
Roughness Factors of Electrodeposited Nanostructured Copper Foams
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Electrodeposition
2.2. S Copper Foam Characterization
2.3. Electrochemical Measurements
3. Results and Discussion
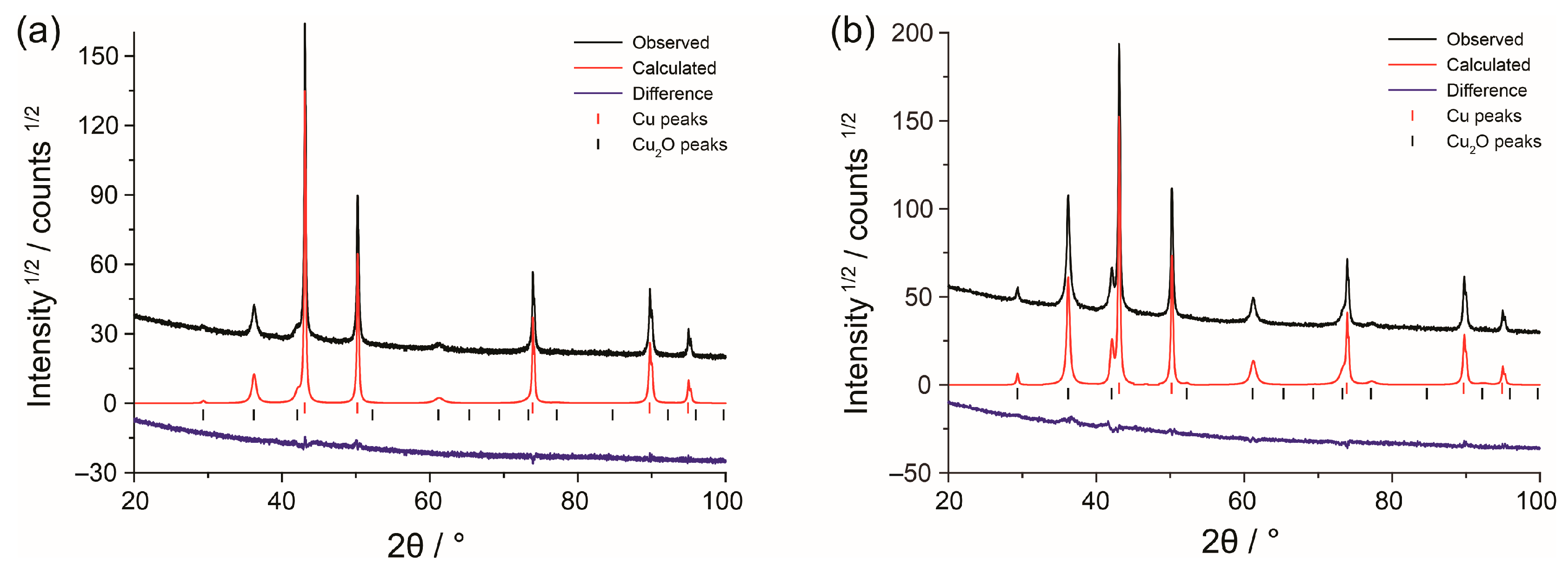
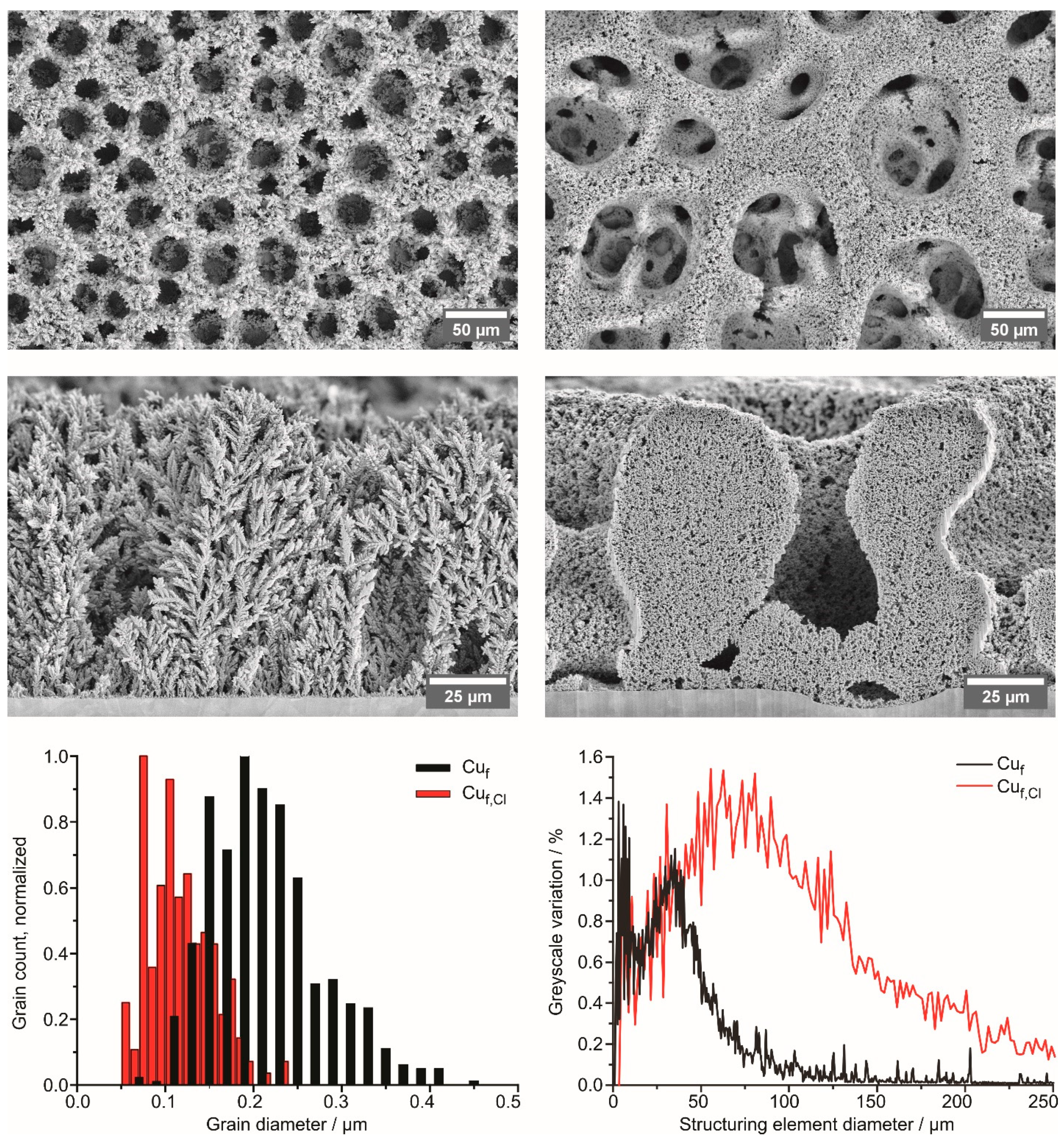
3.1. Structure and Morphology of Copper Foams
3.2. Pb UPD
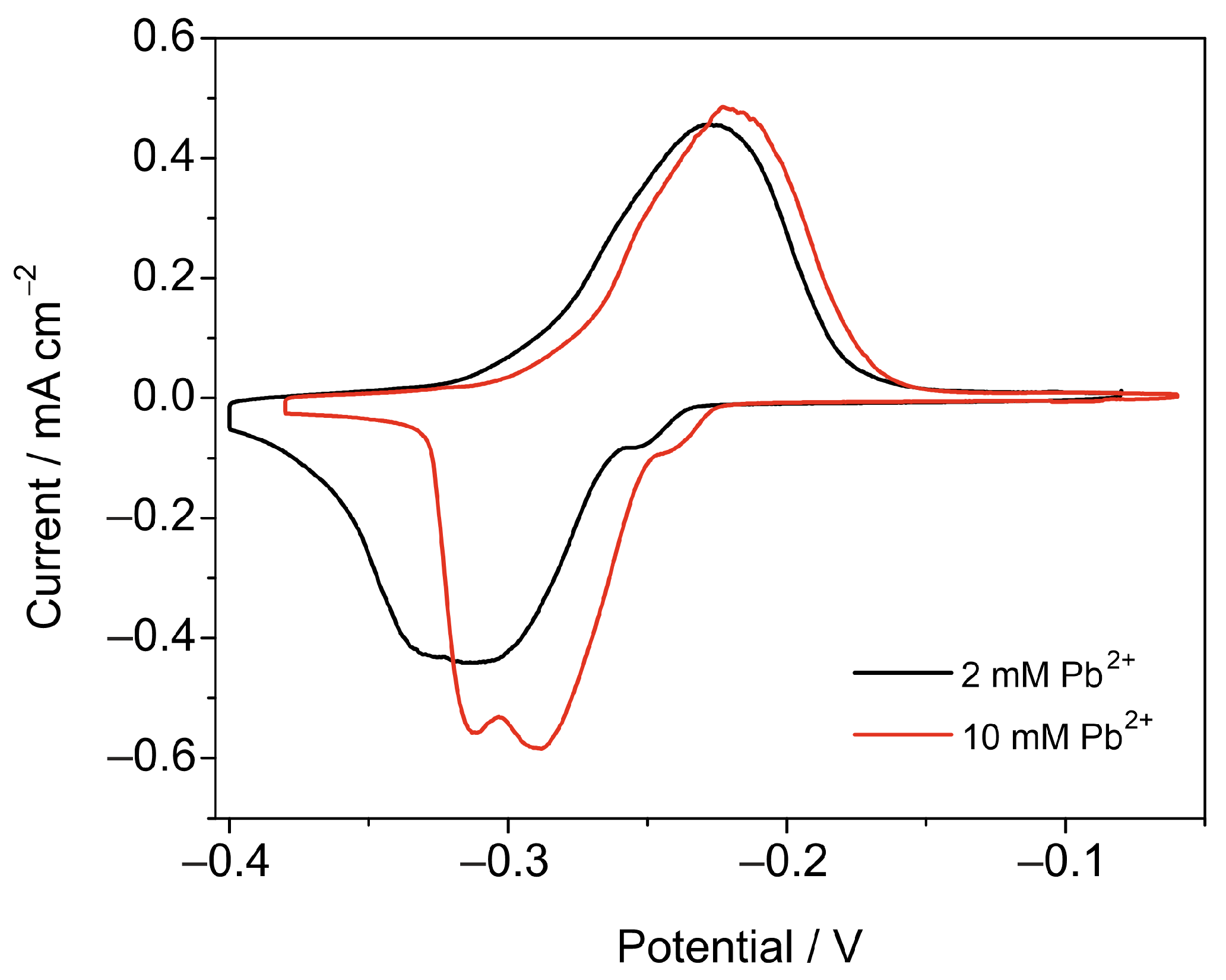
3.2.1. Pb2+ Concentration
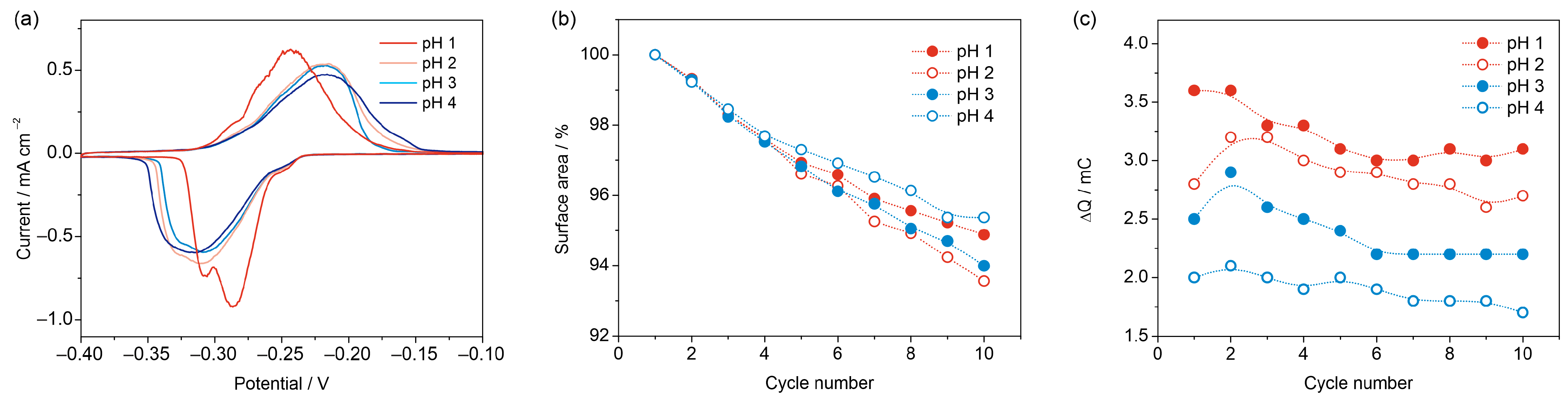
3.2.2. Effect of pH
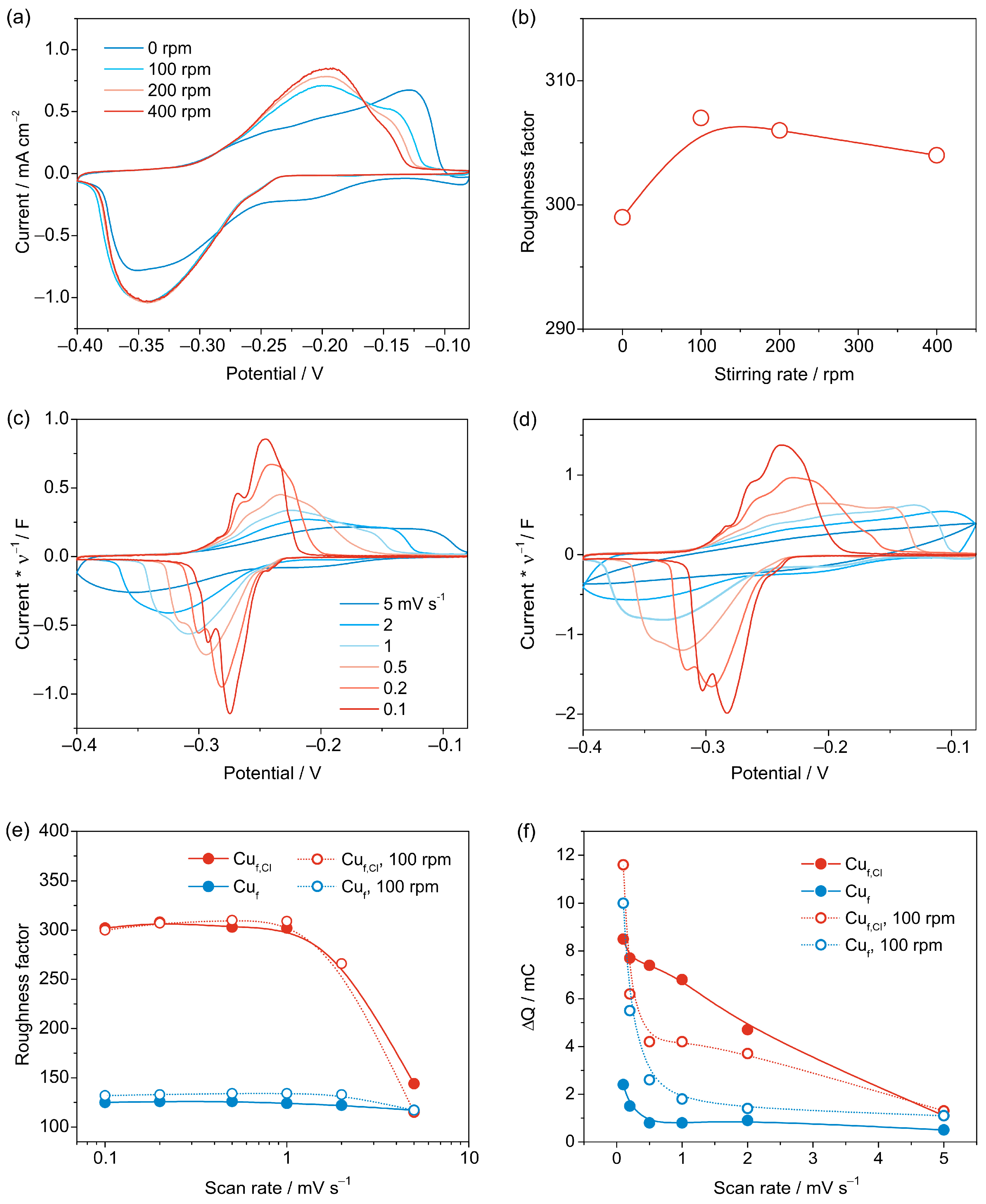
3.2.3. Cyclic Voltammetry
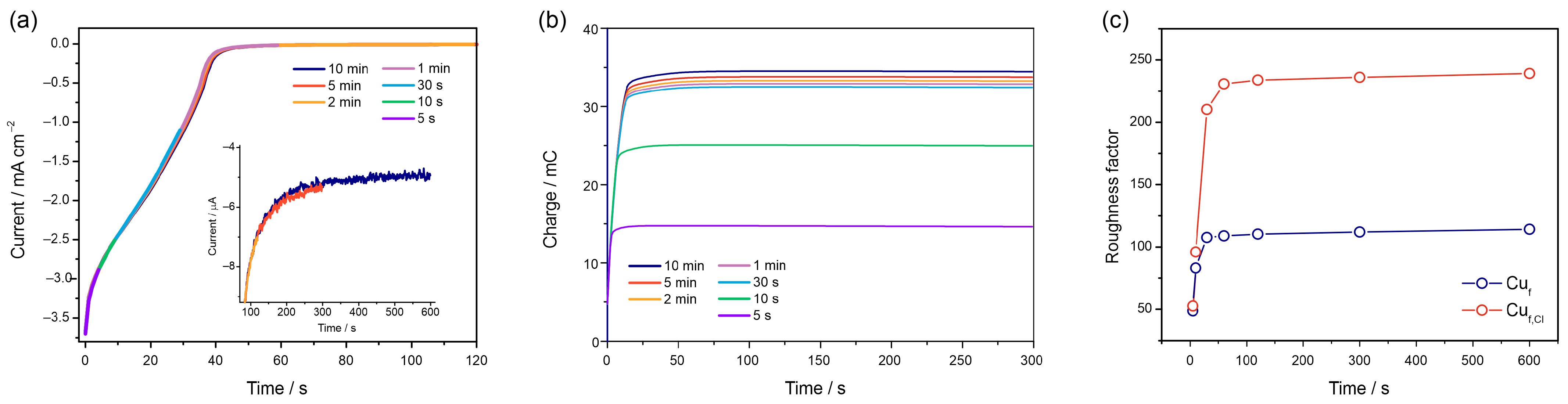
3.2.4. Chronoamperometric Measurements
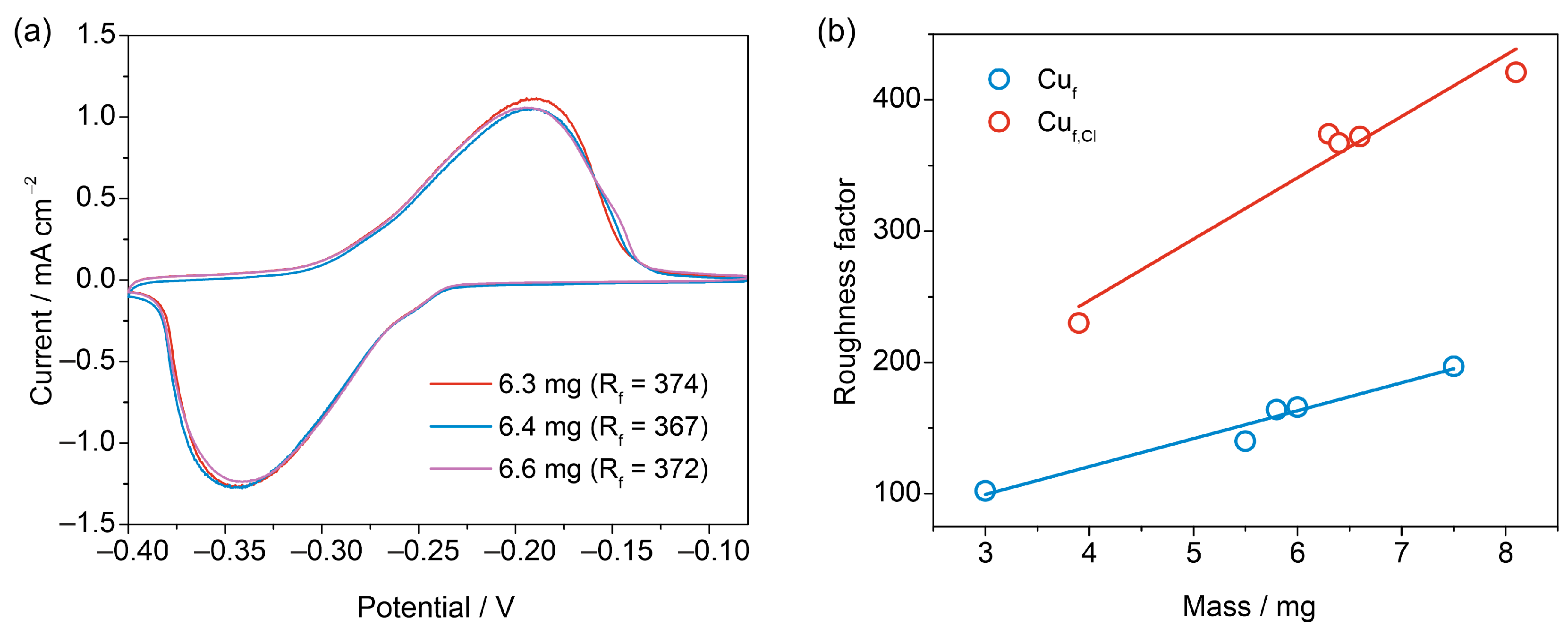
3.3. Reproducibility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Zhu, M.; Zeng, G.; Liu, X.; Fang, C.; Li, C. A three-dimensional Cu nanobelt cathode for highly efficient electrocatalytic nitrate reduction. Nanoscale 2020, 12, 9385–9391. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Ciston, J.; Kanan, M.W. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 2014, 508, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Lum, Y.; Yue, B.; Lobaccaro, P.; Bell, A.T.; Ager, J.W. Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 2017, 121, 14191–14203. [Google Scholar] [CrossRef]
- Lum, Y.; Ager, J.W. Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction. Nat. Catal. 2018, 2, 86–93. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef]
- Serafini, M.; Mariani, F.; Fasolini, A.; Scavetta, E.; Basile, F.; Tonelli, D. Nanostructured Copper-Based Electrodes Electrochemically Synthesized on a Carbonaceous Gas Diffusion Membrane with Catalytic Activity for the Electroreduction of CO2. ACS Appl. Mater. Interfaces 2021, 13, 57451–57461. [Google Scholar] [CrossRef]
- Castillo, E.; Zhang, J.; Dimitrov, N. All-electrochemical synthesis of tunable fine-structured nanoporous copper films. MRS Bull. 2022, 47, 913–925. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, X.; Qi, Z.; Ji, H.; Zhang, Z. On the electrochemical dealloying of Mg–Cu alloys in a NaCl aqueous solution. Corros. Sci. 2010, 52, 3962–3972. [Google Scholar] [CrossRef]
- Jia, F.; Zhao, J.; Yu, X. Nanoporous Cu film/Cu plate with superior catalytic performance toward electro-oxidation of hydrazine. J. Power Sources 2013, 222, 135–139. [Google Scholar] [CrossRef]
- Sen, S.; Liu, D.; Palmore, G.T.R. Electrochemical Reduction of CO2 at Copper Nanofoams. ACS Catal. 2014, 4, 3091–3095. [Google Scholar] [CrossRef]
- Dutta, A.; Rahaman, M.; Mohos, M.; Zanetti, A.; Broekmann, P. Electrochemical CO2 Conversion Using Skeleton (Sponge) Type of Cu Catalysts. ACS Catal. 2017, 7, 5431–5437. [Google Scholar] [CrossRef]
- Dutta, A.; Rahaman, M.; Luedi, N.C.; Mohos, M.; Broekmann, P. Morphology Matters: Tuning the Product Distribution of CO2 Electroreduction on Oxide-Derived Cu Foam Catalysts. ACS Catal. 2016, 6, 3804–3814. [Google Scholar] [CrossRef]
- Vesztergom, S.; Dutta, A.; Rahaman, M.; Kiran, K.; Zelocualtecatl Montiel, I.; Broekmann, P. Hydrogen Bubble Templated Metal Foams as Efficient Catalysts of CO2 Electroreduction. ChemCatChem 2020, 13, 1039–1058. [Google Scholar] [CrossRef]
- Reyter, D.; Odziemkowski, M.; Bélanger, D.; Roué, L. Electrochemically Activated Copper Electrodes. J. Electrochem. Soc. 2007, 154, K36–K44. [Google Scholar] [CrossRef]
- Fang, J.Y.; Fan, J.L.; Liu, S.B.; Sun, S.P.; Lou, Y.Y. Copper-Based Electrocatalysts for Nitrate Reduction to Ammonia. Materials 2023, 16, 4000. [Google Scholar] [CrossRef]
- Lim, M.; Sun, J.; Ma, Z.; Jalili, R.; Daiyan, R.; Lovell, E.C.; Amal, R. Engineering CuOx Nanoparticles on Cu Foam for Acidic Nitrate Reduction to Ammonium. ACS Appl. Nano Mater. 2023, 6, 4936–4945. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Biswas, A.; Das, M.; Dey, R.S. Nanostructured Cu foam and its derivatives: Emerging materials for the heterogeneous conversion of CO2 to fuels. Sustain. Energy Fuels 2021, 5, 2393–2414. [Google Scholar] [CrossRef]
- Giri, S.D.; Mahajani, S.M.; Suresh, A.K.; Sarkar, A. Electrochemical reduction of CO2 on activated copper: Influence of surface area. Mater. Res. Bull. 2020, 123, 110702. [Google Scholar] [CrossRef]
- Voiry, D.; Chhowalla, M.; Gogotsi, Y.; Kotov, N.A.; Li, Y.; Penner, R.M.; Schaak, R.E.; Weiss, P.S. Best Practices for Reporting Electrocatalytic Performance of Nanomaterials. ACS Nano 2018, 12, 9635–9638. [Google Scholar] [CrossRef]
- Li, D.; Batchelor-McAuley, C.; Compton, R.G. Some thoughts about reporting the electrocatalytic performance of nanomaterials. Appl. Mater. Today 2020, 18, 100404. [Google Scholar] [CrossRef]
- Wei, C.; Sun, S.; Mandler, D.; Wang, X.; Qiao, S.Z.; Xu, Z.J. Approaches for measuring the surface areas of metal oxide electrocatalysts for determining their intrinsic electrocatalytic activity. Chem. Soc. Rev. 2019, 48, 2518–2534. [Google Scholar] [CrossRef] [PubMed]
- Batchelor-McAuley, C.; Yang, M.; Hall, E.M.; Compton, R.G. Correction factors for the analysis of voltammetric peak currents measured using staircase voltammetry. J. Electroanal. Chem. 2015, 758, 1–6. [Google Scholar] [CrossRef]
- Vilche, J.R.; Jüttner, K. Anion effects on the underpotential deposition of lead on Cu(111). Electrochim. Acta 1987, 32, 1567–1572. [Google Scholar] [CrossRef]
- Rouya, E.; Cattarin, S.; Reed, M.L.; Kelly, R.G.; Zangari, G. Electrochemical Characterization of the Surface Area of Nanoporous Gold Films. J. Electrochem. Soc. 2012, 159, K97–K102. [Google Scholar] [CrossRef]
- Serapinienė, B.; Gudavičiūtė, L.; Tutlienė, S.; Grigucevičienė, A.; Selskis, A.; Juodkazytė, J.; Ramanauskas, R. On the Electrochemically Active Surface Area Determination of Electrodeposited Porous Cu 3D Nanostructures. Coatings 2023, 13, 1335. [Google Scholar] [CrossRef]
- Cattarin, S.; Kramer, D.; Lui, A.; Musiani, M.M. Preparation and Characterization of Gold Nanostructures of Controlled Dimension by Electrochemical Techniques. J. Phys. Chem. C 2007, 111, 12643–12649. [Google Scholar] [CrossRef]
- Zhu, P.; Zhao, Y. Effects of electrochemical reaction and surface morphology on electroactive surface area of porous copper manufactured by Lost Carbonate Sintering. RSC Adv. 2017, 7, 26392–26400. [Google Scholar] [CrossRef]
- Diao, K.K.; Xiao, Z.; Zhao, Y.Y. Specific surface areas of porous Cu manufactured by Lost Carbonate Sintering: Measurements by quantitative stereology and cyclic voltammetry. Mater. Chem. Phys. 2015, 162, 571–579. [Google Scholar] [CrossRef]
- Liu, Y.; Bliznakov, S.; Dimitrov, N. Comprehensive Study of the Application of a Pb Underpotential Deposition-Assisted Method for Surface Area Measurement of Metallic Nanoporous Materials. J. Phys. Chem. C 2009, 113, 12362–12372. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- Herrero, E.; Buller, L.J.; Abruna, H.D. Underpotential deposition at single crystal surfaces of Au, Pt, Ag and other materials. Chem. Rev. 2001, 101, 1897–1930. [Google Scholar] [CrossRef] [PubMed]
- Brisard, G.M.; Zenati, E.; Gasteiger, H.A.; Marković, N.M.; Ross, P.N. Underpotential Deposition of Lead on Cu(100) in the Presence of Chloride: Ex-Situ Low-Energy Electron Diffraction, Auger Electron Spectroscopy, and Electrochemical Studies. Langmuir 1997, 13, 2390–2397. [Google Scholar] [CrossRef]
- Łukomska, A.; Sobkowski, J. Potential of zero charge of monocrystalline copper electrodes in perchlorate solutions. J. Electroanal. Chem. 2004, 567, 95–102. [Google Scholar] [CrossRef]
- Sebastián-Pascual, P.; Escudero-Escribano, M. Surface characterization of copper electrocatalysts by lead underpotential deposition. J. Electroanal. Chem. 2021, 896, 115446. [Google Scholar] [CrossRef]
- Siegenthaler, H.; Jüttner, K. Voltammetric investigation of lead adsorption on Cu(111) single crystal substrates. J. Electroanal. Chem. 1984, 163, 327–343. [Google Scholar] [CrossRef]
- Rawlings, K.J.; Gibson, M.J.; Dobson, P.J. The epitaxial growth of lead and thallium on (111) silver and copper. J. Phys. D Appl. Phys. 1978, 11, 2059–2070. [Google Scholar] [CrossRef]
- Brisard, G.M.; Zenati, E.; Gasteiger, H.A.; Markovic, N.; Ross, P.N. Underpotential Deposition of Lead on Copper(111): A Study Using a Single-Crystal Rotating Ring Disk Electrode and ex Situ Low-Energy Electron Diffraction and Scanning tunneling Microscopy. Langmuir 1995, 11, 2221–2230. [Google Scholar] [CrossRef]
- Giri, S.D.; Sarkar, A. Estimating surface area of copper powder: A comparison between electrochemical, microscopy and laser diffraction methods. Adv. Powder Technol. 2018, 29, 3520–3526. [Google Scholar] [CrossRef]
- Bewick, A.; Jovićević, J.; Thomas, B. Phase formation in the underpotential deposition of metals. Faraday Symp. Chem. Soc. 1977, 12, 24–35. [Google Scholar] [CrossRef]
- Shin, H.-C.; Liu, M. Copper Foam Structures with Highly Porous Nanostructured Walls. Chem. Mater. 2004, 16, 5460–5464. [Google Scholar] [CrossRef]
- Nikolić, N.D.; Pavlović, L.J.; Pavlović, M.G.; Popov, K.I. Formation of dish-like holes and a channel structure in electrodeposition of copper under hydrogen co-deposition. Electrochim. Acta 2007, 52, 8096–8104. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Soille, P. Texture analysis. In Morphological Image Analysis; Springer: Berlin/Heidelberg, Germany, 2004; pp. 317–346. [Google Scholar]
- Markovic, N.M.; Gasteiger, H.A.; Ross, P.N., Jr. Copper Electrodeposition on Pt(111) in the Presence of Chloride and (Bi)sulfate: Rotating Ring-Pt(111) Disk Electrode Studies. Langmuir 1995, 11, 4098–4108. [Google Scholar] [CrossRef]
- Vukmirovic, M.B.; Vasiljevic, N.; Dimitrov, N.; Sieradzki, K. Diffusion-Limited Current Density of Oxygen Reduction on Copper. J. Electrochem. Soc. 2003, 150, B10. [Google Scholar] [CrossRef]
- Vasilic, R.; Vasiljevic, N.; Dimitrov, N. Open circuit stability of underpotentially deposited Pb monolayer on Cu(111). J. Electroanal. Chem. 2005, 580, 203–212. [Google Scholar] [CrossRef]
- Vasiljevic, N.; Viyannalage, L.T.; Dimitrov, N.; Missert, N.A.; Copeland, R.G. Oxidation of the Cu(100) Surface Induced by Local Alkalization. J. Electrochem. Soc. 2007, 154, C202. [Google Scholar] [CrossRef]
- Darayen, J.; Chailapakul, O.; Praserthdam, P.; Panpranot, J.; Tungasmita, D.N.; Boonyongmaneerat, Y. Porous Electrodeposited Cu as a Potential Electrode for Electrochemical Reduction Reactions of CO2. Appl. Sci. 2021, 11, 11104. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levin, E.E.; Morozov, D.A.; Frolov, V.V.; Arkharova, N.A.; Khmelenin, D.N.; Antipov, E.V.; Nikitina, V.A. Roughness Factors of Electrodeposited Nanostructured Copper Foams. Nanomaterials 2023, 13, 3011. https://doi.org/10.3390/nano13233011
Levin EE, Morozov DA, Frolov VV, Arkharova NA, Khmelenin DN, Antipov EV, Nikitina VA. Roughness Factors of Electrodeposited Nanostructured Copper Foams. Nanomaterials. 2023; 13(23):3011. https://doi.org/10.3390/nano13233011
Chicago/Turabian StyleLevin, Eduard E., Dmitriy A. Morozov, Vsevolod V. Frolov, Natalia A. Arkharova, Dmitry N. Khmelenin, Evgeny V. Antipov, and Victoria A. Nikitina. 2023. "Roughness Factors of Electrodeposited Nanostructured Copper Foams" Nanomaterials 13, no. 23: 3011. https://doi.org/10.3390/nano13233011