1. Introduction
Emissions of volatile organic compounds (VOCs) into air and water from products or processes is considered as serious environmental problem [
1,
2]. According to the classification of the International Agency for Research on Cancer (IARC classification) some of widely used VOCs belong to the three most serious groups; group 1 (carcinogens, e.g., formaldehyde, vinyl chloride), group 2A (probable carcinogens to humans, e.g., halogenated VOCs such as dichloromethane, perchloroethylene) and group 2B (possible carcinogens to humans, e.g., acetaldehyde, halogenated VOCs such as chloroform) [
3]. VOCs such as alcohols (e.g., methanol, 2B group) are toxic to human health and cause serious health problems such as blindness or chronical kidney and liver diseases. Besides primary emissions, emitted VOCs can react with other gases and particulates to form secondary air pollutants that in some cases are even more harmful than the original VOC emissions complicating e.g., their abatement.
From all VOCs, methanol and dichloromethane belong to solvents widely used in pharmaceutical and chemical industry for chemical synthesis or laboratory analytical analyses, being also part of products such disinfectants, degreasers, varnishes, waxes, household cleaners, used in production of paints and furniture materials [
2]. Handling both of them, disposal of used solvents, and the chosen emission abatement technology must be safe and under strict control since even more harmful by-products such as formaldehyde or carbon monoxide may be produced and emitted. Depending on their categorisation, VOCs are regulated by a wide variety of governmental agencies [
2]. Catalytic combustion is a well-accepted and reliable technology for industrial VOCs abatement in environmentally and economically acceptable manner [
2,
4].
From a practical point of view, there are given strict requirements on the catalysts applied for VOCs catalytic combustion, i.e., high activity, high selectivity as well as good durability. Concerning catalytic oxidation of a broad range of individual VOCs over individual types of catalysts some review papers were already reported [
1,
2,
4,
5,
6], also including investigated confined-structured catalysts [
6] and by-products analysis for chlorinated VOCs (CVOCs) oxidation [
7].The main challenges which need to be still further solved concerning the development of catalysts for VOCs oxidation are the development of highly selective and durable catalysts, deeper knowledge and prediction of oxidation mechanisms [
4,
5], catalyst deactivation, development of more complex catalytic systems being able to oxidize a range of VOCs at much lower temperatures [
1] and with respect to character of contaminated sites the coupling process combining different VOCs/CVOCs abatement methods should be applied to enhance the removal efficiency in a more cost effective way [
1,
2,
5]. Concerning the confined-structured catalysts more easy, available and green synthetic methods should be developed and used, including the optimization of chemical composition. There also arise some limitations in their precise physicochemical characterization, which is necessary for understanding of their catalytic performance. There is also a lack of testing of confined-structured catalysts in more realistic working conditions and oxidation of a mixture of VOCs [
6]. For CVOCs oxidation, (i) the examination of Cl migration and evolution pathway via advanced modelling and characterization techniques, providing the information how to prevent unwanted chlorinated by-products formation with working adjustment, (ii) revelation of proper metal combination within catalyst design, (iii) definition of the exact role of each physicochemical property in deep oxidation and chlorination steps with the aim to tailor catalyst surface composition, inhibiting the formation of unwanted chlorinated by-products and (iv) enhancing oxidation efficiency at low temperature that can evade Cl
2 formation and chlorination reactions, are of keen interests. It was suggested that e.g., technological coupling of hydrodechlorination, catalytic oxidation and ozone-assisted oxidation is feasible approach how to enhance total elimination at low temperature without chlorination reactions. (v) Last but not least, investigation of the effect of other additives and oxidizing agents, promoting dechlorination reactions and inducing Cl species into final products, is also necessary [
7]. Based on above mentioned facts the development of industrially-attractive and robust form of catalyst based on mixed metal oxides, which are cheaper and more resistant against metal chlorination than noble metal-based catalysts, using simple chemical and deposition route, is the logical motivation for the research. Moreover, the developed form of catalysts should be examined in oxidation of more model VOCs and in the presence of water to reveal the catalyst selectivity and be closer to realistic adjustment, yielding more desired HCl than Cl
2.
The TiO
2-CeO
2-CuO ternary mixture is attractive oxidation catalyst in terms of good temperature stability, adjustable acid-base properties, easy reducibility and the ability to store oxygen in its crystal structure [
8]. CuO-based materials are suitable catalysts for environmental remediation, but their disadvantage is easy sintering and deactivation due to blockage of active sites e.g., by carbon deposits and the associated reduced catalyst stability [
9]. Cerium, respectively, CeO
2, serves as a suitable CuO promoter due to the promotion of redox properties (there can be an interaction between the redox pairs Cu
2+/Cu
0 and Ce
3+/Ce
4+) and oxygen storage capacity due to the presence of Ce
3+/Ce
4+ [
10,
11]. Furthermore, the presence of CeO
2 helps the thermal stability of the oxide mixture and reduces the formation of coke on its surface [
12]. Nano CeO
2 and Cu-doped nano CeO
2 are even effective in the oxidation of particulate matter/soot [
13,
14]. The CeO
2 and CuO also interacts strongly with TiO
2 support [
15]. Ce doped TiO
2 increases Cu activity and stabilizes the Cu/TiO
2 surface [
15]. Concerning TiO
2-CeO
2 mixtures, the addition of cerium/CeO
2 to TiO
2 decreases the TiO
2 anatase crystallite-size and increases its thermal stability [
16,
17], increases reducibility [
18] as well as Lewis and Brønsted acidity [
19].
Concerning methanol oxidation (oxygenated VOC), the existing literature reveals that TiO
2, CeO
2 and CuO-based catalysts are effective catalysts in the decomposition of methanol to H
2, respectively to a mixture of H
2 and CO (so-called syngas) which can be an alternative fuel [
11,
20,
21], or have been studied in the oxidation of ethanol, ethylacetate and CO [
11,
15,
22,
23] (
Table 1). These catalysts have been rarely studied in the deep methanol oxidation, where the required reaction products are CO
2 and H
2O [
23,
24], whereas being cheaper and more efficient equivalent to catalysts doped with noble metals.
Concerning dichloromethane oxidation (chlorinated VOC, CVOC), a few interesting studies indicating good potential of TiO
2, CeO
2 and CuO-based catalysts instead of more expensive noble metals-based catalysts were reported [
18,
25,
26,
27,
28] (
Table 1). However, published research from Cao et al. [
18,
25] announced interestingly some existing limitations related to the design of the mixed TiO
2-CeO
2-CuO catalyst and its higher tendency to deactivation compared to a separate two step catalytic system Ce/TiO
2-Cu/CeO
2 applied in a series. Cao et al. studied the dichloromethane oxidation in dry conditions, firstly over TiO
2 and Ce/TiO
2 [
18], followed by a study on the Ce/TiO
2-Cu/CeO
2 in a series [
25]. Within their first study [
18], they suggested a two-step mechanism of dichloromethane oxidation over dehydrated TiO
2 and Ce/TiO
2, explaining the production of unwanted by-products such as methylchloride (CH
3Cl), formic acid (HCOOH) and carbon monoxide (CO) over TiO
2. Ce/TiO
2 showed significantly improved catalytic activity as well as selectivity to CO and CO
2 in dichloromethane oxidation compared to TiO
2. Moreover, Ce/TiO
2 showed to be stable during the 100 h test. They proved the rapid removal of Cl on the surface of CeO
2 (on oxygen vacancies of ceria), preventing TiO
2 (Ti
4+ sites) deactivation by adsorbed chlorine, accompanied by CeO
2′s capability to enhance further deep oxidation of C–H from by-products and retain a certain oxidation of CO to CO
2 [
18]. In their second study Cao et al. [
25] suggested a two-step catalytic system Ce/TiO
2-Cu/CeO
2 in a series to reach total oxidation of dichloromethane. The investigated catalytic system achieved improved activity as well as CO
2 selectivity with less undesired CO, Cl
2 and by-products (C
xH
yCl
z) in dry air. It showed good stability in long-term test in the presence of water. For the Ce/TiO
2-Cu/CeO
2 in a series they suggested a three-step dichloromethane degradation mechanism consisting of (i) dichloromethane adsorption and rupture of the C–Cl bonds, (ii) deep oxidation of C–H from by-products and (iii) total oxidation of CO to CO
2. The Ce/TiO
2-Cu/CeO
2 system in a series ensures that the rupture of C–Cl and the total oxidation of CO are physically separated, avoiding the decrease in acid sites to happen on a (Ce + Cu)/TiO
2 catalyst and the chlorine poisoning of TiO
2 due to the strong adsorption of Cl on CuO. Moreover, CuO itself had a high resistance to the toxicity of Cl [
25]. El Assal et al. [
26] have examined the efficiency of V, Mn and Cu supported on TiO
2, MgO and CeO
2 in dichloromethane oxidation. The highest selectivity to HCl was reached by Cu/TiO
2, followed by V
2O
5/MgO. Pure TiO
2 support was the most acidic support from all investigated and its highest acidity correlated well with the best activity and selectivity in dichloromethane oxidation. Impregnation of Cu onto TiO
2 increased the total catalyst acidity, resulting in better dichloromethane adsorption and cleavage of the C–Cl bond, leading to the best HCl selectivity, the least formation of by-products and the highest catalytic activity. Concerning the selectivity of the examined catalysts, both undesired chlorinated and oxygenated by-products such as methyl chloride (CH
3Cl), chloroform (CHCl
3), formaldehyde (CH
2O) and carbon monoxide (CO) were identified in dichloromethane oxidation experiments. From this point of view Cu/TiO
2 produced the lowest concentrations of the mentioned undesirable by-products [
26]. Wang et al. [
28] tested M/TiO
2 and Pd-M/TiO
2, where M was Ce, V or Mn in dichloromethane oxidation. While Ce/TiO
2 and Pd-Ce/TiO
2 showed the best catalytic activity, Ce/TiO
2 showed very poor selectivity to CO
2. Pd modification of Ce/TiO
2 led to the improvement of the catalyst redox ability and surface adsorbed oxygen species which was reflected to significant improvement of selectivity to CO
2 of Pd-Ce/TiO
2, but still with detected high concentrations of CO (100 ppm) at 100% dichlromethane conversion. Concerning unwanted chlorinated by-products, they detected traces of methyl chloride (CH
3Cl) over the tested catalysts. The performed DFT calculations and in-situ DRIFT studies over the best Pd-V/TiO
2 catalyst helped to reveal the mechanism of dichloromethane oxidation: C–Cl bonds and C–H bonds are dissociated by the surface lattice oxygen species after the adsorption of molecular dichloromethane on the acid sites, producing intermediates. After that, the oxidation of CO to CO
2 takes place by the surface adsorbed oxygen species.
Table 1.
Overview of TiO2, CeO2 and CuO-based catalysts investigated in methanol oxidation/decomposition, dichloromethane oxidation or oxidation of their possible reaction by-products.
Table 1.
Overview of TiO2, CeO2 and CuO-based catalysts investigated in methanol oxidation/decomposition, dichloromethane oxidation or oxidation of their possible reaction by-products.
| Catalyst | VOC/Reaction | Reference |
|---|
| 20%CuO/CeO2 | methanol oxidation | [24] |
| TiO2-CeO2 | methanol decomposition
ethylacetate oxidation | [11] |
| TiO2-CeO2-CuO | methanol decomposition
ethylacetate oxidation | [20] |
| TiO2-CeO2-CuO | ethanol, ethylacetate and CO oxidation | [22] |
| 5% CuOx-TiO2 | CO oxidation | [15] |
| Pd, Pt-Pd/CeO2-Al2O3-TiO2 | methanol oxidation | [23] |
| CuO-TiO2, CuO-CeO2 | dichloromethane oxidation | [26] |
| TiO2, Ce/TiO2 | dichloromethane oxidation | [18] |
| Ce/TiO2-Cu/CeO2 | dichloromethane oxidation | [25] |
| Cu-Mn/Al2O3 | ethanol and dichloromethane oxidation | [27] |
| M/TiO2, Pd-M/TiO2 (M-Ce, V, Mn) | dichloromethane oxidation | [28] |
The aim of this study was to develop the application-attractive forms of TiO
2, CeO
2 and CuO-based open-cell foam supported catalysts and investigate their activity, selectivity and stability in oxidation of two model VOCs—dichloromethane (chlorinated VOC) and methanol (oxygenated VOC). Thus, binary TiO
2-CeO
2 and TiO
2-CuO catalysts and ternary TiO
2-CeO
2-CuO catalyst as thin films deposited on VUKOPOR
®A ceramic Al
2O
3-based foam were prepared by using a reverse micelles-controlled sol-gel method in combination with dip-coating and calcination. This preparation method is feasible in industrial scale due to high chemical stability of the sol from the reverse micelles-mediated sol-gel synthesis and leads to crystallization of metal oxides of small crystallite size, which does not change significantly with rising temperature [
29,
30], crystallite size is typically <18 nm, which may be positively reflected to their catalytic performance and stability. Using dip-coating method and calcination the catalytic thin film is very well-fixed on the foam due to its roughness and chemical bonding. These catalytic foams were prepared for testing in these two oxidation reactions for the first time. The activity and selectivity of 3 catalytic foams in dichloromethane and methanol oxidation were studied and compared based on light-off tests and the best catalytic foam was then exposed to 20 h stability test. Special attention was dedicated to studying the selectivity over the studied catalytic foams in both oxidation reactions since this issue is very important in catalyst development for VOC oxidation in industrial scale.
2. Materials and Methods
2.1. Preparation of TiO2, CeO2 and CuO-Supported VUKOPOR®A Ceramic Foams
The sol-gel method controlled in reverse micelles environment of nonionic surfactant Triton X-114 in cyclohexane combined with dip-coating and calcination was selected for preparation and deposition of TiO2-CeO2, TiO2-CeO2-CuO and TiO2-CuO catalysts (4 layers) on VUKOPOR®A ceramic foams (85% Al2O3 + 14% SiO2 +1% MgO, porosity 40 ppi, diameter × length = 8 × 20 mm, LANIK s.r.o., Boskovice, Czech Republic). Firstly, the sols were prepared by mixing cyclohexane, Triton X-114, water and Ti-Ce-Cu precursors dissolved in anhydrous ethanol. The metal precursors used for synthesis were Ce(NO3)3·6H2O, Cu(NO3)2·3H2O and titanium (IV) isopropoxide (Ti(OCH(CH3)2)4, purity < 97%) from Aldrich (St. Louis, MO, USA).
For the preparation of TiO2-CeO2-CuO-supported VUKOPOR®A ceramic foam, the synthesis was as follows. 3.26 g of Ce(NO3)3·6H2O and 0.55 g of Cu(NO3)2·3H2O were individually dissolved in 5 mL of anhydrous ethanol. After complete dissolution of both precursors these two solutions were mixed, and the beakers were rinsed additionally with 5 mL of ethanol. The CeCu-based solution was stirred for 10 min. Meanwhile, 91.9 mL of cyclohexane, 37.6 mL of Triton X-114 and 0.46 mL of water was mixed to form Triton-based sol, which was stirred for 20 min. Then, the CeCu-based solution was added to the Triton-based sol and the formed micellar sol was stirred for 15 min. After that, 20 mL of titanium (IV) isopropoxide was added dropwise and the final Ti/Ce/Cu-based micellar sol was mixed for further 20 min. The transparent homogeneous sol was left standing in closed flask for minimally 24 h. Then, the washed VUKOPOR®A ceramic foams were immersed into the Ti/Ce/Cu-based micellar sol using dip-coater iDLab, using the following conditions of dip-coating (immersion velocity—150 mm/min, delay in the sol—60 s, emergency velocity—60 mm/min). The deposited foams were left to dry on air for 4 h and then were calcined at 500 °C for 4 h with the heating rate 3 °C·min−1.
For the preparation of TiO2-CeO2-supported VUKOPOR®A ceramic foam, the synthesis steps were identical as in the case of TiO2-CeO2-CuO foams, but the used amounts of precursors were following: 89 mL of cyclohexane, 36.5 mL of Triton X-114, 0.54 mL of H2O, 3.26 g of Ce(NO3)3·6H2O and 20 mL of titanium (IV) isopropoxide.
For the preparation of TiO2-CuO-supported VUKOPOR®A ceramic foam, the synthesis steps were identical as in the case of both previous foams, but the used amounts of precursors were following: 83 mL of cyclohexane, 34 mL of Triton X-114, 1.136 mL of H2O, 0.547 g of Cu(NO3)3·3H2O and 20 mL of titanium (IV) isopropoxide.
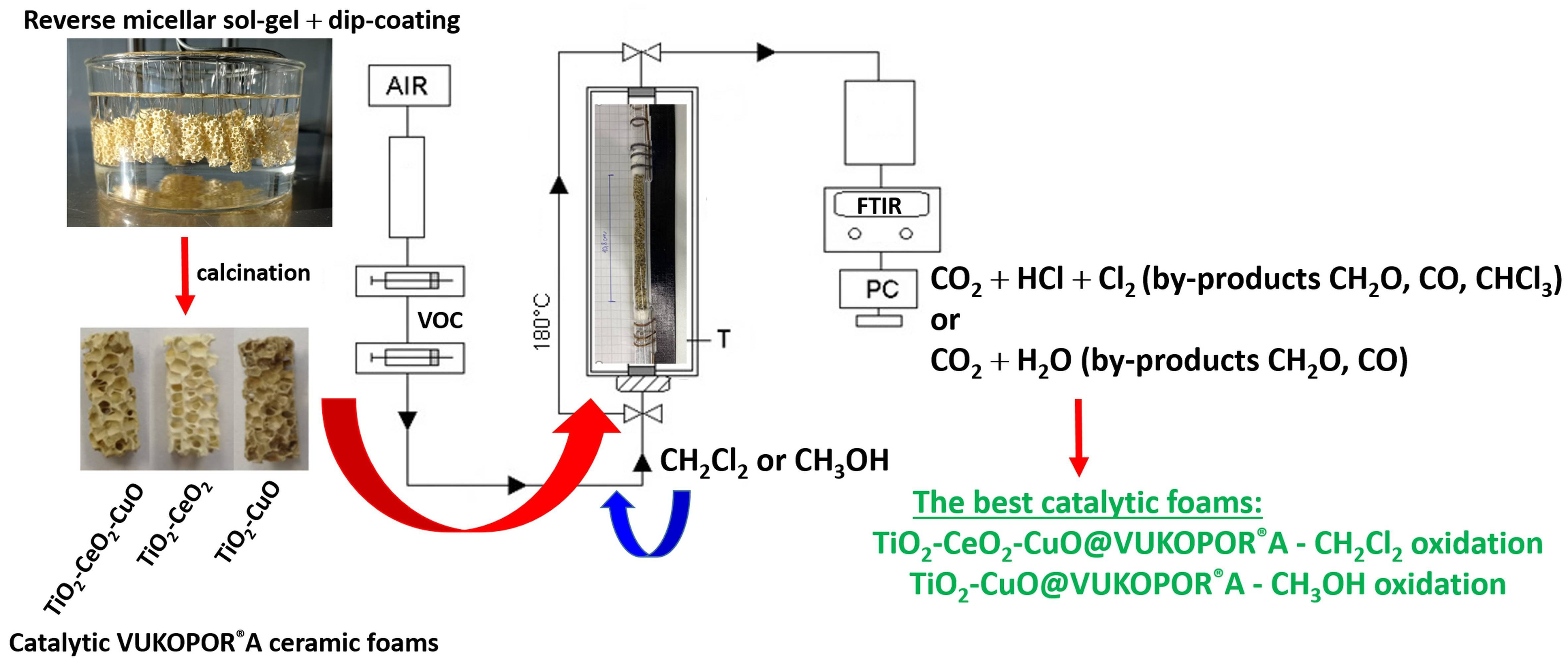
The appearance of parent VUKOPOR
®A ceramic foam and the prepared catalytic foams is shown in
Figure 1.
2.2. Physicochemical Characterization of TiO2, CeO2 and CuO-Supported VUKOPOR®A Ceramic Foams
X-ray fluorescence spectrometry (XRF) was implemented in order to determine the real chemical composition of the catalytic foams. The mass content of TiO2, CeO2 and CuO (in wt.%) was determined semi-quantitatively. XRF measurements were made on a dispersion spectrometer XEPOS (Spectro, Kleve, Germany) in a protective atmosphere (He), using a powder produced from the crashed catalytic foam, processed into a tablet with a diameter of 24 mm.
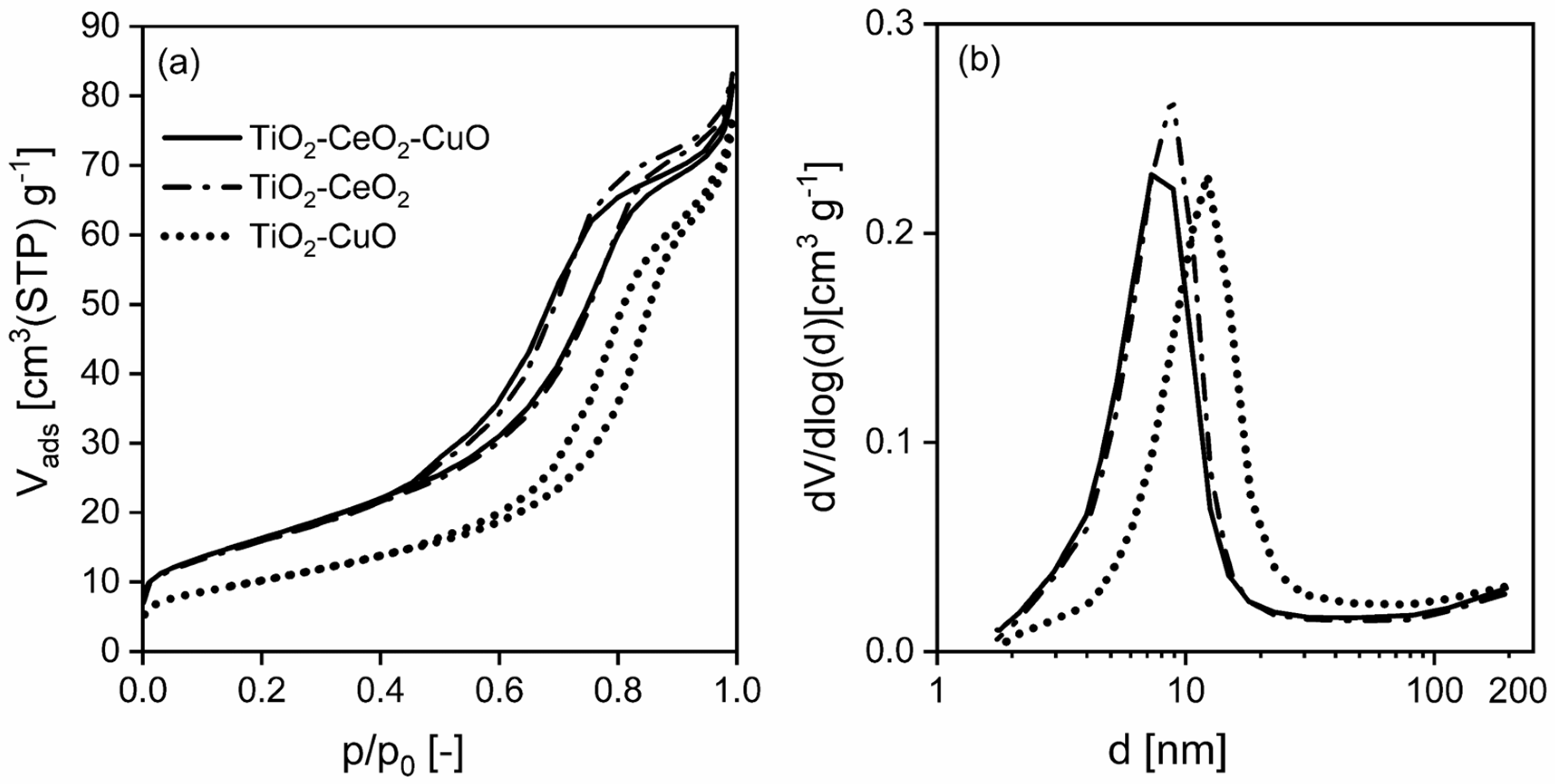
Textural properties of the prepared catalytic foams (i.e., specific surface area, S
BET, total pore volume, V
total, mesopore and macropore-size distribution) were evaluated on the basis of measurements of physical nitrogen adsorption at 77 K. Physisorption measurements were carried out on the 3Flex instrument (Micromeritics, Norcross, GA, USA). Prior to the physisorption measurement, the foams (4 pieces, containing ~0.14 g of deposited oxidic catalyst) were degassed under vacuum (<1 Pa) for ~24 h at 105 °C to remove the physically adsorbed water. The dried foams were then subjected to nitrogen physisorption at 77 K. The specific surface area, S
BET, was evaluated using the Brunauer-Emmett-Teller (BET) theory [
31] for the range of relative pressures p/p
0 = 0.05–0.30. The total pore volume, V
total, was evaluated from the adsorption branch of the nitrogen adsorption-desorption isotherm for the maximum relative pressure p/p
0 = 0.99. The mesopore-macropore-size distributions were evaluated using the Barret-Joyner-Halenda (BJH) method [
32], using the Roberts algorithm [
33] and the standard Broekhoff-deBoer isotherm with the Faas correction, using a Micromeritics software (3Flex, V6.02, Norcross, GA, USA).
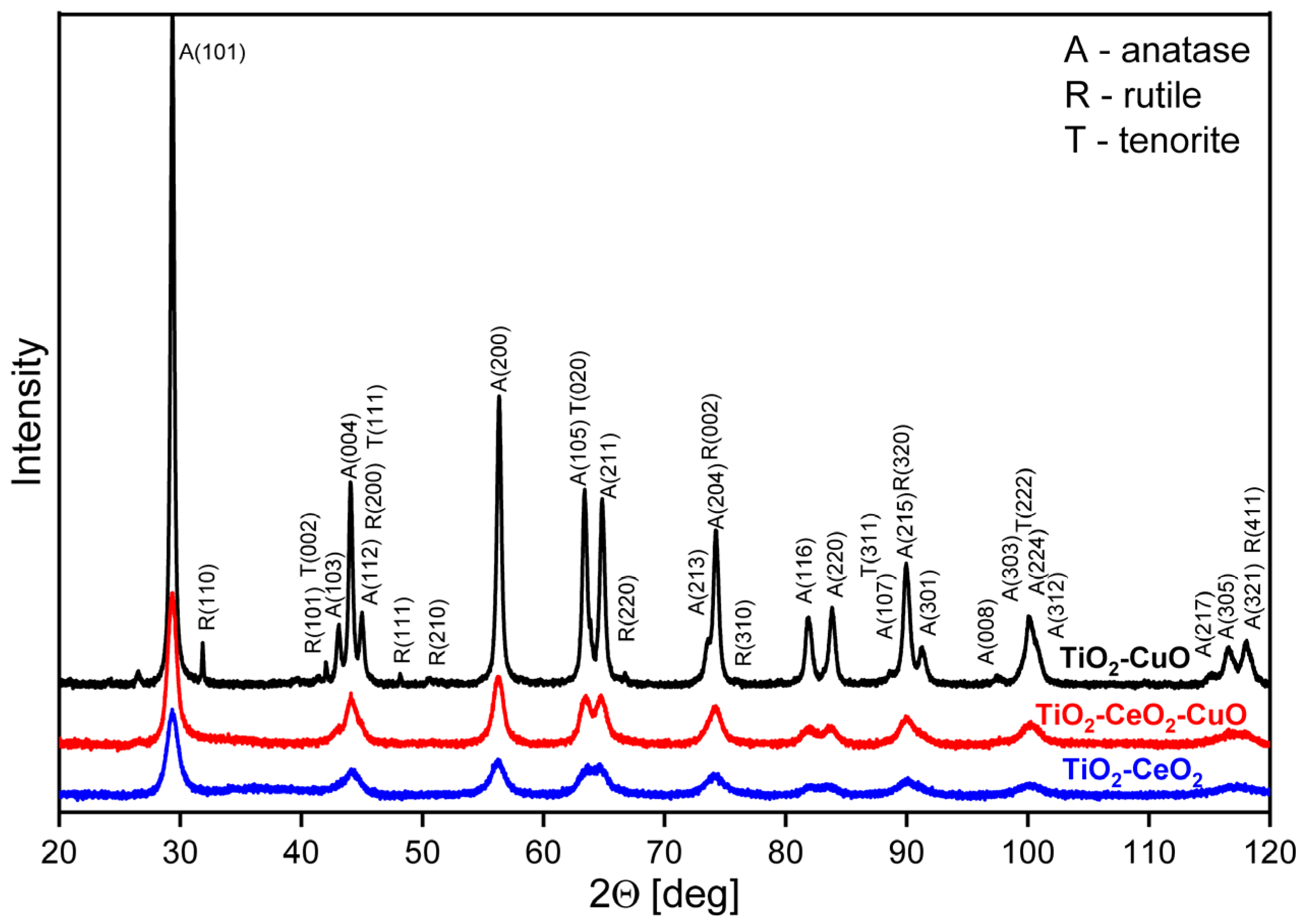
Structural properties of catalytic foams were determined by X-ray diffraction (XRD), specifically the crystalline phases of oxides present and the size of the crystallites. The diffraction patterns were measured on a Rigaku SmartLab powder diffractometer using the Bragg-Brentano Θ–2Θ geometry of the goniometer (Rigaku, Wilmington, MA, USA). For the XRD measurements, the powder equivalents of the catalysts deposited on the foams (i.e., oxides’ powders without foam) were used. Each powder catalyst in a thin layer (0.5 mm) on a silicon wafer in the form of a tablet with a diameter of 24 mm was prepared for the XRD measurement. Diffraction recording was made in the range of 2Θ = 5–90° with a step of 0.01° using a D/teX Ultra 250 detector (Rigaku Technologies, Austin, TX, USA) in a continuous scanning mode and a CoKα lamp (λ1 = 1.78892 nm, λ2 = 1.79278 nm). Phase composition was determined using the reference diffractograms database ICDD (PDF-2). The size of the crystallites was calculated based on the Scherrer equation with a diffractometer resolution correction to LaB6 standard.
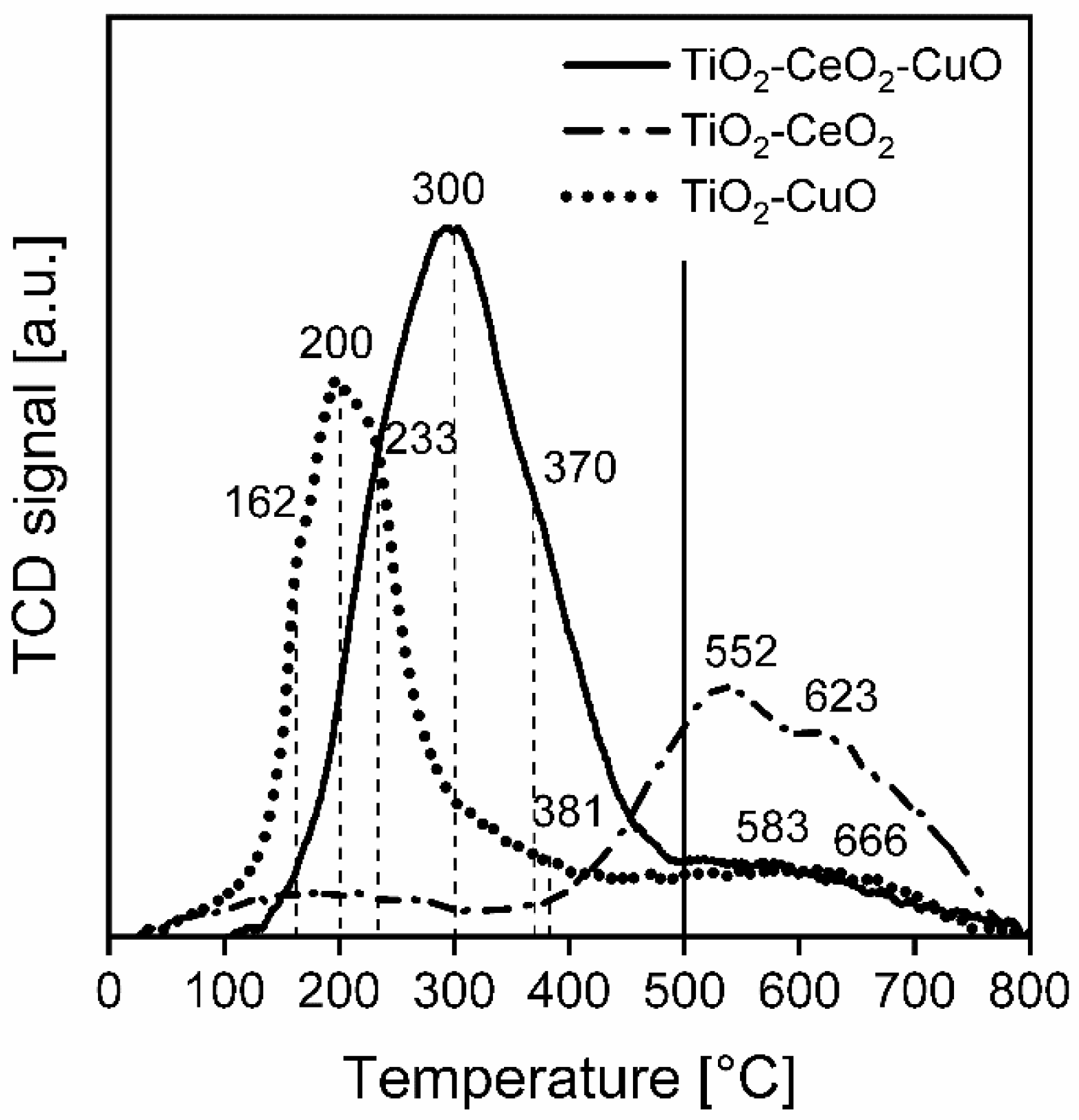
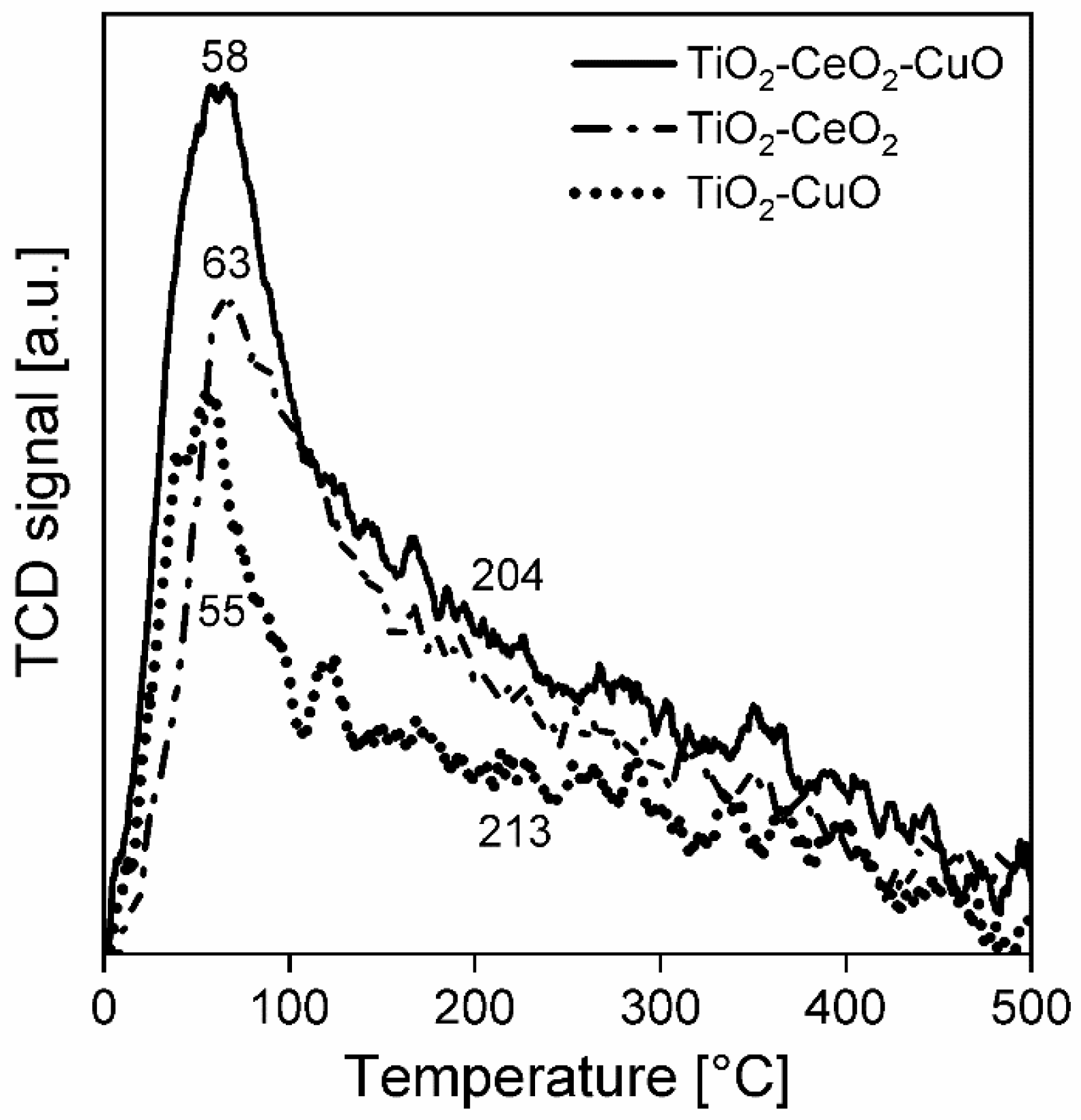
Temperature-programmed desorption of ammonia (NH3-TPD) was performed on an adsorption analyser AutoChem II (Micromeritics, Norcross, GA, USA) to investigate acid properties of the surface of the catalytic foams. The measurements were accomplished with two pieces of catalytic foams (mass of deposited catalyst ~60–66 mg) in the temperature range of 0–500 °C, with He as a carrier gas and NH3 as an adsorbing gas. Prior to the measurement, each catalytic foam was activated in He (50 mL/min) at 500 °C for 1 h, then cooled to 0 °C and saturated by 5% NH3/He for 30 min at 0 °C. Then, the catalytic foams were flushed with He (50 mL/min) to remove physically adsorbed NH3 up to the stable baseline (it took 110 min), with control using the mass spectrometer (MS-Hiden, Warrington, WA, United Kingdom). After that, the catalytic foams were heated in a He flow (50 mL/min) with a heating rate of 10 °C/min from 0 °C up to 500 °C (keeping at 500 °C for 5 min). A change in NH3 concentration was detected by a thermal conductivity detector (TCD, Norcross, GA, USA) and a mass spectrometer (MS). During the measurements, the following mass contributions were detected: 2-H2, 18-H2O, 16-NH3 and 44-CO2. TPD experiments were evaluated based on the calibration using a Micromeritics software (AutoChem II 2920MA, V6.01).
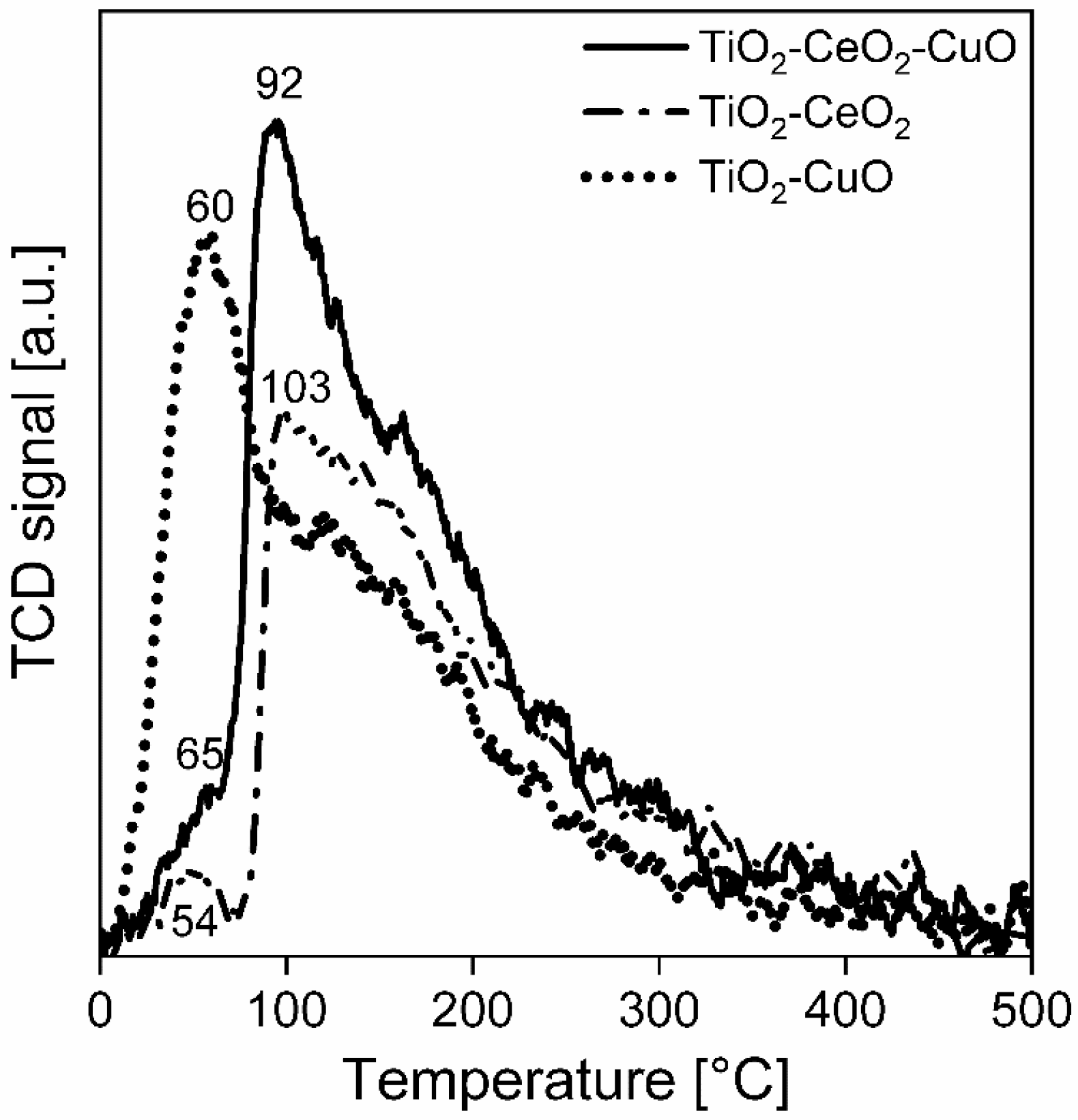
Temperature-programmed desorption of carbon dioxide (CO2-TPD) was performed on an adsorption analyser AutoChem II (Micromeritics, Norcross, GA, USA) as well to investigate basic properties of the surface of the catalytic foams. The measurements were accomplished with two pieces of catalytic foams (mass of deposited catalyst ~57–68 mg) in the temperature range of 0–500 °C, with He as a carrier gas and CO2 as an adsorbing gas. Prior to the measurement, each catalytic foam was activated in He (50 mL/min) at 500 °C for 1 h, then cooled to 0 °C and saturated by 50% CO2/He for 30 min at 0 °C. Then, the catalytic foams were flushed with He (50 mL/min) to remove physically adsorbed CO2 up to the stable baseline (it took 65 min), with control using a mass spectrometer (MS-Hiden). After that, the catalytic foams were heated in a He flow (50 mL/min) with a heating rate of 10 °C/min from 0 °C up to 500 °C (keeping at 500 °C for 5 min). A change in CO2 concentration was detected by a thermal conductivity detector (TCD) and a mass spectrometer (MS). During the measurements, the following mass contributions were detected: 2-H2, 18-H2O, 16-NH3 and 44-CO2. TPD experiments were evaluated based on the calibration using a Micromeritics software.
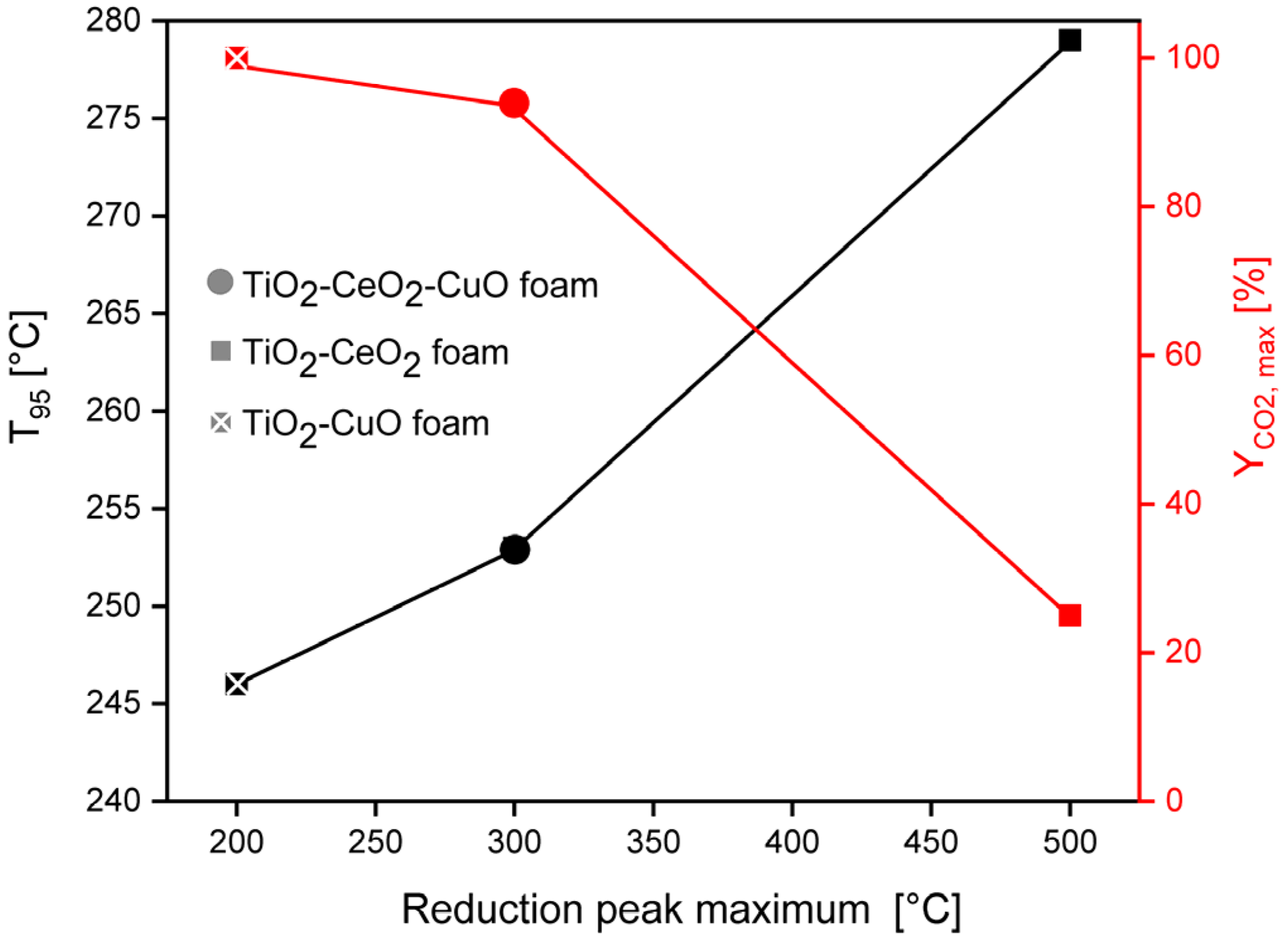
Temperature-programmed reduction by hydrogen (H2-TPR) was performed on an adsorption analyser AutoChem II (Micromeritics, Norcross, GA, USA) to investigate reducibility of the catalystic foams. The measurements were accomplished with two pieces of catalytic foams (mass of deposited catalyst ~58–65 mg) in the temperature range of 0–800 °C, with 10% H2/Ar mixture, Ar as a reference gas and H2 as a reducing gas. Prior to the measurement, each catalytic foam was dried at 250 °C for 30 min in Ar (30 mL/min), cooled to 28 °C and after setting the baseline the experiment started. The catalyst was heated in a 10% H2/Ar mixture flow (30 mL/min) with a heating rate of 10 °C/min from 28 °C up to 800 °C (keeping at 800 °C for 15 min). A change in the H2 concentration was detected by TCD. The TPR experiments were evaluated based on the calibration using a Micromeritics software.
For all measured temperature-programmed techniques (NH3-TPD, CO2-TPD and H2-TPR) the experimental error of measurements of temperature expressed as a relative standard deviation, determined on the basis of repeated tests of standards (Co3O4 and CoO), is 1.4%. The peaks area in NH3-TPD, CO2-TPD and H2-TPR spectra was calculated as the area under the curve in the region directly defined by the temperature without any deconvolution procedure.
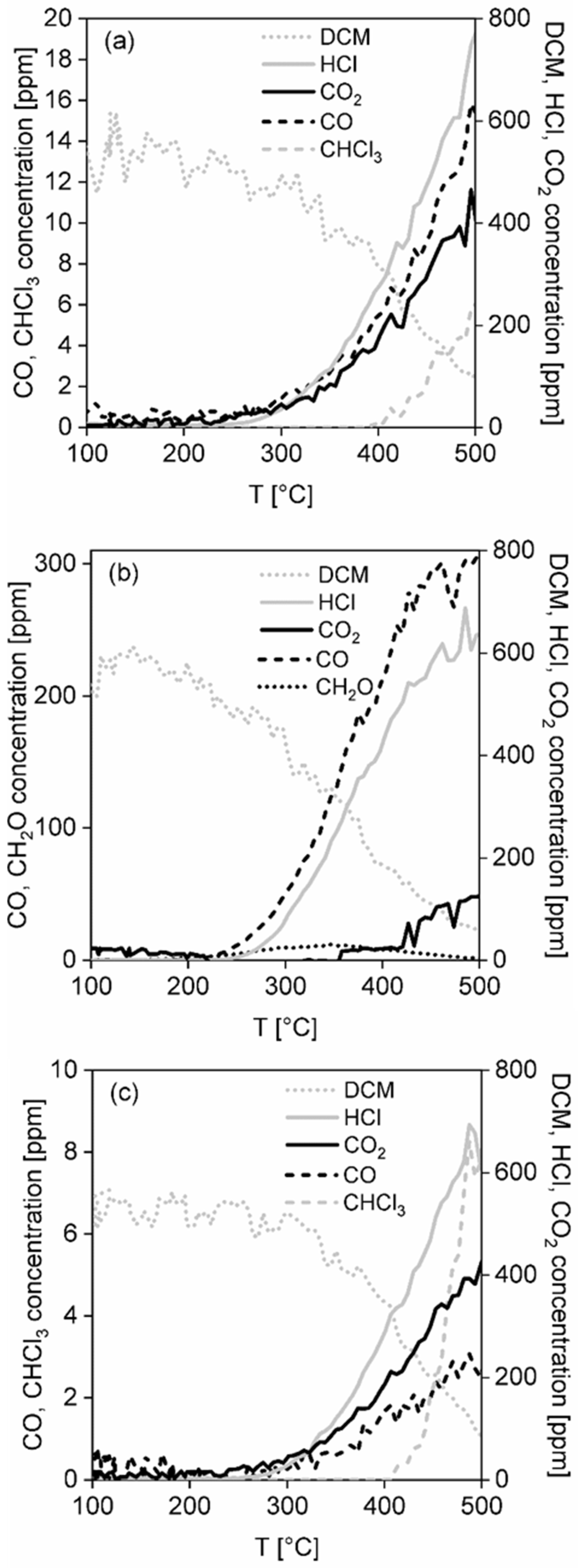
To determine chlorine in the catalytic foams after dichloromethane oxidation the catalytic foam was crushed in a friction bowl, placed in an ampoule and weighed. After that 5 mL H2O was added and the ampoule was placed in an ultrasound bath for 15min. After centrifugation, 4 mL of liquid was taken and diluted to 1:1 with water in an IC sample box. The sample was analysed using an ion chromatograph Metrohm (type ECO IC, a carbonate system with a suppressor, Metrosep A5 250/4.4 mm column, Herisau, Switzerland). This analysis is assigned as DisMC (join dissolution and mass chromatography).
Scanning electron microscope (SEM) Tescan Vega with a Tungsten cathode and an energy-dispersive X-ray spectroscopy (EDS-EDAX) (Tescan, Brno, Czech Republic) were used for morphological investigations. Micrographs were obtained using the modes of secondary electrons (SE) and backscattered electrons (BSE) with an acceleration voltage of 30 KeV. Before imaging, materials were gold sputtered to ensure adequate electron conductivity.
2.3. TiO2, CeO2 and CuO-Supported VUKOPOR®A Ceramic Foams Testing in Methanol and Dichloromethane Oxidation
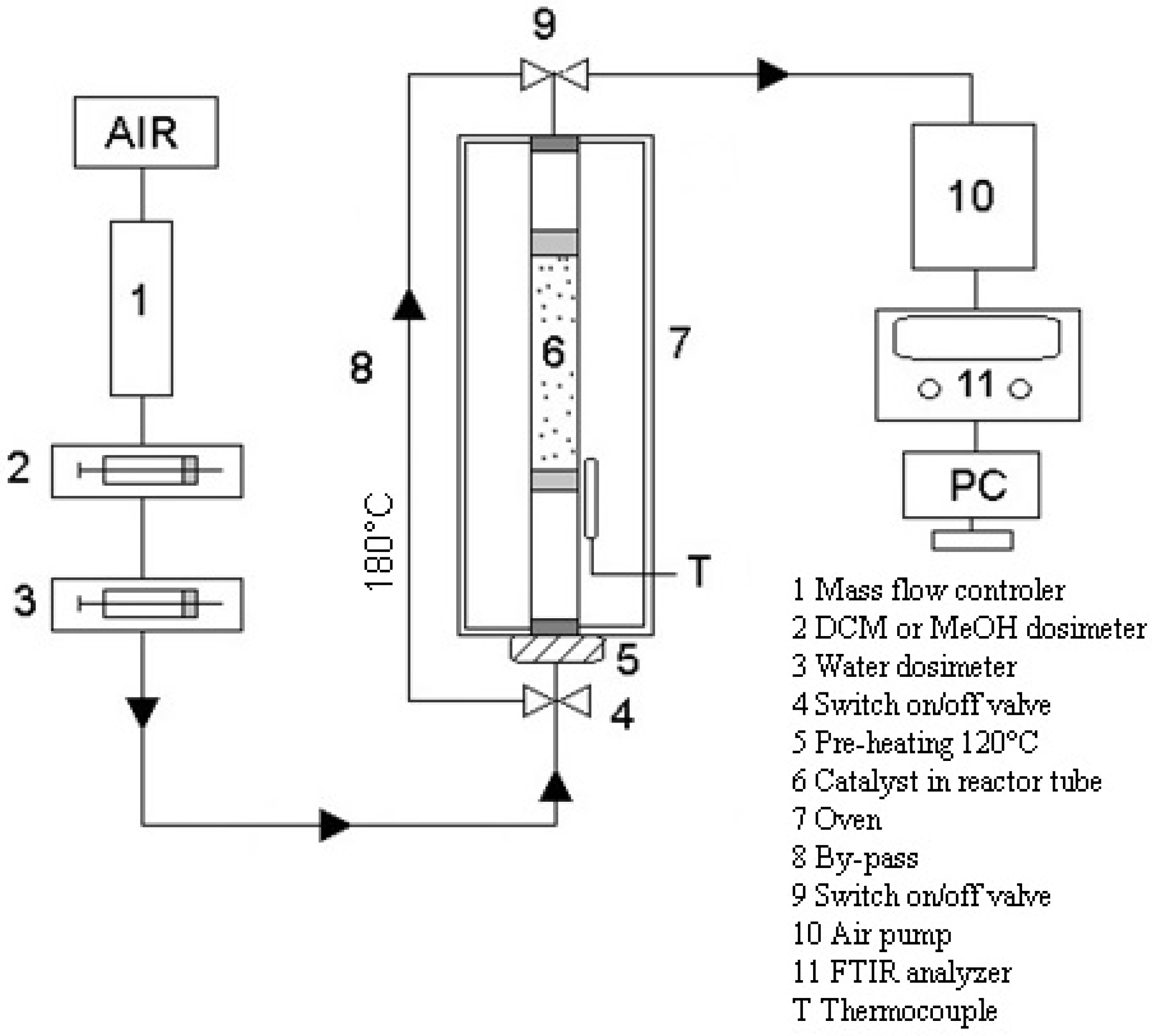
Testing of catalytic foams in methanol and dichloromethane oxidation was carried out in a fixed-bed flow tubular quartz reactor (inner diameter 8 mm) placed in a vertically situated ceramic tubular oven, operating under atmospheric pressure. The scheme of catalytic testing apparatus is shown in
Figure 2. Temperature during the catalytic experiments was measured on the reactor wall before the catalyst bed and all catalytic tests were evaluated with respect to this measured temperature. The length of the catalyst bed was kept constant 10.8 cm. The gas phase analysis was performed using an online Gasmet DX-4000N FTIR gas analyser (Gasmet, Helsinki, Finland) which was calibrated to detect chlorinated hydrocarbons such as C
2Cl
4, C
2HCl
3, CH
3Cl, CHCl
3, CH
2Cl
2, COCl
2, HCl, and oxyderivatives of hydrocarbons such as CO
2, CO, HCOOH, HCOOCH
3, CH
2O, CH
3OH, CH
4 and C
2H
4. The measured IR spectra were analysed with a Calcmet software. The FTIR analyser is not able to detect noble gases and diatomic homonuclear compounds such as O
2, N
2, H
2, and Cl
2.
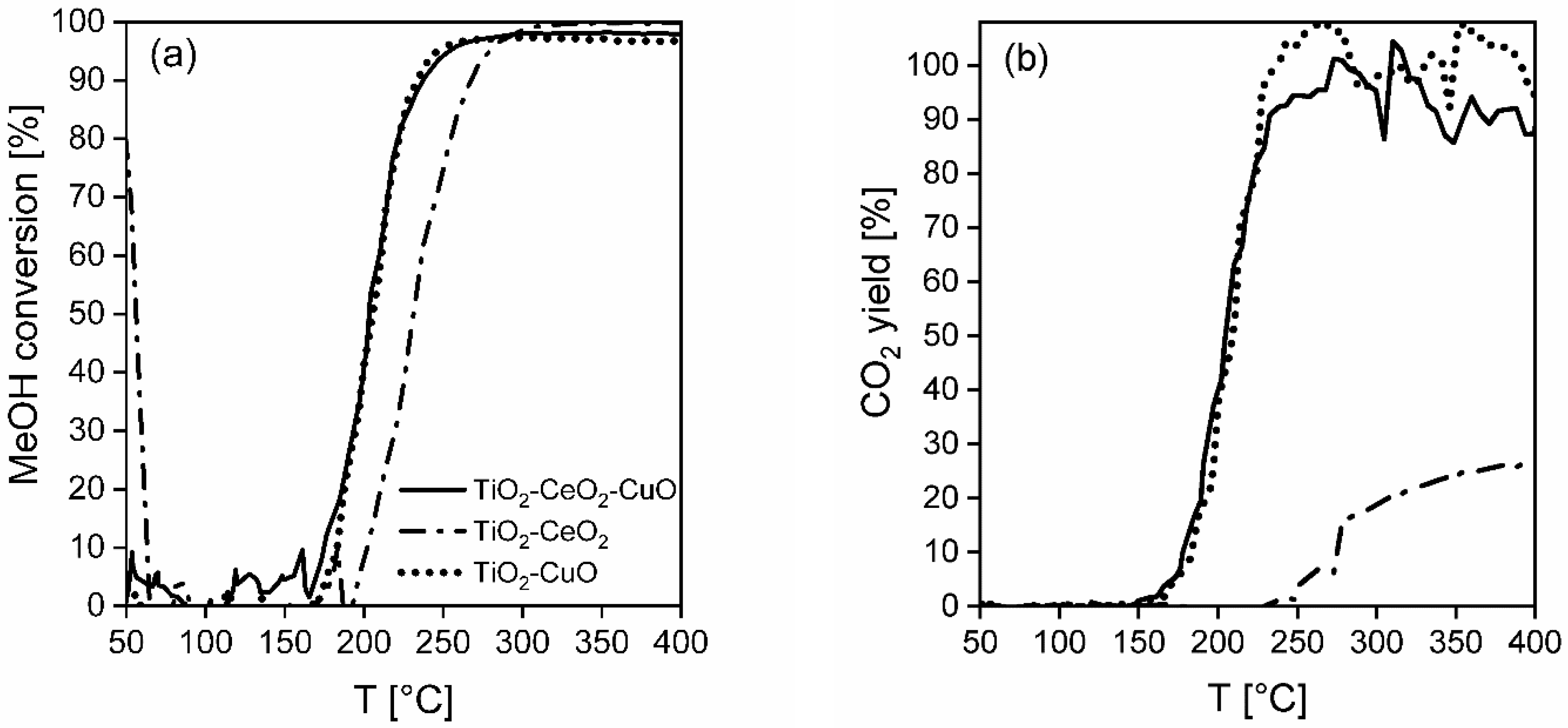
The initial methanol concentration was 500 ppm in all tests; i.e., ~0.715 g m−3 of methanol. Methanol was injected into an air flow by using automatic syringe pump and immediately evaporated to the air stream. The catalytic activity tests were carried out with fresh ceramic foams, keeping the constant length in the catalyst bed, i.e., 10.8 cm (the mass of deposited catalysts were as follows: TiO2-CeO2-CuO 0.1819 g, TiO2-CeO2 0.1985 g, TiO2-CuO 0.1748 g), which were pretreated prior to each catalytic test. The pretreatment of the fresh catalytic foam consisted of heating up the catalytic foam in an air flow from laboratory temperature up to 500 °C and cooling down to 45 °C, when the catalytic test with methanol started. The catalytic test with the same catalytic foam was repeated 2–4 times. Air flow was 1.05 L min−1, which corresponded to the space velocity (GHSV) of 11, 600 h−1. During the catalytic experiment the reaction temperature was increased from 45 °C up to 400 °C with a heating rate 5 °C min−1.
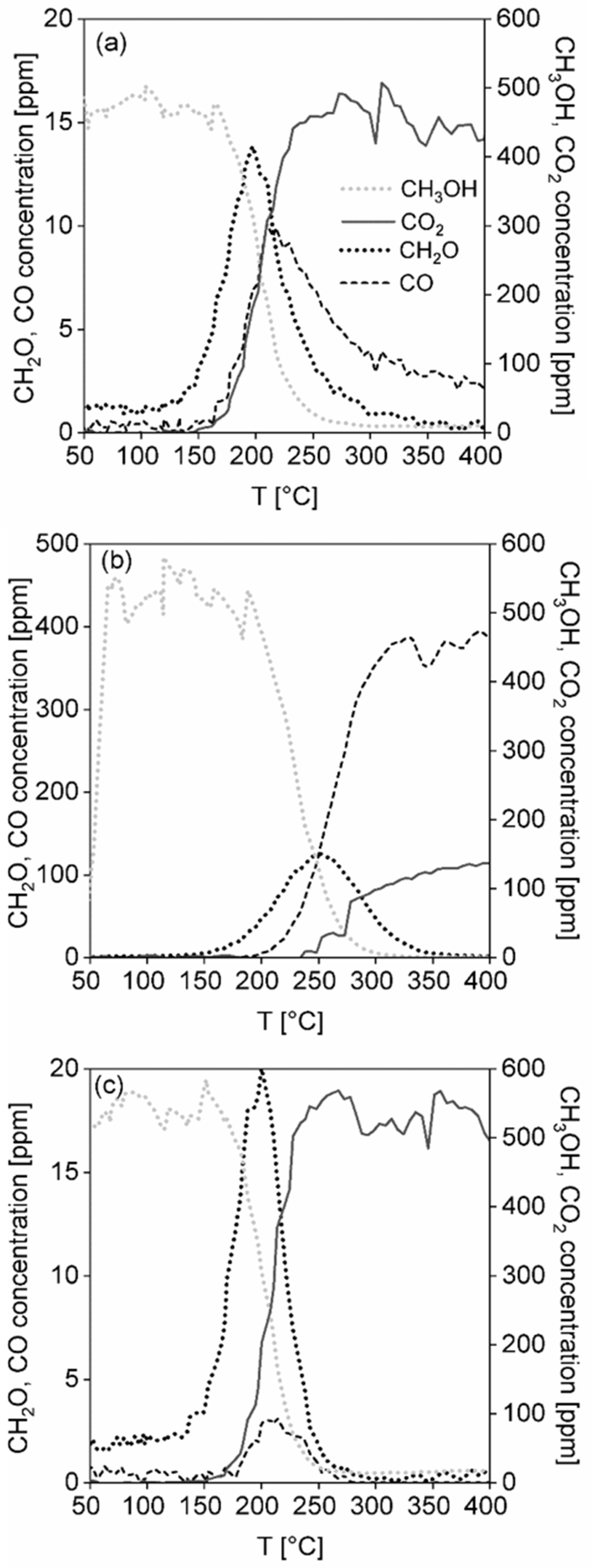
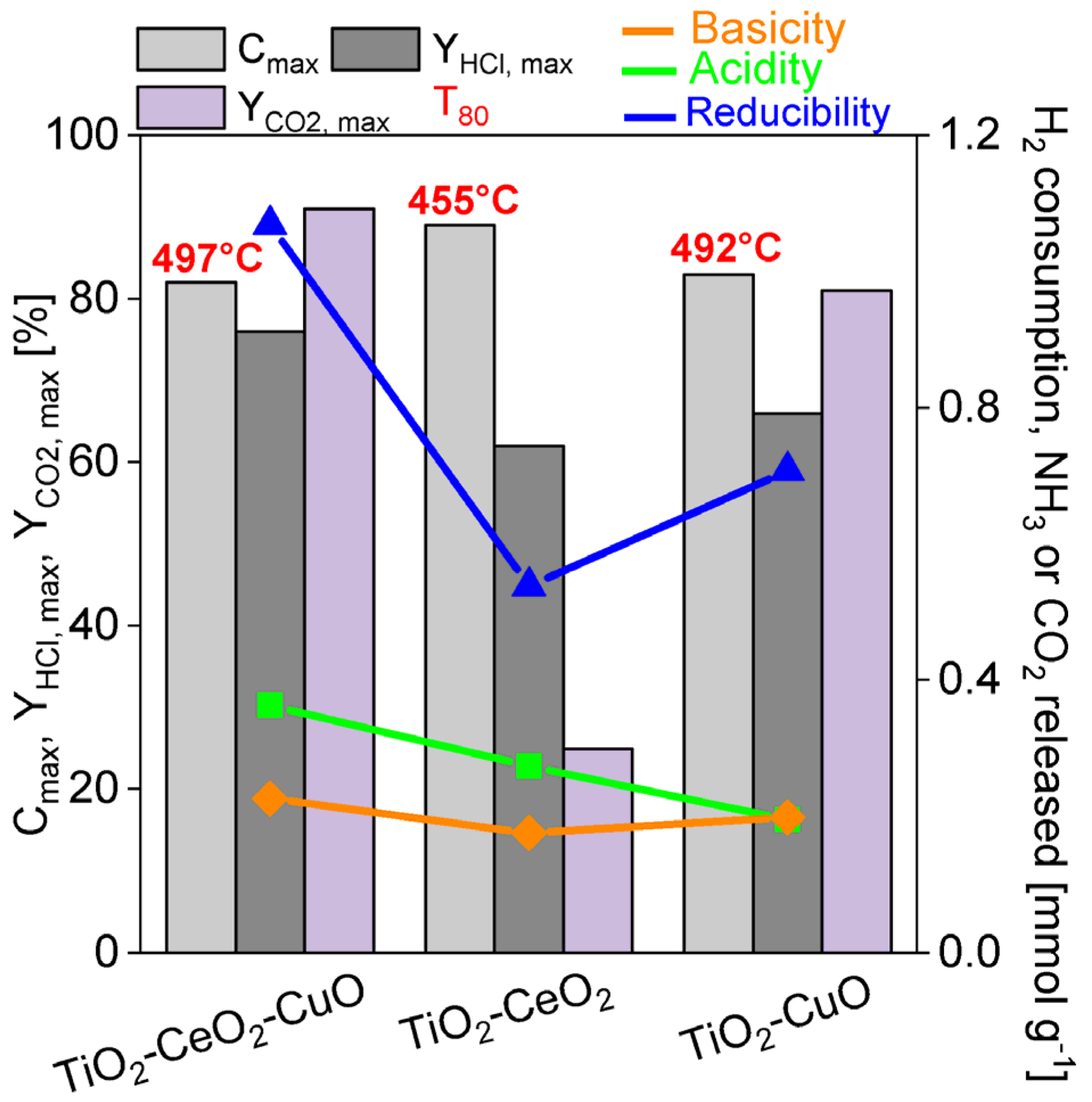
Temperatures T50, T90, T95 and Cmax corresponding to 50, 90, 95% and the maximum achieved methanol conversion were chosen as comparative measures of the catalyst activity. The CO2 and H2O yields were chosen as measures of catalyst selectivity.
The methanol conversion, C
MeOH, was calculated according to Equation (1):
where C
MeOH is the methanol conversion (%), c
MeOH,0 is the initial concentration of CH
3OH (volume ppm) and c
MeOH is the measured output concentration of methanol (volume ppm).
The CO
2 yield, Y
CO2, was calculated according to Equation (2):
where Y
CO2 is the CO
2 yield (%), c
CO2 is the measured output concentration of CO
2 (volume ppm) and c
MeOH,0 is the initial concentration of methanol (volume ppm).
The H
2O yield, Y
H2O, was calculated according to Equation (3):
where Y
H20 is the water yield (%), c
H2O is the measured output concentration of water (volume ppm) and c
MeOH,0 is the initial concentration of methanol (volume ppm).
The initial dichloromethane concentration was 500 ppm in all tests, i.e., ~1.895 g m−3 of dichloromethane. The catalytic activity measurement was performed in the presence of 1.5 wt.% of water. Dichloromethane and water were simultaneously injected into an air flow by using automatic syringe pumps and were evaporated immediately. The catalytic activity tests were carried out with fresh catalytic foams, keeping the constant length of catalyst bed in 10.8 cm (the mass of the deposited catalyst was 0.1650 g for TiO2-CeO2-CuO, 0.2029 g for TiO2-CeO2, 0.1737 g for TiO2-CuO), which was pretreated prior to each catalytic test. The pretreatment of the fresh catalytic foam consisted of heating up the catalyst in an air flow from room temperature to 500 °C and cooling down to 100 °C when the catalytic test started. The catalytic test with the same catalytic foam was repeated 2–4 times. The air flow was 1.05 L min−1, which corresponded to the space velocity (GHSV) of 11, 600 h−1. During the catalytic experiment the reaction temperature was increased from 100 °C up to 500 °C with a heating rate 5 °C min−1.
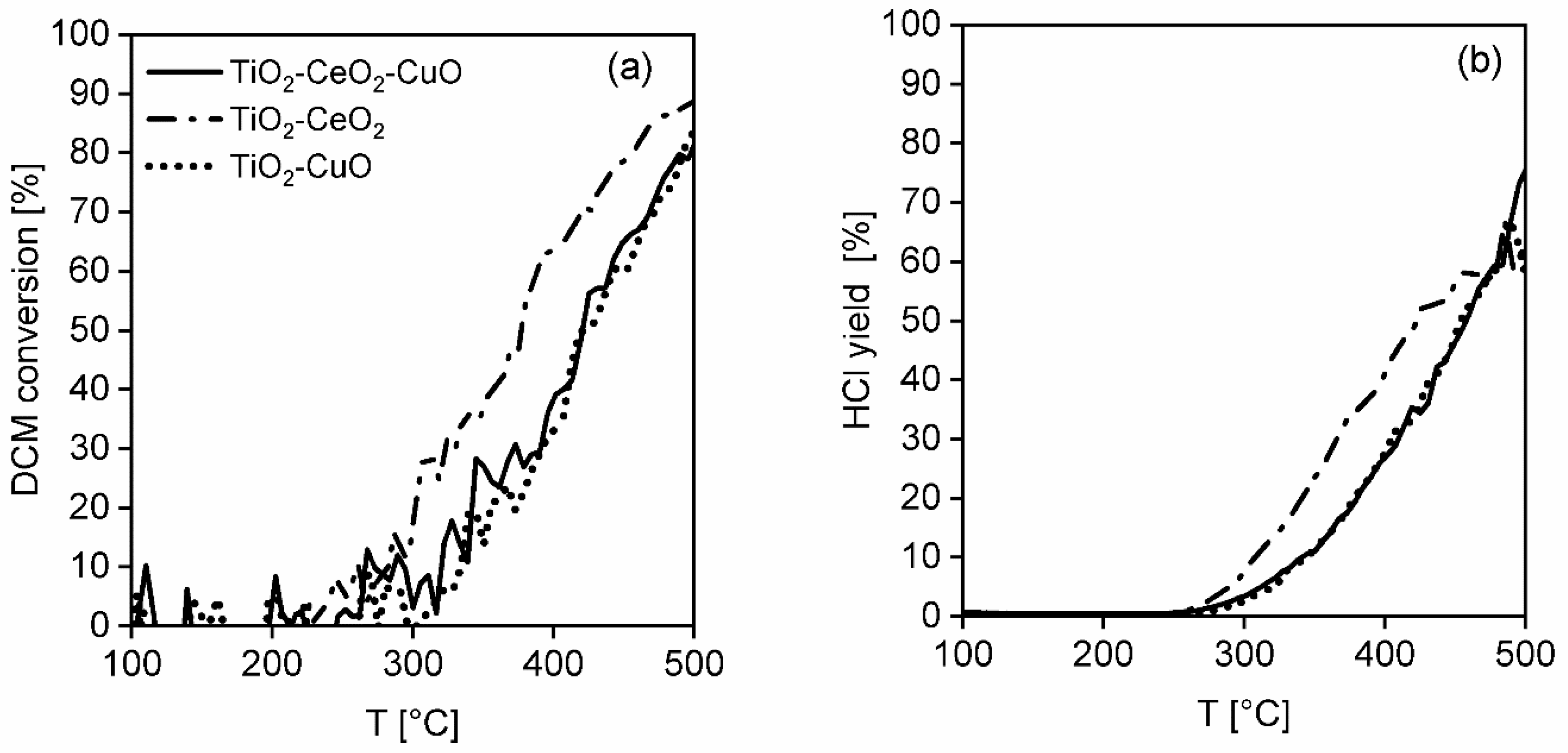
Temperatures T50 and T80 and Cmax corresponding to 50 and 80% dichloromethane conversion and the achieved maximum dichloromethane conversion were chosen as comparative measures of the catalyst activity. The maximum reached CO2 concentration (in ppm) and HCl yield were chosen as measures of selectivity.
The dichloromethane conversion, C
DCM, was calculated according to Equation (4):
where C
DCM is dichloromethane conversion (%), c
DCM,0 is the initial concentration of dichloromethane (volume ppm) and c
DCM in the measured output dichloromethane concentration (volume ppm).
The HCl yield, Y
HCl, was calculated according to Equation (5):
where Y
HCl is the HCl yield (%), c
HCl is the measured output concentration of HCl (volume ppm) and c
DCM,0 is the initial dichloromethane concentration (volume ppm).
The CO
2 yield, Y
CO2, was calculated according to Equation (6):
where Y
CO2 is the CO
2 yield (%), c
CO2 is the measured output concentration of CO
2 (volume ppm) and c
DCM,0 is the initial concentration of dichloromethane (volume ppm).
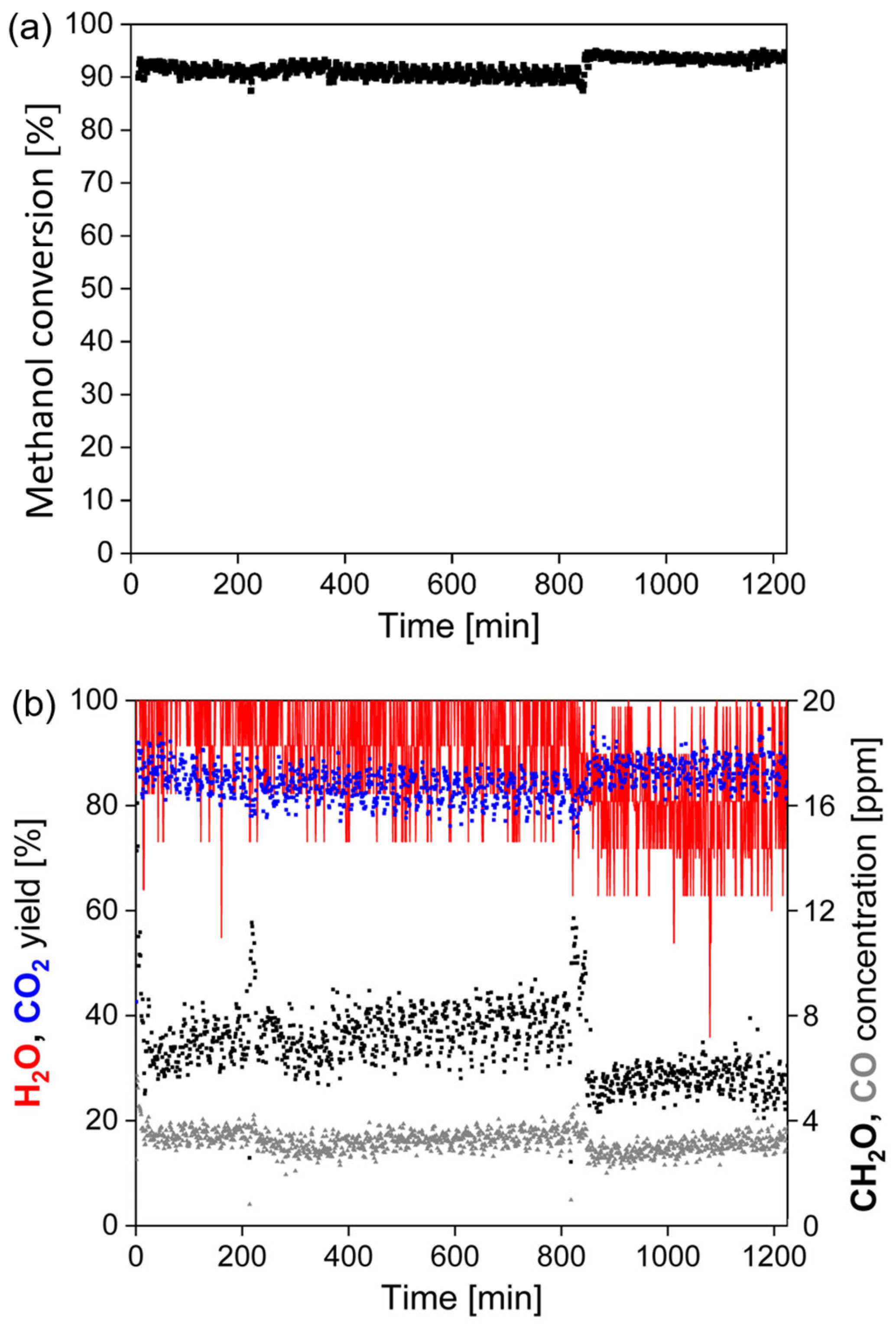
The stability test of the catalytic foams in methanol oxidation was performed with 6 pieces of the best catalytic foams, which created the catalyst bed of ~10.5 cm (corresponding to similar space velocity as in the light-off tests). Foams were first pre-treated in an air flow by heating-up them from room temperature up to 400 °C with heating rate of 5 °C min−1 and then cooled down to the starting temperature (i.e., to T90). After reaching T90 ~233 °C, the 20 h stability test was started with the methanol concentration of 500 ppm in an air flow of 1.05 L min−1. Based on the knowledge from the light-off tests, the main reaction products (CO2, H2O) and expected undesired by-products (CH2O, CO, HCOOH, CH4, HCOOCH3) were followed. The data was saved for each 60 s.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















