

Unravelling Dynamics Involving Multiple Charge Carriers in Semiconductor Nanocrystals


Abstract
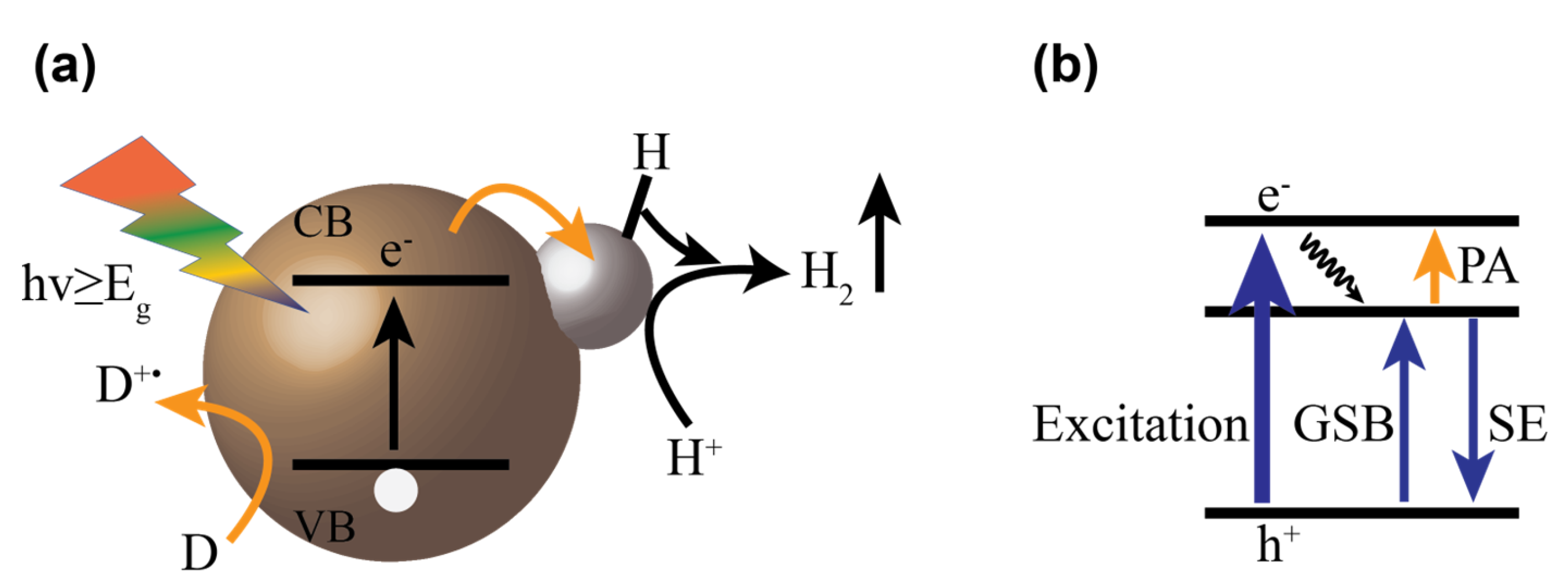
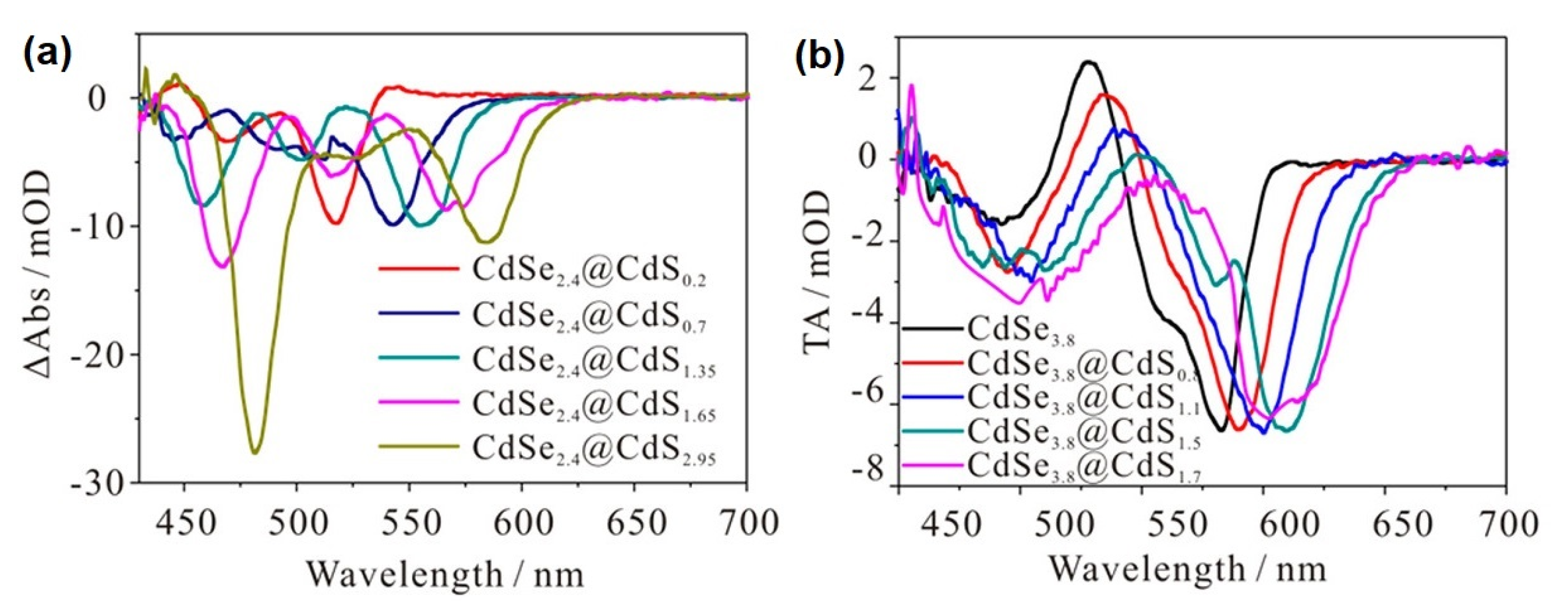
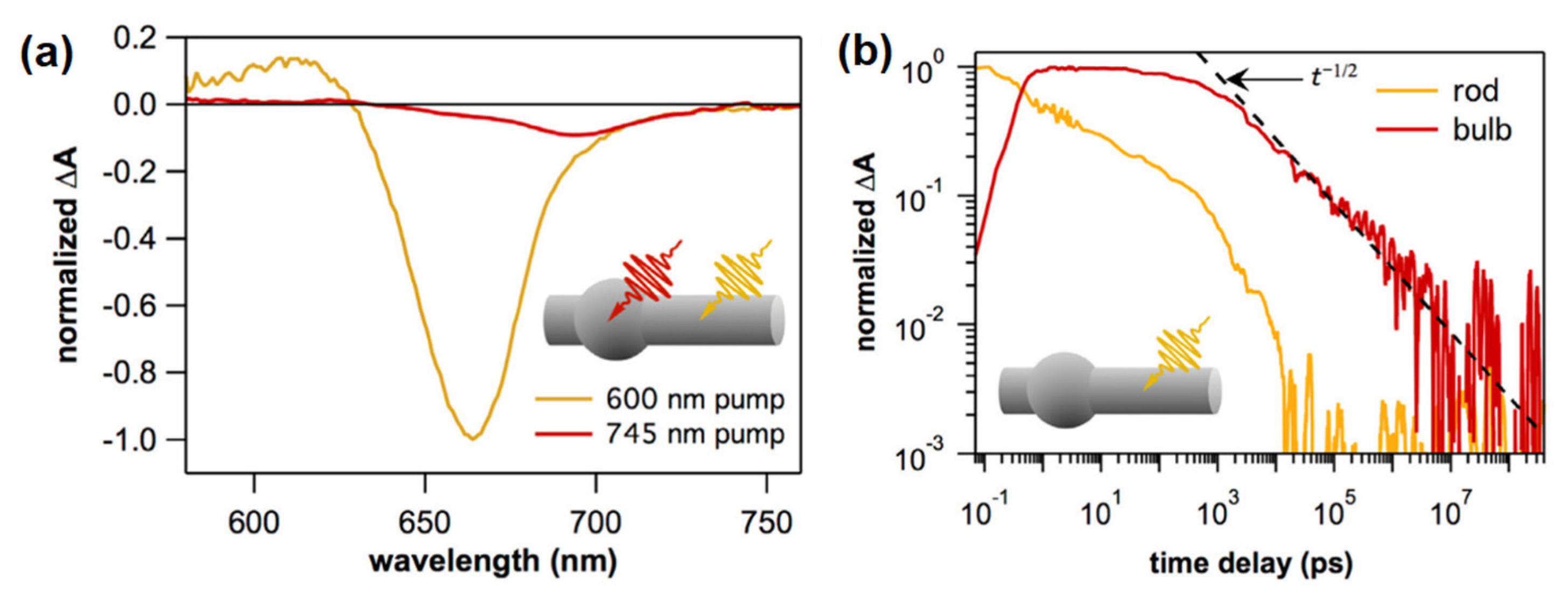
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Probing Electronic Structure of Heterostructures
3. Charge Carrier Dynamics in Nanorods
4. Charge Transfer Dynamics in Nanorods
5. Multiple Charge Carrier Accumulation and Transfer
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lesnyak, V.; Gaponik, N.; Eychmüller, A. Colloidal Semiconductor Nanocrystals: The Aqueous Approach. Chem. Soc. Rev. 2013, 42, 2905–2929. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.G.; Paria, S. Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Lee, J.S.; Kovalenko, M.V.; Shevchenko, E.V. Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications. Chem. Rev. 2010, 110, 389–458. [Google Scholar] [CrossRef] [PubMed]
- Pietryga, J.M.; Park, Y.-S.; Lim, J.; Fidler, A.F.; Bae, W.K.; Brovelli, S.; Klimov, V.I. Spectroscopic and Device Aspects of Nanocrystal Quantum Dots. Chem. Rev. 2016, 116, 10513–10622. [Google Scholar] [CrossRef] [PubMed]
- Gaponik, N.; Hickey, S.G.; Dorfs, D.; Rogach, A.L.; Eychmüller, A. Progress in the Light Emission of Colloidal Semiconductor Nanocrystals. Small 2010, 6, 1364–1378. [Google Scholar] [CrossRef]
- Vanmaekelbergh, D.; Liljeroth, P. Electron-Conducting Quantum Dot Solids: Novel Materials Based on Colloidal Semiconductor Nanocrystals. Chem. Soc. Rev. 2005, 34, 299–312. [Google Scholar] [CrossRef]
- Li, X.-B.; Tung, C.-H.; Wu, L.-Z. Semiconducting Quantum Dots for Artificial Photosynthesis. Nat. Rev. Chem. 2018, 2, 160–173. [Google Scholar] [CrossRef]
- Stolarczyk, J.K.; Bhattacharyya, S.; Polavarapu, L.; Feldmann, J. Challenges and Prospects in Solar Water Splitting and CO2 Reduction with Inorganic and Hybrid Nanostructures. ACS Catal. 2018, 8, 3602–3635. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. Photocatalytic Water Splitting: Recent Progress and Future Challenges. J. Phys. Chem. Lett. 2010, 1, 2655–2661. [Google Scholar] [CrossRef]
- Izadpanah Ostad, M.; Niknam Shahrak, M.; Galli, F. Photocatalytic Carbon Dioxide Reduction to Methanol Catalyzed by ZnO, Pt, Au, and Cu Nanoparticles Decorated Zeolitic Imidazolate Framework-8. J. CO2 Util. 2021, 43, 101373. [Google Scholar] [CrossRef]
- Kumar, S.; Regue, M.; Isaacs, M.A.; Freeman, E.; Eslava, S. All-Inorganic CsPbBr 3 Nanocrystals: Gram-Scale Mechanochemical Synthesis and Selective Photocatalytic CO2 Reduction to Methane. ACS Appl. Energy Mater. 2020, 3, 4509–4522. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lim, C.K.; Park, B.; Kim, M.; Jamal, A.; Song, H. Surface Activation of Cobalt Oxide Nanoparticles for Photocatalytic Carbon Dioxide Reduction to Methane. J. Mater. Chem. A 2019, 7, 15068–15072. [Google Scholar] [CrossRef]
- Feng, Y.-X.; Wang, H.-J.; Wang, J.-W.; Zhang, W.; Zhang, M.; Lu, T.-B. Stand-Alone CdS Nanocrystals for Photocatalytic CO2 Reduction with High Efficiency and Selectivity. ACS Appl. Mater. Interfaces 2021, 13, 26573–26580. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Domen, K. New Non-Oxide Photocatalysts Designed for Overall Water Splitting under Visible Light. J. Phys. Chem. C 2007, 111, 7851–7861. [Google Scholar] [CrossRef]
- Moroz, P.; Boddy, A.; Zamkov, M. Challenges and Prospects of Photocatalytic Applications Utilizing Semiconductor Nanocrystals. Front. Chem. 2018, 6, 353. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, G.; Liu, R.; Zhou, S.; Sheehan, S.W.; Wang, D. Semiconductor Nanostructure-Based Photoelectrochemical Water Splitting: A Brief Review. Chem. Phys. Lett. 2011, 507, 209–215. [Google Scholar] [CrossRef]
- Vaneski, A.; Schneider, J.; Susha, A.S.; Rogach, A.L. Colloidal Hybrid Heterostructures Based on II-VI Semiconductor Nanocrystals for Photocatalytic Hydrogen Generation. J. Photochem. Photobiol. C Photochem. Rev. 2014, 19, 52–61. [Google Scholar] [CrossRef]
- Wu, K.; Lian, T. Quantum Confined Colloidal Nanorod Heterostructures for Solar-to-Fuel Conversion. Chem. Soc. Rev. 2016, 45, 3781–3810. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial Photosynthesis: Opportunities and Challenges of Molecular Catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Low, J.; Fang, Y.; Xiao, J.; Chen, X. Engineering Heterogeneous Semiconductors for Solar Water Splitting. J. Mater. Chem. A 2015, 3, 2485–2534. [Google Scholar] [CrossRef]
- Maeda, K. Z-Scheme Water Splitting Using Two Different Semiconductor Photocatalysts. ACS Catal. 2013, 3, 1486–1503. [Google Scholar] [CrossRef]
- Wilker, M.B.; Schnitzenbaumer, K.J.; Dukovic, G. Recent Progress in Photocatalysis Mediated by Colloidal II-VI Nanocrystals. Isr. J. Chem. 2012, 52, 1002–1015. [Google Scholar] [CrossRef]
- Peng, X.; Schlamp, M.C.; Kadavanich, A.V.; Alivisatos, A.P. Epitaxial Growth of Highly Luminescent CdSe/CdS Core/Shell Nanocrystals with Photostability and Electronic Accessibility. J. Am. Chem. Soc. 1997, 119, 7019–7029. [Google Scholar] [CrossRef]
- Talapin, D.V.; Koeppe, R.; Götzinger, S.; Kornowski, A.; Lupton, J.M.; Rogach, A.L.; Benson, O.; Feldmann, J.; Weller, H. Highly Emissive Colloidal CdSe/CdS Heterostructures of Mixed Dimensionality. Nano Lett. 2003, 3, 1677–1681. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, R. Nickel-Based Cocatalysts for Photocatalytic Hydrogen Production. Appl. Surf. Sci. 2015, 351, 779–793. [Google Scholar] [CrossRef]
- Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S.Z. Earth-Abundant Cocatalysts for Semiconductor-Based Photocatalytic Water Splitting. Chem. Soc. Rev. 2014, 43, 7787–7812. [Google Scholar] [CrossRef]
- Han, B.; Hu, Y.H. MoS2 as a Co-catalyst for Photocatalytic Hydrogen Production from Water. Energy Sci. Eng. 2016, 4, 285–304. [Google Scholar] [CrossRef]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-Based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef]
- Wu, K.; Du, Y.; Tang, H.; Chen, Z.; Lian, T. Efficient Extraction of Trapped Holes from Colloidal CdS Nanorods. J. Am. Chem. Soc. 2015, 137, 10224–10230. [Google Scholar] [CrossRef]
- Wolff, C.M.; Frischmann, P.D.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F.; et al. All-in-One Visible-Light-Driven Water Splitting by Combining Nanoparticulate and Molecular Co-Catalysts on CdS Nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Spanhel, L.; Haase, M.; Weller, H.; Henglein, A. Photochemistry of Colloidal Semiconductors. 20. Surface Modification and Stability of Strong Luminescing CdS Particles. J. Am. Chem. Soc. 1987, 109, 5649–5655. [Google Scholar] [CrossRef]
- Chen, H.; Gai, H.; Yeung, E.S. Inhibition of Photobleaching and Blue Shift in Quantum Dots. Chem. Commun. 2009, 1676–1678. [Google Scholar] [CrossRef] [PubMed]
- Pietra, F.; van Dijk-Moes, R.J.A.; Ke, X.; Bals, S.; Van Tendeloo, G.; de Mello Donega, C.; Vanmaekelbergh, D. Synthesis of Highly Luminescent Silica-Coated CdSe/CdS Nanorods. Chem. Mater. 2013, 25, 3427–3434. [Google Scholar] [CrossRef]
- Reiss, P.; Bleuse, J.; Pron, A. Highly Luminescent CdSe/ZnSe Core/Shell Nanocrystals of Low Size Dispersion. Nano Lett. 2002, 2, 781–784. [Google Scholar] [CrossRef]
- Wen, X.; Sitt, A.; Yu, P.; Ko, H.; Toh, Y.-R.; Tang, J. Studies of the Photostability of CdSe/CdS Dot-in-Rod Nanoparticles. J. Nanoparticle Res. 2012, 14, 1278. [Google Scholar] [CrossRef]
- Amirav, L.; Alivisatos, A.P. Photocatalytic Hydrogen Production with Tunable Nanorod Heterostructures. J. Phys. Chem. Lett. 2010, 1, 1051–1054. [Google Scholar] [CrossRef]
- Perry, D.; Waiskopf, N.; Verbitsky, L.; Remennik, S.; Banin, U. Shell Stabilization of Photocatalytic ZnSe Nanorods. ChemCatChem 2019, 11, 6208–6212. [Google Scholar] [CrossRef]
- Berr, M.J.; Wagner, P.; Fischbach, S.; Vaneski, A.; Schneider, J.; Susha, A.S.; Rogach, A.L.; Jäckel, F.; Feldmann, J. Hole Scavenger Redox Potentials Determine Quantum Efficiency and Stability of Pt-Decorated CdS Nanorods for Photocatalytic Hydrogen Generation. Appl. Phys. Lett. 2012, 100, 223903. [Google Scholar] [CrossRef]
- Bang, J.U.; Lee, S.J.; Jang, J.S.; Choi, W.; Song, H. Geometric Effect of Single or Double Metal-Tipped CdSe Nanorods on Photocatalytic H2 Generation. J. Phys. Chem. Lett. 2012, 3, 3781–3785. [Google Scholar] [CrossRef]
- Nakibli, Y.; Kalisman, P.; Amirav, L. Less Is More: The Case of Metal Cocatalysts. J. Phys. Chem. Lett. 2015, 6, 2265–2268. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, W.-W.; Park, B.; Sul, S.; Kwon, O.-H.; Song, H. Optimal Length of Hybrid Metal–Semiconductor Nanorods for Photocatalytic Hydrogen Generation. ACS Catal. 2021, 11, 13303–13311. [Google Scholar] [CrossRef]
- Simon, T.; Carlson, M.T.; Stolarczyk, J.K.; Feldmann, J. Electron Transfer Rate vs Recombination Losses in Photocatalytic H2 Generation on Pt-Decorated CdS Nanorods. ACS Energy Lett. 2016, 1, 1137–1142. [Google Scholar] [CrossRef]
- Chen, W.; Li, X.; Wang, F.; Javaid, S.; Pang, Y.; Chen, J.; Yin, Z.; Wang, S.; Li, Y.; Jia, G. Nonepitaxial Gold-Tipped ZnSe Hybrid Nanorods for Efficient Photocatalytic Hydrogen Production. Small 2020, 16, 1902231. [Google Scholar] [CrossRef]
- Acharya, K.P.; Khnayzer, R.S.; O’Connor, T.; Diederich, G.; Kirsanova, M.; Klinkova, A.; Roth, D.; Kinder, E.; Imboden, M.; Zamkov, M. The Role of Hole Localization in Sacrificial Hydrogen Production by Semiconductor-Metal Heterostructured Nanocrystals. Nano Lett. 2011, 11, 2919–2926. [Google Scholar] [CrossRef]
- Boecker, M.; Micheel, M.; Mengele, A.K.; Neumann, C.; Herberger, T.; Marchesi D’Alvise, T.; Liu, B.; Undisz, A.; Rau, S.; Turchanin, A.; et al. Rhodium-Complex-Functionalized and Polydopamine-Coated CdSe@CdS Nanorods for Photocatalytic NAD+ Reduction. ACS Appl. Nano Mater. 2021, 4, 12913–12919. [Google Scholar] [CrossRef]
- Bie, C.; Fu, J.; Cheng, B.; Zhang, L. Ultrathin CdS Nanosheets with Tunable Thickness and Efficient Photocatalytic Hydrogen Generation. Appl. Surf. Sci. 2018, 462, 606–614. [Google Scholar] [CrossRef]
- Jia, J.; Sun, W.; Zhang, Q.; Zhang, X.; Hu, X.; Liu, E.; Fan, J. Inter-Plane Heterojunctions within 2D/2D FeSe2/g-C3N4 Nanosheet Semiconductors for Photocatalytic Hydrogen Generation. Appl. Catal. B Environ. 2020, 261, 118249. [Google Scholar] [CrossRef]
- Wu, K.; Zhu, H.; Lian, T. Ultrafast Exciton Dynamics and Light-Driven H2 Evolution in Colloidal Semiconductor Nanorods and Pt-Tipped Nanorods. Acc. Chem. Res. 2015, 48, 851–859. [Google Scholar] [CrossRef]
- Banin, U.; Ben-Shahar, Y.; Vinokurov, K. Hybrid Semiconductor-Metal Nanoparticles: From Architecture to Function. Chem. Mater. 2014, 26, 97–110. [Google Scholar] [CrossRef]
- Giblin, J.; Kuno, M. Nanostructure Absorption: A Comparative Study of Nanowire and Colloidal Quantum Dot Absorption Cross Sections. J. Phys. Chem. Lett. 2010, 1, 3340–3348. [Google Scholar] [CrossRef]
- Carey, C.R.; LeBel, T.; Crisostomo, D.; Giblin, J.; Kuno, M.; Hartland, G.V. Imaging and Absolute Extinction Cross-Section Measurements of Nanorods and Nanowires through Polarization Modulation Microscopy. J. Phys. Chem. C 2010, 114, 16029–16036. [Google Scholar] [CrossRef]
- Berr, M.; Vaneski, A.; Susha, A.S.; Rodríguez-Fernández, J.; Döblinger, M.; Jäckel, F.; Rogach, A.L.; Feldmann, J. Colloidal CdS Nanorods Decorated with Subnanometer Sized Pt Clusters for Photocatalytic Hydrogen Generation. Appl. Phys. Lett. 2010, 97, 093108. [Google Scholar] [CrossRef]
- Vaneski, A.; Susha, A.S.; Rodríguez-Fernández, J.; Berr, M.; Jäckel, F.; Feldmann, J.; Rogach, A.L. Hybrid Colloidal Heterostructures of Anisotropic Semiconductor Nanocrystals Decorated with Noble Metals: Synthesis and Function. Adv. Funct. Mater. 2011, 21, 1547–1556. [Google Scholar] [CrossRef]
- Wu, K.; Chen, Z.; Lv, H.; Zhu, H.; Hill, C.L.; Lian, T. Hole Removal Rate Limits Photodriven H2 Generation Efficiency in CdS-Pt and CdSe/CdS-Pt Semiconductor Nanorod-Metal Tip Heterostructures. J. Am. Chem. Soc. 2014, 136, 7708–7716. [Google Scholar] [CrossRef]
- Kalisman, P.; Nakibli, Y.; Amirav, L. Perfect Photon-to-Hydrogen Conversion Efficiency. Nano Lett. 2016, 16, 1776–1781. [Google Scholar] [CrossRef]
- Wächtler, M.; Kalisman, P.; Amirav, L. Charge-Transfer Dynamics in Nanorod Photocatalysts with Bimetallic Metal Tips. J. Phys. Chem. C 2016, 120, 24491–24497. [Google Scholar] [CrossRef]
- Kong, D.; Jia, Y.; Ren, Y.; Xie, Z.; Wu, K.; Lian, T. Shell-Thickness-Dependent Biexciton Lifetime in Type I and Quasi-Type II CdSe@CdS Core/Shell Quantum Dots. J. Phys. Chem. C 2018, 122, 14091–14098. [Google Scholar] [CrossRef]
- Müller, J.; Lupton, J.M.; Lagoudakis, P.G.; Schindler, F.; Koeppe, R.; Rogach, A.L.; Feldmann, J.; Talapin, D.V.; Weller, H. Wave Function Engineering in Elongated Semiconductor Nanocrystals with Heterogeneous Carrier Confinement. Nano Lett. 2005, 5, 2044–2049. [Google Scholar] [CrossRef]
- Irfan, R.M.; Jiang, D.; Sun, Z.; Lu, D.; Du, P. Enhanced Photocatalytic H2 Production on CdS Nanorods with Simple Molecular Bidentate Cobalt Complexes as Cocatalysts under Visible Light. Dalton Trans. 2016, 45, 12897–12905. [Google Scholar] [CrossRef]
- Simon, T.; Bouchonville, N.; Berr, M.J.; Vaneski, A.; Adrović, A.; Volbers, D.; Wyrwich, R.; Döblinger, M.; Susha, A.S.; Rogach, A.L.; et al. Redox Shuttle Mechanism Enhances Photocatalytic H2 Generation on Ni-Decorated CdS Nanorods. Nat. Mater. 2014, 13, 1013–1018. [Google Scholar] [CrossRef]
- Dong, K.; Le, T.; Nakibli, Y.; Schleusener, A.; Wächtler, M.; Amirav, L. Molecular Metallocorrole–Nanorod Photocatalytic System for Sustainable Hydrogen Production. ChemSusChem 2022, 15, e202200804. [Google Scholar] [CrossRef]
- Ruhman, S. Solving Quantum-Dot Excitonic Riddles with Absolute Pump–Probe Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 9336–9343. [Google Scholar] [CrossRef]
- Knowles, K.E.; McArthur, E.A.; Weiss, E.A. A Multi-Timescale Map of Radiative and Nonradiative Decay Pathways for Excitons in CdSe Quantum Dots. ACS Nano 2011, 5, 2026–2035. [Google Scholar] [CrossRef]
- Zhang, C.; Do, T.N.; Ong, X.; Chan, Y.; Tan, H.-S. Understanding the Features in the Ultrafast Transient Absorption Spectra of CdSe Quantum Dots. Chem. Phys. 2016, 481, 157–164. [Google Scholar] [CrossRef]
- Klimov, V.I. Spectral and Dynamical Properties of Multiexcitons in Semiconductor Nanocrystals. Annu. Rev. Phys. Chem. 2007, 58, 635–673. [Google Scholar] [CrossRef]
- Klimov, V.I. Optical Nonlinearities and Ultrafast Carrier Dynamics in Semiconductor Nanocrystals. J. Phys. Chem. B 2000, 104, 6112–6123. [Google Scholar] [CrossRef]
- Dana, J.; Haggag, O.S.; Dehnel, J.; Mor, M.; Lifshitz, E.; Ruhman, S. Testing the Fate of Nascent Holes in CdSe Nanocrystals with Sub-10 Fs Pump–Probe Spectroscopy. Nanoscale 2021, 13, 1982–1987. [Google Scholar] [CrossRef]
- Morgan, D.P.; Kelley, D.F. What Does the Transient Absorption Spectrum of CdSe Quantum Dots Measure? J. Phys. Chem. C 2020, 124, 8448–8455. [Google Scholar] [CrossRef]
- Hunsche, S.; Dekorsy, T.; Klimov, V.; Kurz, H. Ultrafast Dynamics of Carrier-Induced Absorption Changes in Highly-Excited CdSe Nanocrystals. Appl. Phys. B Laser Opt. 1996, 62, 3–10. [Google Scholar] [CrossRef]
- Wu, K.; Zhu, H.; Liu, Z.; Rodríguez-Córdoba, W.; Lian, T. Ultrafast Charge Separation and Long-Lived Charge Separated State in Photocatalytic CdS-Pt Nanorod Heterostructures. J. Am. Chem. Soc. 2012, 134, 10337–10340. [Google Scholar] [CrossRef] [PubMed]
- Jasrasaria, D.; Philbin, J.P.; Yan, C.; Weinberg, D.; Alivisatos, A.P.; Rabani, E. Sub-Bandgap Photoinduced Transient Absorption Features in CdSe Nanostructures: The Role of Trapped Holes. J. Phys. Chem. C 2020, 124, 17372–17378. [Google Scholar] [CrossRef]
- Reiss, P.; Protière, M.; Li, L. Core/Shell Semiconductor Nanocrystals. Small 2009, 5, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Zavelani-Rossi, M.; Lupo, M.G.; Tassone, F.; Manna, L.; Lanzani, G. Suppression of Biexciton Auger Recombination in CdSe/CdS Dot/Rods: Role of the Electronic Structure in the Carrier Dynamics. Nano Lett. 2010, 10, 3142–3150. [Google Scholar] [CrossRef]
- Smith, E.R.; Luther, J.M.; Johnson, J.C. Ultrafast Electronic Delocalization in CdSe/CdS Quantum Rod Heterostructures. Nano Lett. 2011, 11, 4923–4931. [Google Scholar] [CrossRef]
- Steiner, D.; Dorfs, D.; Banin, U.; Della Sala, F.; Manna, L.; Millo, O. Determination of Band Offsets in Heterostructured Colloidal Nanorods Using Scanning Tunneling Spectroscopy. Nano Lett. 2008, 8, 2954–2958. [Google Scholar] [CrossRef]
- She, C.; Demortière, A.; Shevchenko, E.V.; Pelton, M. Using Shape to Control Photoluminescence from CdSe/CdS Core/Shell Nanorods. J. Phys. Chem. Lett. 2011, 2, 1469–1475. [Google Scholar] [CrossRef]
- Sift, A.; Sala, F.D.; Menagen, G.; Banin, U. Multiexciton Engineering in Seeded Core/Shell Nanorods: Transfer from Type-I to Quasi-Type-Ll Regimes. Nano Lett. 2009, 9, 3470–3476. [Google Scholar] [CrossRef]
- Rainò, G.; Stöferle, T.; Moreels, I.; Gomes, R.; Kamal, J.S.; Hens, Z.; Mahrt, R.F. Probing the Wave Function Delocalization in CdSe/CdS Dot-in-Rod Nanocrystals by Time- and Temperature-Resolved Spectroscopy. ACS Nano 2011, 5, 4031–4036. [Google Scholar] [CrossRef]
- Eshet, H.; Grünwald, M.; Rabani, E. The Electronic Structure of CdSe/CdS Core/Shell Seeded Nanorods: Type-I or Quasi-Type-II? Nano Lett. 2013, 13, 5880–5885. [Google Scholar] [CrossRef]
- Wu, K.; Hill, L.J.; Chen, J.; McBride, J.R.; Pavlopolous, N.G.; Richey, N.E.; Pyun, J.; Lian, T. Universal Length Dependence of Rod-to-Seed Exciton Localization Efficiency in Type I and Quasi-Type II CdSe@CdS Nanorods. ACS Nano 2015, 9, 4591–4599. [Google Scholar] [CrossRef]
- Wang, L.; Nonaka, K.; Okuhata, T.; Katayama, T.; Tamai, N. Quasi-Type II Carrier Distribution in CdSe/CdS Core/Shell Quantum Dots with Type I Band Alignment. J. Phys. Chem. C 2018, 122, 12038–12046. [Google Scholar] [CrossRef]
- Rosner, T.; Pavlopoulos, N.G.; Shoyhet, H.; Micheel, M.; Wächtler, M.; Adir, N.; Amirav, L. The Other Dimension—Tuning Hole Extraction via Nanorod Width. Nanomaterials 2022, 12, 3343. [Google Scholar] [CrossRef]
- Utterback, J.K.; Grennell, A.N.; Wilker, M.B.; Pearce, O.M.; Eaves, J.D.; Dukovic, G. Observation of Trapped-Hole Diffusion on the Surfaces of CdS Nanorods. Nat. Chem. 2016, 8, 1061–1066. [Google Scholar] [CrossRef]
- Robinson, R.D.; Sadtler, B.; Demchenko, D.O.; Erdonmez, C.K.; Wang, L.-W.; Alivisatos, A.P. Spontaneous Superlattice Formation in Nanorods Through Partial Cation Exchange. Science 2007, 317, 355–358. [Google Scholar] [CrossRef]
- Wu, K.; Rodríguez-Córdoba, W.; Lian, T. Exciton Localization and Dissociation Dynamics in CdS and CdS–Pt Quantum Confined Nanorods: Effect of Nonuniform Rod Diameters. J. Phys. Chem. B 2014, 118, 14062–14069. [Google Scholar] [CrossRef]
- Utterback, J.K.; Hamby, H.; Pearce, O.M.; Eaves, J.D.; Dukovic, G. Trapped-Hole Diffusion in Photoexcited CdSe Nanorods. J. Phys. Chem. C 2018, 122, 16974–16982. [Google Scholar] [CrossRef]
- Utterback, J.K.; Ruzicka, J.L.; Hamby, H.; Eaves, J.D.; Dukovic, G. Temperature-Dependent Transient Absorption Spectroscopy Elucidates Trapped-Hole Dynamics in CdS and CdSe Nanorods. J. Phys. Chem. Lett. 2019, 10, 2782–2787. [Google Scholar] [CrossRef]
- Kuno, M.; Fromm, D.P.; Hamann, H.F.; Gallagher, A.; Nesbitt, D.J. Nonexponential “Blinking” Kinetics of Single CdSe Quantum Dots: A Universal Power Law Behavior. J. Chem. Phys. 2000, 112, 3117–3120. [Google Scholar] [CrossRef]
- Grennell, A.N.; Utterback, J.K.; Pearce, O.M.; Wilker, M.B.; Dukovic, G. Relationships between Exciton Dissociation and Slow Recombination within ZnSe/CdS and CdSe/CdS Dot-in-Rod Heterostructures. Nano Lett. 2017, 17, 3764–3774. [Google Scholar] [CrossRef]
- Talapin, D.V.; Nelson, J.H.; Shevchenko, E.V.; Aloni, S.; Sadtler, B.; Alivisatos, A.P. Seeded Growth of Highly Luminescent CdSe/CdS Nanoheterostructures with Rod and Tetrapod Morphologies. Nano Lett. 2007, 7, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- Borys, N.J.; Walter, M.J.; Huang, J.; Talapin, D.V.; Lupton, J.M. The Role of Particle Morphology in Interfacial Energy Transfer in CdSe/CdS Heterostructure Nanocrystals. Science 2010, 330, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Rodríguez-Córdoba, W.E.; Liu, Z.; Zhu, H.; Lian, T. Beyond Band Alignment: Hole Localization Driven Formation of Three Spatially Separated Long-Lived Exciton States in CdSe/CdS Nanorods. ACS Nano 2013, 7, 7173–7185. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shahar, Y.; Scotognella, F.; Waiskopf, N.; Kriegel, I.; Conte, S.D.; Cerullo, G.; Banin, U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS-Au Nanorods. Small 2015, 11, 462–471. [Google Scholar] [CrossRef]
- O’Connor, T.; Panov, M.S.; Mereshchenko, A.; Tarnovsky, A.N.; Lorek, R.; Perera, D.; Diederich, G.; Lambright, S.; Moroz, P.; Zamkov, M. The Effect of the Charge-Separating Interface on Exciton Dynamics in Photocatalytic Colloidal Heteronanocrystals. ACS Nano 2012, 6, 8156–8165. [Google Scholar] [CrossRef]
- Ben-Shahar, Y.; Scotognella, F.; Kriegel, I.; Moretti, L.; Cerullo, G.; Rabani, E.; Banin, U. Optimal Metal Domain Size for Photocatalysis with Hybrid Semiconductor-Metal Nanorods. Nat. Commun. 2016, 7, 10413. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, W.; Chen, Q.; Cullen, D.A.; Xie, Z.; Lian, T. Pt Particle Size Affects Both the Charge Separation and Water Reduction Efficiencies of CdS–Pt Nanorod Photocatalysts for Light Driven H2 Generation. J. Am. Chem. Soc. 2022, 144, 2705–2715. [Google Scholar] [CrossRef]
- Kalisman, P.; Houben, L.; Aronovitch, E.; Kauffmann, Y.; Bar-Sadan, M.; Amirav, L. The Golden Gate to Photocatalytic Hydrogen Production. J. Mater. Chem. A 2015, 3, 19679–19682. [Google Scholar] [CrossRef]
- Huang, J.; Huang, Z.; Yang, Y.; Zhu, H.; Lian, T. Multiple Exciton Dissociation in CdSe Quantum Dots by Ultrafast Electron Transfer to Adsorbed Methylene Blue. J. Am. Chem. Soc. 2010, 132, 4858–4864. [Google Scholar] [CrossRef]
- Benndorf, S.; Schleusener, A.; Müller, R.; Micheel, M.; Baruah, R.; Dellith, J.; Undisz, A.; Neumann, C.; Turchanin, A.; Leopold, K.; et al. Covalent Functionalization of CdSe Quantum Dot Films with Molecular [FeFe] Hydrogenase Mimics for Light-Driven Hydrogen Evolution. ACS Appl Mater Interfaces 2023, 15, 18889–18897. [Google Scholar] [CrossRef]
- Matylitsky, V.V.; Dworak, L.; Breus, V.V.; Basché, T.; Wachtveitl, J. Ultrafast Charge Separation in Multiexcited CdSe Quantum Dots Mediated by Adsorbed Electron Acceptors. J. Am. Chem. Soc. 2009, 131, 2424–2425. [Google Scholar] [CrossRef]
- Jiang, Z.-J.; Kelley, D.F. Hot and Relaxed Electron Transfer from the CdSe Core and Core/Shell Nanorods. J. Phys. Chem. C 2011, 115, 4594–4602. [Google Scholar] [CrossRef]
- Schleusener, A.; Micheel, M.; Benndorf, S.; Rettenmayr, M.; Weigand, W.; Wächtler, M. Ultrafast Electron Transfer from CdSe Quantum Dots to an [FeFe]-Hydrogenase Mimic. J. Phys. Chem. Lett. 2021, 12, 4385–4391. [Google Scholar] [CrossRef]
- Thomas, A.; Sandeep, K.; Somasundaran, S.M.; Thomas, K.G. How Trap States Affect Charge Carrier Dynamics of CdSe and InP Quantum Dots: Visualization through Complexation with Viologen. ACS Energy Lett. 2018, 3, 2368–2375. [Google Scholar] [CrossRef]
- Tseng, H.-W.; Wilker, M.B.; Damrauer, N.H.; Dukovic, G. Charge Transfer Dynamics between Photoexcited CdS Nanorods and Mononuclear Ru Water-Oxidation Catalysts. J. Am. Chem. Soc. 2013, 135, 3383–3386. [Google Scholar] [CrossRef]
- Huang, J.; Huang, Z.; Jin, S.; Lian, T. Exciton Dissociation in CdSe Quantum Dots by Hole Transfer to Phenothiazine. J. Phys. Chem. C 2008, 112, 19734–19738. [Google Scholar] [CrossRef]
- Lian, S.; Weinberg, D.J.; Harris, R.D.; Kodaimati, M.S.; Weiss, E.A. Subpicosecond Photoinduced Hole Transfer from a CdS Quantum Dot to a Molecular Acceptor Bound Through an Exciton-Delocalizing Ligand. ACS Nano 2016, 10, 6372–6382. [Google Scholar] [CrossRef]
- Pearce, O.M.; Duncan, J.S.; Damrauer, N.H.; Dukovic, G. Ultrafast Hole Transfer from CdS Quantum Dots to a Water Oxidation Catalyst. J. Phys. Chem. C 2018, 122, 17559–17565. [Google Scholar] [CrossRef]
- Berr, M.J.; Vaneski, A.; Mauser, C.; Fischbach, S.; Susha, A.S.; Rogach, A.L.; Jäckel, F.; Feldmann, J. Delayed Photoelectron Transfer in Pt-Decorated CdS Nanorods under Hydrogen Generation Conditions. Small 2012, 8, 291–297. [Google Scholar] [CrossRef]
- Beckwith, J.S.; Lang, B.; Grilj, J.; Vauthey, E. Ion-Pair Dynamics upon Photoinduced Electron Transfer Monitored by Pump-Pump-Probe Spectroscopy. J. Phys. Chem. Lett. 2019, 10, 3688–3693. [Google Scholar] [CrossRef]
- Pagès, S.; Lang, B.; Vauthey, E. Ultrafast Excited State Dynamics of the Perylene Radical Cation Generated upon Bimolecular Photoinduced Electron Transfer Reaction. J. Phys. Chem. A 2006, 110, 7547–7553. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Petermann, M.; Wenger, O.S. Pump-Pump-Probe Spectroscopy of a Molecular Triad Monitoring Detrimental Processes for Photoinduced Charge Accumulation. Helv. Chim. Acta 2017, 100, e1600283. [Google Scholar] [CrossRef]
- Tran, T.T.; Pino, T.; Ha-Thi, M.H. Watching Intermolecular Light-Induced Charge Accumulation on Naphthalene Diimide by Tris(Bipyridyl)Ruthenium(II) Photosensitizer. J. Phys. Chem. C 2019, 123, 28651–28658. [Google Scholar] [CrossRef]
- Ha-Thi, M.H.; Pham, V.T.; Pino, T.; Maslova, V.; Quaranta, A.; Lefumeux, C.; Leibl, W.; Aukauloo, A. Photoinduced Electron Transfer in a Molecular Dyad by Nanosecond Pump-Pump-Probe Spectroscopy. Photochem. Photobiol. Sci. 2018, 17, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Kerzig, C.; Wenger, O.S. Quantitative Insights into Charge-Separated States from One- and Two-Pulse Laser Experiments Relevant for Artificial Photosynthesis. Chem. Sci. 2019, 10, 5624–5633. [Google Scholar] [CrossRef]
- Wang, J.; Ding, T.; Leng, J.; Jin, S.; Wu, K. “Intact” Carrier Doping by Pump-Pump-Probe Spectroscopy in Combination with Interfacial Charge Transfer: A Case Study of CsPbBr3 Nanocrystals. J. Phys. Chem. Lett. 2018, 9, 3372–3377. [Google Scholar] [CrossRef]
- Wang, J.; Ding, T.; Wu, K. Coulomb Barrier for Sequential Two-Electron Transfer in a Nanoengineered Photocatalyst. J. Am. Chem. Soc. 2020, 142, 13934–13940. [Google Scholar] [CrossRef]
- Scheidt, R.A.; Samu, G.F.; Janáky, C.; Kamat, P.V. Modulation of Charge Recombination in CsPbBr3 Perovskite Films with Electrochemical Bias. J. Am. Chem. Soc. 2018, 140, 86–89. [Google Scholar] [CrossRef]
- Qin, W.; Guyot-Sionnest, P. Evidence for the Role of Holes in Blinking: Negative and Oxidized CdSe/CdS Dots. ACS Nano 2012, 6, 9125–9132. [Google Scholar] [CrossRef]
- Kambhampati, P. Multiexcitons in Semiconductor Nanocrystals: A Platform for Optoelectronics at High Carrier Concentration. J. Phys. Chem. Lett. 2012, 3, 1182–1190. [Google Scholar] [CrossRef]
- Klimov, V.I.; McGuire, J.A.; Schaller, R.D.; Rupasov, V.I. Scaling of Multiexciton Lifetimes in Semiconductor Nanocrystals. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 195324. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, Y.; Lian, T. Multiexciton Annihilation and Dissociation in Quantum Confined Semiconductor Nanocrystals. Acc. Chem. Res. 2013, 46, 1270–1279. [Google Scholar] [CrossRef]
- Schaller, R.D.; Sykora, M.; Jeong, S.; Klimov, V.I. High-Efficiency Carrier Multiplication and Ultrafast Charge Separation in Semiconductor Nanocrystals Studied via Time-Resolved Photoluminescence. J. Phys. Chem. B 2006, 110, 25332–25338. [Google Scholar] [CrossRef]
- Schaller, R.D.; Klimov, V.I. High Efficiency Carrier Multiplication in PbSe Nanocrystals: Implications for Solar Energy Conversion. Phys. Rev. Lett. 2004, 92, 186601. [Google Scholar] [CrossRef]
- Ellingson, R.J.; Beard, M.C.; Johnson, J.C.; Yu, P.; Micic, O.I.; Nozik, A.J.; Shabaev, A.; Efros, A.L. Highly Efficient Multiple Exciton Generation in Colloidal PbSe and PbS Quantum Dots. Nano Lett. 2005, 5, 865–871. [Google Scholar] [CrossRef]
- Ben-Shahar, Y.; Philbin, J.P.; Scotognella, F.; Ganzer, L.; Cerullo, G.; Rabani, E.; Banin, U. Charge Carrier Dynamics in Photocatalytic Hybrid Semiconductor-Metal Nanorods: Crossover from Auger Recombination to Charge Transfer. Nano Lett. 2018, 18, 5211–5216. [Google Scholar] [CrossRef]
- Lupo, M.G.; Sala, F.D.; Carbone, L.; Zavelani-Rossi, M.; Fiore, A.; Lüer, L.; Polli, D.; Cingolani, R.; Manna, L.; Lanzani, G. Ultrafast Electron-Hole Dynamics in Core/Shell CdSe/CdS Dot/Rod Nanocrystals. Nano Lett. 2008, 8, 4582–4587. [Google Scholar] [CrossRef]
- Zhu, H.; Song, N.; Rodríguez-Córdoba, W.; Lian, T. Wave Function Engineering for Efficient Extraction of up to Nineteen Electrons from One CdSe/CdS Quasi-Type II Quantum Dot. J. Am. Chem. Soc. 2012, 134, 4250–4257. [Google Scholar] [CrossRef]
- Yan, C.; Weinberg, D.; Jasrasaria, D.; Kolaczkowski, M.A.; Liu, Z.; Philbin, J.P.; Balan, A.D.; Liu, Y.; Schwartzberg, A.M.; Rabani, E.; et al. Uncovering the Role of Hole Traps in Promoting Hole Transfer from Multiexcitonic Quantum Dots to Molecular Acceptors. ACS Nano 2021, 15, 2281–2291. [Google Scholar] [CrossRef]
- Zhu, H.; Lian, T. Enhanced Multiple Exciton Dissociation from CdSe Quantum Rods: The Effect of Nanocrystal Shape. J. Am. Chem. Soc. 2012, 134, 11289–11297. [Google Scholar] [CrossRef]
- Young, R.M.; Jensen, S.C.; Edme, K.; Wu, Y.; Krzyaniak, M.D.; Vermeulen, N.A.; Dale, E.J.; Stoddart, J.F.; Weiss, E.A.; Wasielewski, M.R.; et al. Ultrafast Two-Electron Transfer in a CdS Quantum Dot–Extended-Viologen Cyclophane Complex. J. Am. Chem. Soc. 2016, 138, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cullen, D.A.; Lian, T. Slow Auger Recombination of Trapped Excitons Enables Efficient Multiple Electron Transfer in CdS–Pt Nanorod Heterostructures. J. Am. Chem. Soc. 2021, 143, 20264–20273. [Google Scholar] [CrossRef] [PubMed]
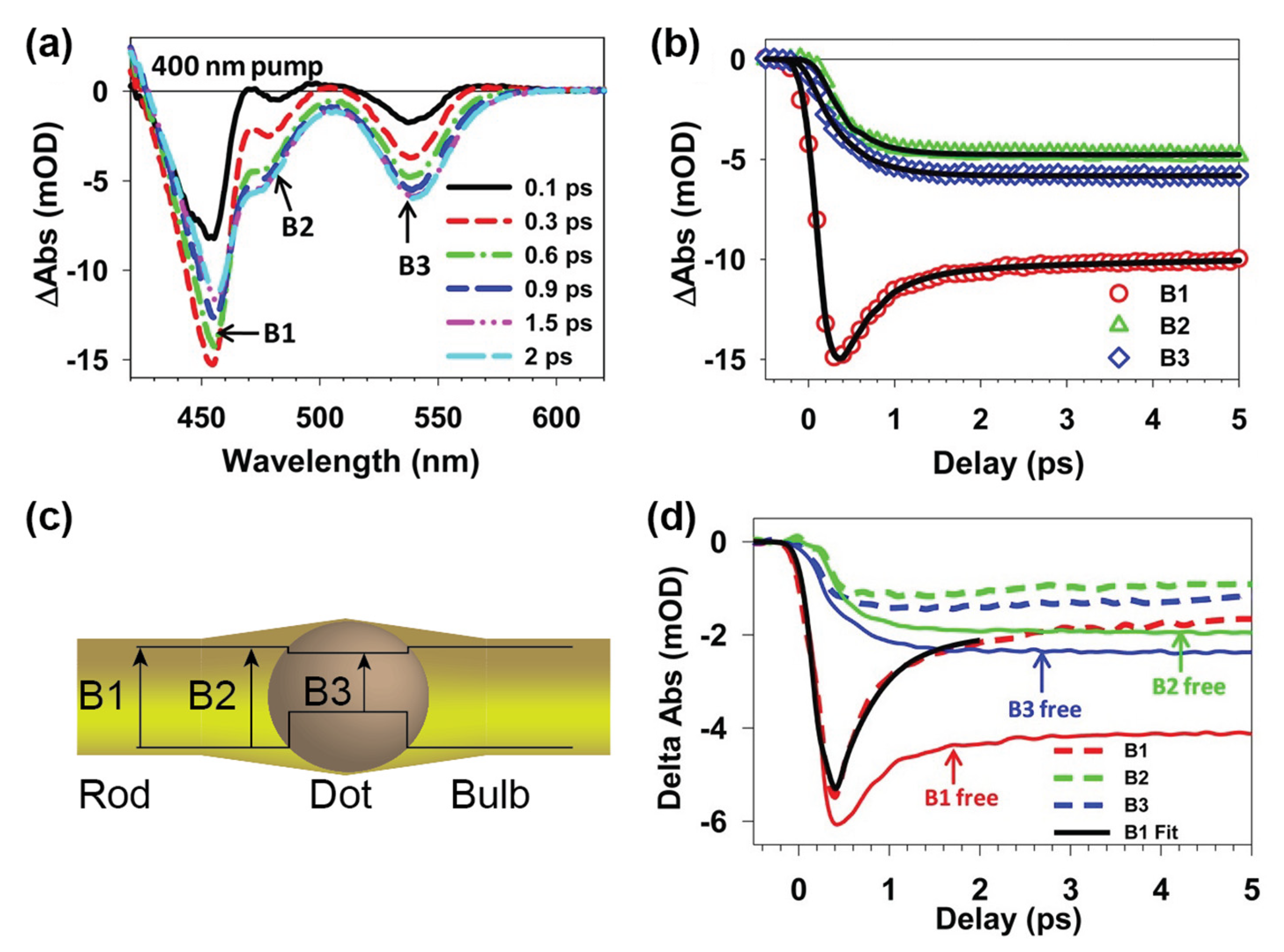
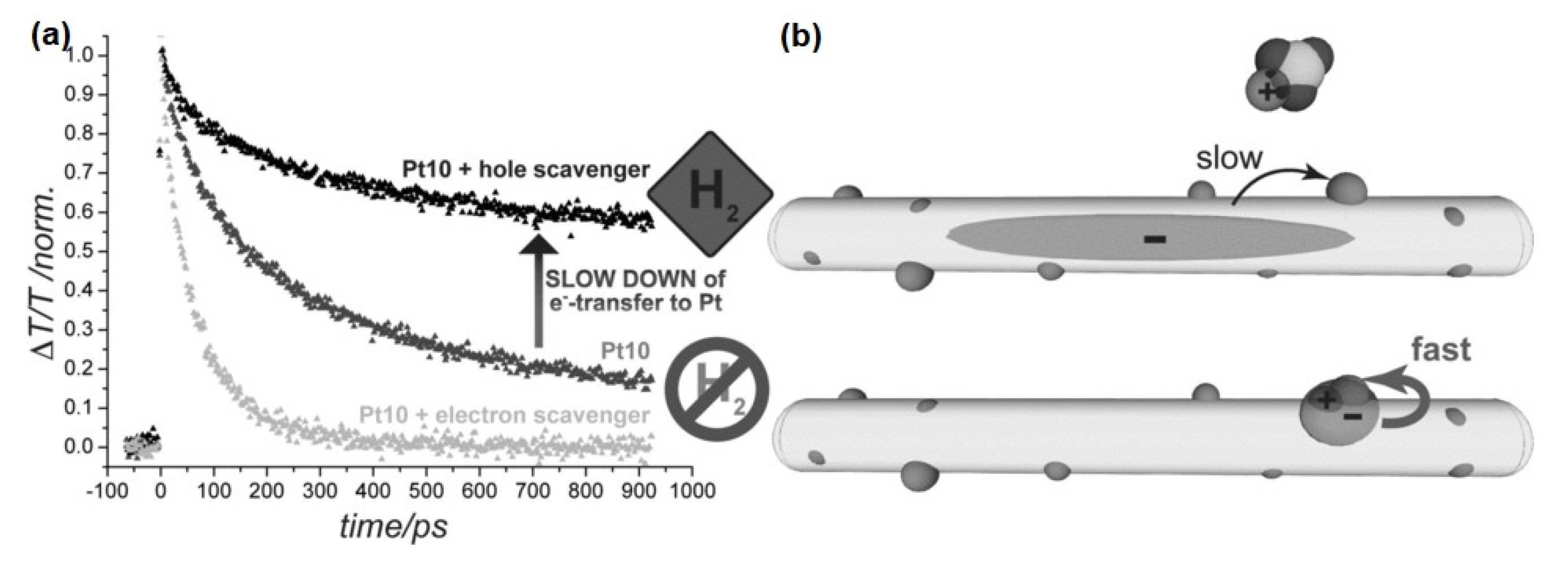


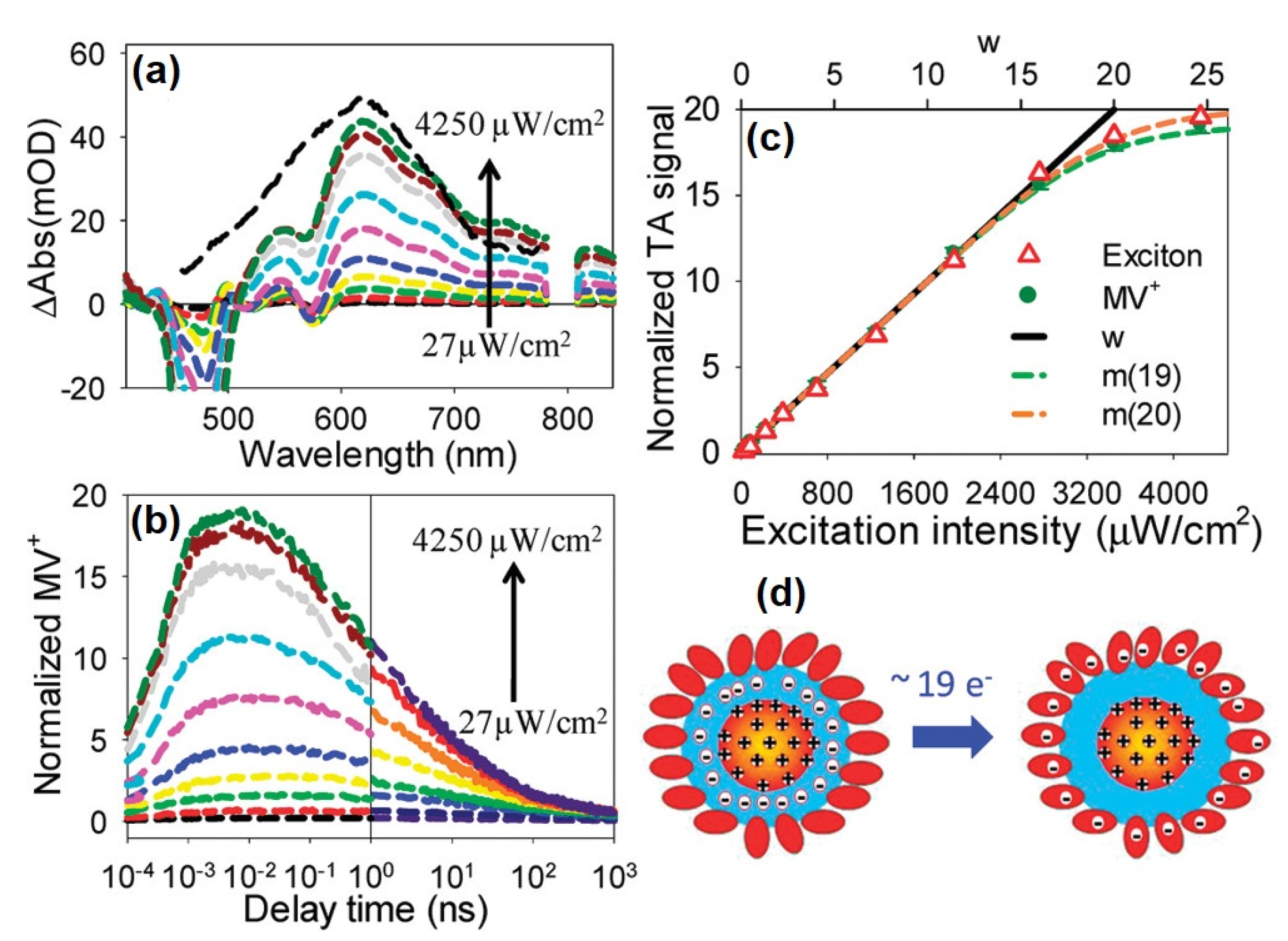
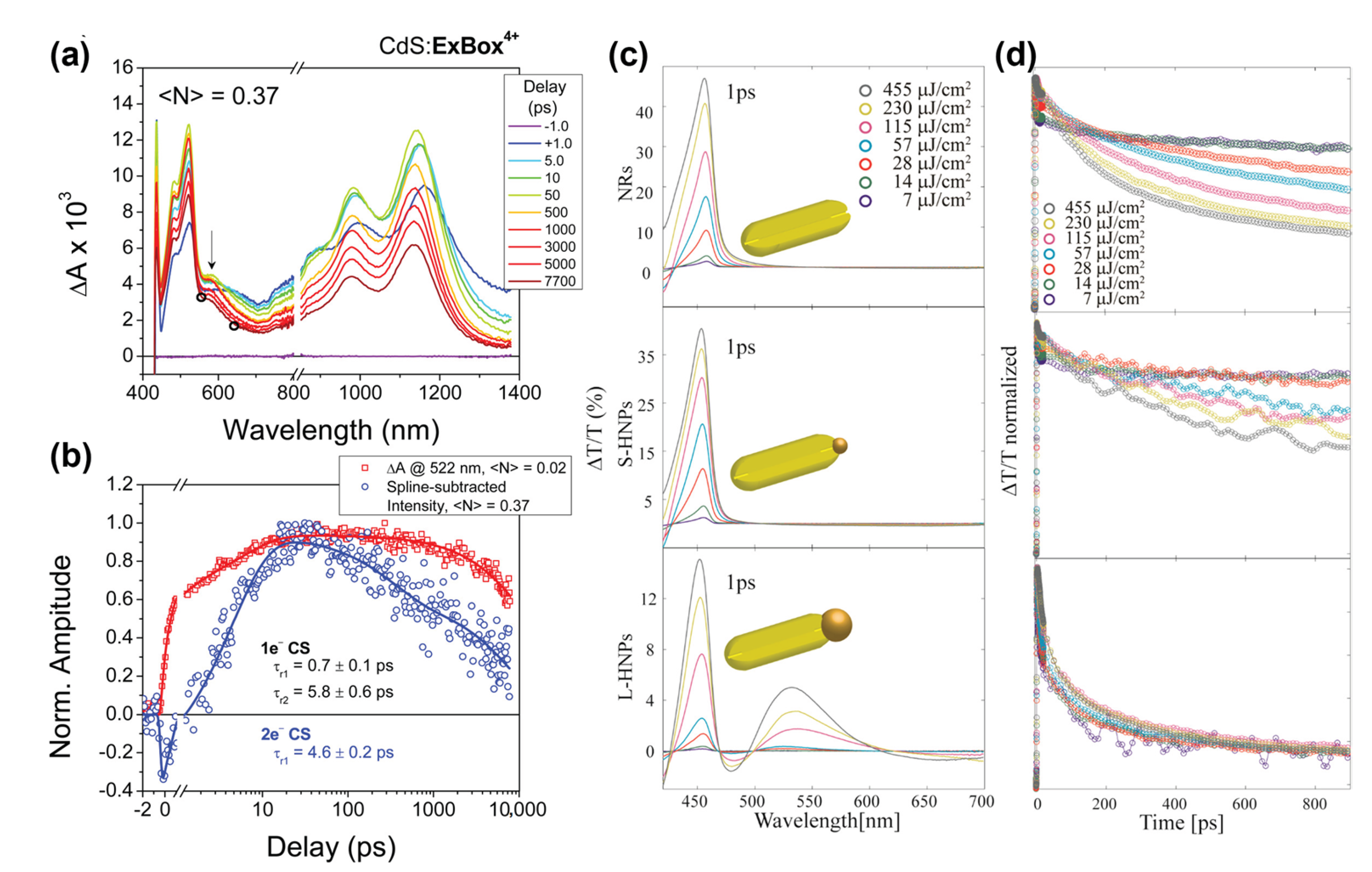
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, K.; Wächtler, M. Unravelling Dynamics Involving Multiple Charge Carriers in Semiconductor Nanocrystals. Nanomaterials 2023, 13, 1579. https://doi.org/10.3390/nano13091579
Kumar K, Wächtler M. Unravelling Dynamics Involving Multiple Charge Carriers in Semiconductor Nanocrystals. Nanomaterials. 2023; 13(9):1579. https://doi.org/10.3390/nano13091579
Chicago/Turabian StyleKumar, Krishan, and Maria Wächtler. 2023. "Unravelling Dynamics Involving Multiple Charge Carriers in Semiconductor Nanocrystals" Nanomaterials 13, no. 9: 1579. https://doi.org/10.3390/nano13091579
APA StyleKumar, K., & Wächtler, M. (2023). Unravelling Dynamics Involving Multiple Charge Carriers in Semiconductor Nanocrystals. Nanomaterials, 13(9), 1579. https://doi.org/10.3390/nano13091579