, J.; Shabani, S.; et al. Coupling between magnetic order and charge transport in a two-dimensional magnetic semiconductor. Nat. Mater. 2022, 21, 754–760. [Google Scholar] [CrossRef]
- Tahir, R.; Fatima, S.; Zahra, S.A.; Akinwande, D.; Li, H.; Jafri, S.H.M.; Rizwan, S. Multiferroic and ferroelectric phases revealed in 2D Ti3C2Tx MXene film for high performance resistive data storage devices. Npj 2D Mater. Appl. 2023, 7, 7. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, J.; Wang, S.S.; An, M.; Dong, S. Multiferroic properties of oxygen-functionalized magnetic i-MXene. Phys. Rev. Mater. 2021, 5, 094408. [Google Scholar] [CrossRef]
- Zhang, J.J.; Lin, L.; Zhang, Y.; Wu, M.; Yakobson, B.I.; Dong, S. Type-II Multiferroic Hf2VC2F2 MXene Monolayer with High Transition Temperature. J. Am. Chem. Soc. 2018, 140, 9768–9773. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.C.; Bandyopadhyay, A.; Kumar, H.; Anasori, B.; Gogotsi, Y.; Shenoy, V.B. Surface-Engineered MXenes: Electric Field Control of Magnetism and Enhanced Magnetic Anisotropy. ACS Nano 2019, 13, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, J.; Luo, K.; Li, Y.; Chang, K.; Chen, K.; Zhou, J.; Rosen, J.; Hultman, L.; Eklund, P.; et al. Element Replacement Approach by Reaction with Lewis Acidic Molten Salts to Synthesize Nanolaminated MAX Phases and MXenes. J. Am. Chem. Soc. 2019, 141, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Hassan, A.; Neher, G.; Nepal, D.; Pachter, R.; Kennedy, W.J.; Ramakrishnan, S.; Vaia, R.A. Halogen Etch of Ti3AlC2 MAX Phase for MXene Fabrication. ACS Nano 2021, 15, 2771–2777. [Google Scholar] [CrossRef]
- Sun, Z.; Yuan, M.; Lin, L.; Yang, H.; Nan, C.; Li, H.; Sun, G.; Yang, X. Selective Lithiation–Expansion–Microexplosion Synthesis of Two-Dimensional Fluoride-Free Mxene. ACS Mater. Lett. 2019, 1, 628–632. [Google Scholar] [CrossRef]
- Huang, X.; Wu, P. A Facile, High-Yield, and Freeze-and-Thaw-Assisted Approach to Fabricate MXene with Plentiful Wrinkles and Its Application in On-Chip Micro-Supercapacitors. Adv. Funct. Mater. 2020, 30, 1910048. [Google Scholar] [CrossRef]
- Urbankowski, P.; Anasori, B.; Hantanasirisakul, K.; Yang, L.; Zhang, L.; Haines, B.; May, S.J.; Billinge, S.J.L.; Gogotsi, Y. 2D molybdenum and vanadium nitrides synthesized by ammoniation of 2D transition metal carbides (MXenes). Nanoscale 2017, 9, 17722–17730. [Google Scholar] [CrossRef]
- Soundiraraju, B.; George, B.K. Two-Dimensional Titanium Nitride (Ti2N) MXene: Synthesis, Characterization, and Potential Application as Surface-Enhanced Raman Scattering Substrate. ACS Nano 2017, 11, 8892–8900. [Google Scholar] [CrossRef]
- Urbankowski, P.; Anasori, B.; Makaryan, T.; Er, D.; Kota, S.; Walsh, P.L.; Zhao, M.; Shenoy, V.B.; Barsoum, M.W.; Gogotsi, Y. Synthesis of two-dimensional titanium nitride Ti4N3 (MXene). Nanoscale 2016, 8, 11385–11391. [Google Scholar] [CrossRef]
- Kumar, H.; Frey, N.C.; Dong, L.; Anasori, B.; Gogotsi, Y.; Shenoy, V.B. Tunable Magnetism and Transport Properties in Nitride MXenes. ACS Nano 2017, 11, 7648–7655. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [PubMed]
- da Silva Neto, E.H.; Aynajian, P.; Frano, A.; Comin, R.; Schierle, E.; Weschke, E.; Gyenis, A.; Wen, J.; Schneeloch, J.; Xu, Z.; et al. Ubiquitous Interplay between Charge Ordering and High-Temperature Superconductivity in Cuprates. Science 2014, 343, 393–396. [Google Scholar] [CrossRef]
- Bednorz, J.G.; Müller, K.A.; Takashige, M. Superconductivity in Alkaline Earth-Substituted La2CuO4-y. Science 1987, 236, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Bednorz, J.G.; Müller, K.A. Perovskite-type oxides—The new approach to high-Tc superconductivity. Rev. Mod. Phys. 1988, 60, 585–600. [Google Scholar] [CrossRef]
- Labbé, J.; Bok, J. Superconductivity in Alcaline-Earth-Substituted La2CuO4: A Theoretical Model. Europhys. Lett. 1987, 3, 1225. [Google Scholar] [CrossRef]
- Chahara, K.; Ohno, T.; Kasai, M.; Kozono, Y. Magnetoresistance in magnetic manganese oxide with intrinsic antiferromagnetic spin structure. Appl. Phys. Lett. 1993, 63, 1990–1992. [Google Scholar] [CrossRef]
- von Helmolt, R.; Wecker, J.; Holzapfel, B.; Schultz, L.; Samwer, K. Giant negative magnetoresistance in perovskitelike La2/3Ba1/3MnOx ferromagnetic films. Phys. Rev. Lett. 1993, 71, 2331–2333. [Google Scholar] [CrossRef]
- Raveau, B.; Hervieu, M.; Maignan, A.; Martin, C. The route to CMR manganites: What about charge ordering and phase separation? J. Mater. Chem. 2001, 11, 29–36. [Google Scholar] [CrossRef]
- Varignon, J.; Bristowe, N.C.; Ghosez, P. Electric Field Control of Jahn-Teller Distortions in Bulk Perovskites. Phys. Rev. Lett. 2016, 116, 057602. [Google Scholar] [CrossRef]
- Goodenough, J.B. Jahn-teller phenomena in solids. Annu. Rev. Mater. Sci. 1998, 28, 1–27. [Google Scholar] [CrossRef]
- Chen, L.; Xu, C.; Tian, H.; Xiang, H.; Íñiguez, J.; Yang, Y.; Bellaiche, L. Electric-Field Control of Magnetization, Jahn-Teller Distortion, and Orbital Ordering in Ferroelectric Ferromagnets. Phys. Rev. Lett. 2019, 122, 247701. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Sun, W.; Xie, Y.; Kent, P.R.C. Double transition metal MXenes with wide band gaps and novel magnetic properties. Nanoscale 2018, 10, 11962–11968. [Google Scholar] [CrossRef]
- Li, Y.; Deng, J.; Zhang, Y.F.; Jin, X.; Dong, W.H.; Sun, J.T.; Pan, J.; Du, S. Nonvolatile electrical control of spin polarization in the 2D bipolar magnetic semiconductor VSeF. Npj Comput. Mater. 2023, 9, 50. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, Z.; Sa, B.; Miao, N.; Zhou, J.; Sun, Z. Computational design of double transition metal MXenes with intrinsic magnetic properties. Nanoscale Horiz. 2022, 7, 276–287. [Google Scholar] [CrossRef]
- Reshak, A.H. Microcrystalline β-RbNd(MoO4)2: Spin polarizing DFT+U. RSC Adv. 2015, 5, 44960–44968. [Google Scholar] [CrossRef]
- Aykol, M.; Wolverton, C. Local environment dependent GGA+U method for accurate thermochemistry of transition metal compounds. Phys. Rev. B 2014, 90, 115105. [Google Scholar] [CrossRef]
- Franchini, C.; Podloucky, R.; Paier, J.; Marsman, M.; Kresse, G. Ground-state properties of multivalent manganese oxides: Density functional and hybrid density functional calculations. Phys. Rev. B 2007, 75, 195128. [Google Scholar] [CrossRef]
- Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef]
- King-Smith, R.D.; Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B 1993, 47, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Stokes, H.T.; Hatch, D.M. Introduction to Isotropy Subgroups and Displacive Phase Transitions; Brigham Young University: Provo, Utah, 2006. [Google Scholar]
- Pizzi, G.; Vitale, V.; Arita, R.; Blügel, S.; Freimuth, F.; Géranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T.; et al. Wannier90 as a community code: New features and applications. J. Phys. Condens. Matter 2020, 32, 165902. [Google Scholar] [CrossRef]
- Mostofi, A.A.; Yates, J.R.; Lee, Y.S.; Souza, I.; Vanderbilt, D.; Marzari, N. wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 2008, 178, 685–699. [Google Scholar] [CrossRef]
- Liechtenstein, A.; Katsnelson, M.; Antropov, V.; Gubanov, V. Local spin density functional approach to the theory of exchange interactions in ferromagnetic metals and alloys. J. Magn. Magn. Mater. 1987, 67, 65–74. [Google Scholar] [CrossRef]
- He, X.; Helbig, N.; Verstraete, M.J.; Bousquet, E. TB2J: A python package for computing magnetic interaction parameters. Comput. Phys. Commun. 2021, 264, 107938. [Google Scholar] [CrossRef]
- Müller, G.P.; Hoffmann, M.; Dißelkamp, C.; Schürhoff, D.; Mavros, S.; Sallermann, M.; Kiselev, N.S.; Jónsson, H.; Blügel, S. Spirit: Multifunctional framework for atomistic spin simulations. Phys. Rev. B 2019, 99, 224414. [Google Scholar] [CrossRef]
- Hatch, D.M.; Stokes, H.T. Complete listing of order parameters for a crystalline phase transition: A solution to the generalized inverse Landau problem. Phys. Rev. B 2001, 65, 014113. [Google Scholar] [CrossRef]
- Iqbal, M.; Fatheema, J.; Noor, Q.; Rani, M.; Mumtaz, M.; Zheng, R.K.; Khan, S.A.; Rizwan, S. Co-existence of magnetic phases in two-dimensional MXene. Mater. Today Chem. 2020, 16, 100271. [Google Scholar] [CrossRef]
- Zinke, R.; Richter, J.; Drechsler, S.L. Spiral correlations in frustrated one-dimensional spin-1/2 Heisenberg J1–J2–J3 ferromagnets. J. Phys. Condens. Matter 2010, 22, 446002. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Sun, J.; Liu, Z.; Yang, J. BP5 monolayer with multiferroicity and negative Poisson’s ratio: A prediction by global optimization method. 2D Mater. 2017, 4, 045020. [Google Scholar] [CrossRef]
- Kou, L.; Ma, Y.; Tang, C.; Sun, Z.; Du, A.; Chen, C. Auxetic and Ferroelastic Borophane: A Novel 2D Material with Negative Possion’s Ratio and Switchable Dirac Transport Channels. Nano Lett. 2016, 16, 7910–7914. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zeng, X.C. Intrinsic Ferroelasticity and/or Multiferroicity in Two-Dimensional Phosphorene and Phosphorene Analogues. Nano Lett. 2016, 16, 3236–3241. [Google Scholar] [CrossRef]
- Xu, X.; Ma, Y.; Zhang, T.; Lei, C.; Huang, B.; Dai, Y. Prediction of two-dimensional antiferromagnetic ferroelasticity in an AgF2 monolayer. Nanoscale Horiz. 2020, 5, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xiang, H.; Yin, J.; Xia, Y.; Liu, Z. A two-dimensional tetragonal yttrium nitride monolayer: A ferroelastic semiconductor with switchable anisotropic properties. Nanoscale 2018, 10, 215–221. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.; Li, Z.; Yang, J.; Hou, J.G. Bipolar magnetic semiconductors: A new class of spintronics materials. Nanoscale 2012, 4, 5680–5685. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Yang, J. A review of bipolar magnetic semiconductors from theoretical aspects. Fundam. Res. 2022, 2, 511–521. [Google Scholar] [CrossRef]
- Liu, F.; Hao, Y.; Ni, J.; Zhao, Y.; Zhang, D.; Fabbris, G.; Haskel, D.; Cheng, S.; Xu, X.; Yin, L.; et al. Pressure-induced charge orders and their postulated coupling to magnetism in hexagonal multiferroic LuFe2O4. npj Quantum Mater. 2023, 8, 1. [Google Scholar] [CrossRef]
- Lejman, M.; Paillard, C.; Juvé, V.; Vaudel, G.; Guiblin, N.; Bellaiche, L.; Viret, M.; Gusev, V.E.; Dkhil, B.; Ruello, P. Magnetoelastic and magnetoelectric couplings across the antiferromagnetic transition in multiferroic BiFeO3. Phys. Rev. B 2019, 99, 104103. [Google Scholar] [CrossRef]
- Lu, X.Z.; Wu, X.; Xiang, H.J. General microscopic model of magnetoelastic coupling from first principles. Phys. Rev. B 2015, 91, 100405. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MXene | Band Gap/eV | Magnetic Moment/µB | Magnetic Order (MC) | Transition Temperature/K | E (AFM1)/meV | E (AFM2)/meV | E (AFM3)/meV | E (AFM4)/meV | E (FM)/meV |
|---|---|---|---|---|---|---|---|---|---|
| V2NO2 | 0.78 | 1.9 (1.2) | AFM3 | 140 | 106 | 131 | 0 | - | 44 |
| V2NF2 | 0.85 | 2.7 (2.1) | AFM3 | 380 | 87 | 18 | 0 | 814 | 1304 |
| Cr2NF2 | 2.1 | 3.9 (3.1) | Néel spin spiral | 120 | 394 | 399 | 0 | 340 | 549 |
| Mo2NO2 | 0.95 | 2.4 (1.9) | AFM3 | 350 | 1320 | 1586 | 0 | 1298 | 2796 |
| Mo2NF2 | 1.75 | 3.9 (3.4) | AFM3 | 630 | 1195 | 1863 | 0 | - | 1266 |
| Mn2NO2 | 0.20 | 3.9 (3.4) | Conical spin spiral | 110 | 440 | 334 | 336 | 59 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teh, S.; Jeng, H.-T. Magnetoelastic and Magnetoelectric Coupling in Two-Dimensional Nitride MXenes: A Density Functional Theory Study. Nanomaterials 2023, 13, 2644. https://doi.org/10.3390/nano13192644
Teh S, Jeng H-T. Magnetoelastic and Magnetoelectric Coupling in Two-Dimensional Nitride MXenes: A Density Functional Theory Study. Nanomaterials. 2023; 13(19):2644. https://doi.org/10.3390/nano13192644
Chicago/Turabian StyleTeh, Sukhito, and Horng-Tay Jeng. 2023. "Magnetoelastic and Magnetoelectric Coupling in Two-Dimensional Nitride MXenes: A Density Functional Theory Study" Nanomaterials 13, no. 19: 2644. https://doi.org/10.3390/nano13192644
APA StyleTeh, S., & Jeng, H.-T. (2023). Magnetoelastic and Magnetoelectric Coupling in Two-Dimensional Nitride MXenes: A Density Functional Theory Study. Nanomaterials, 13(19), 2644. https://doi.org/10.3390/nano13192644