
Chemical Orderings in CuCo Nanoparticles: Topological Modeling Using DFT Calculations



Abstract
1. Introduction
2. Methods and Models
3. Results and Discussion
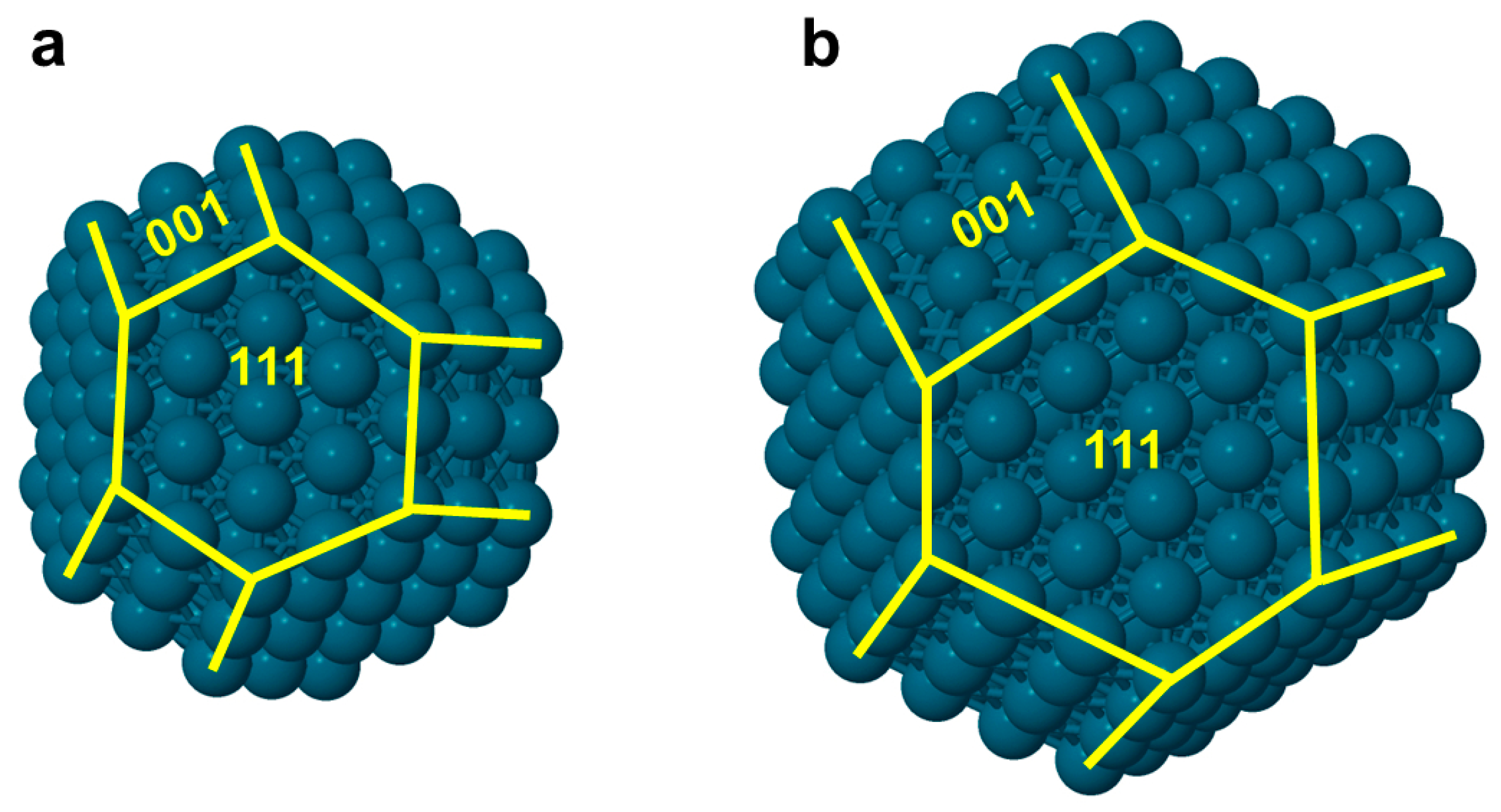
3.1. Topological Equations for CuCo Nanoparticles
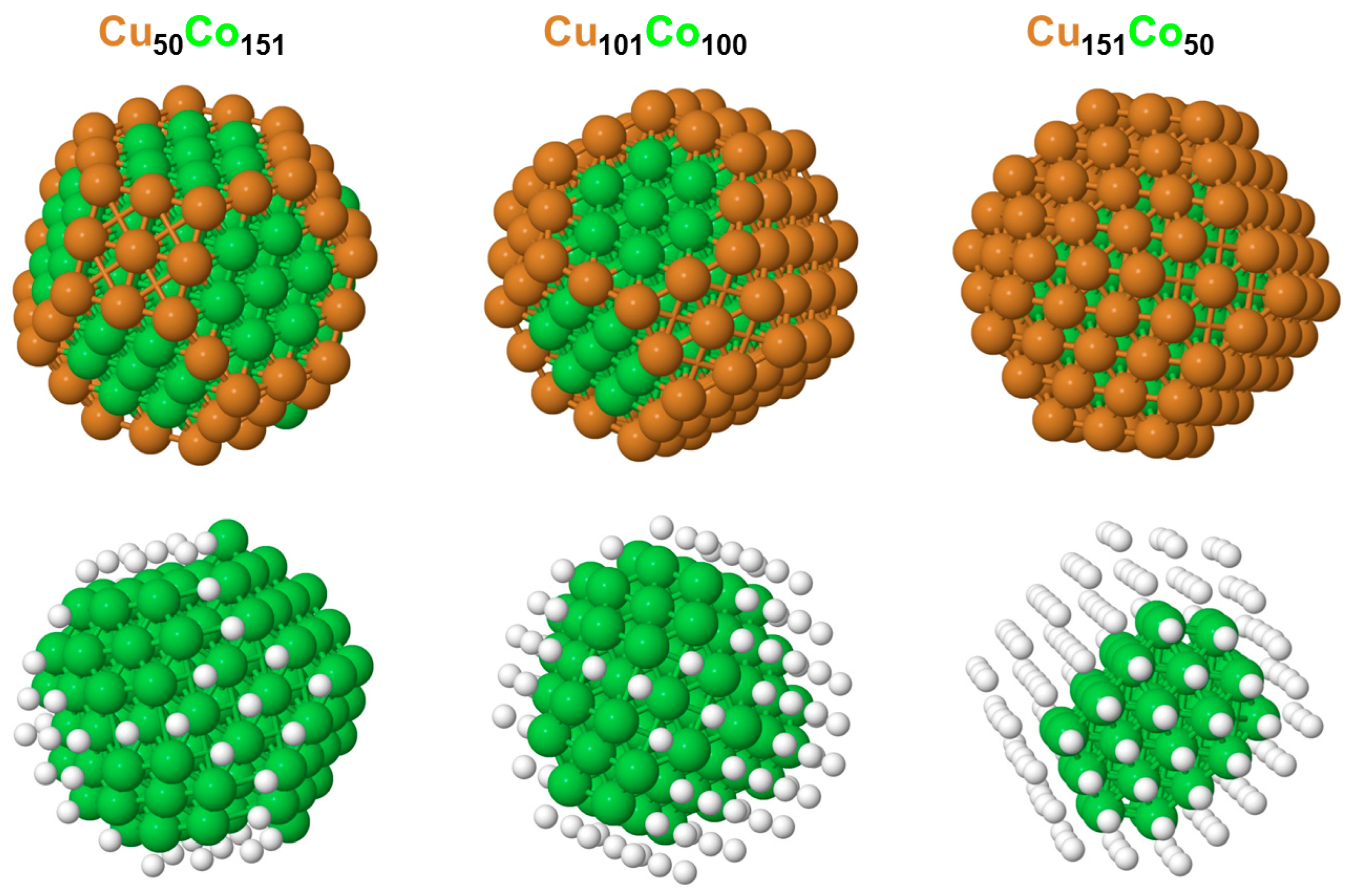
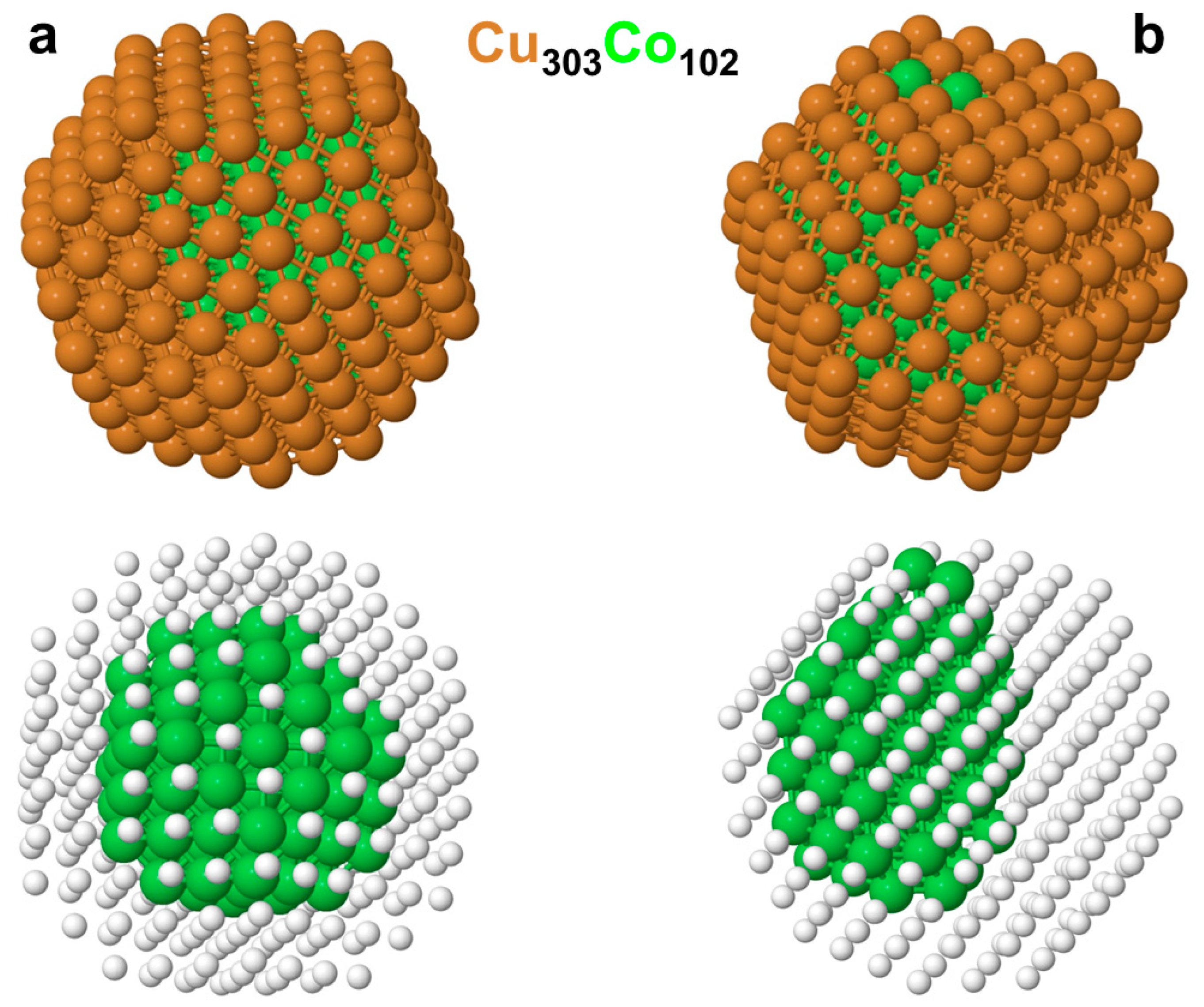
3.2. Chemical Orderings in the Cu201-kCok and Cu303Co102 Nanoparticles
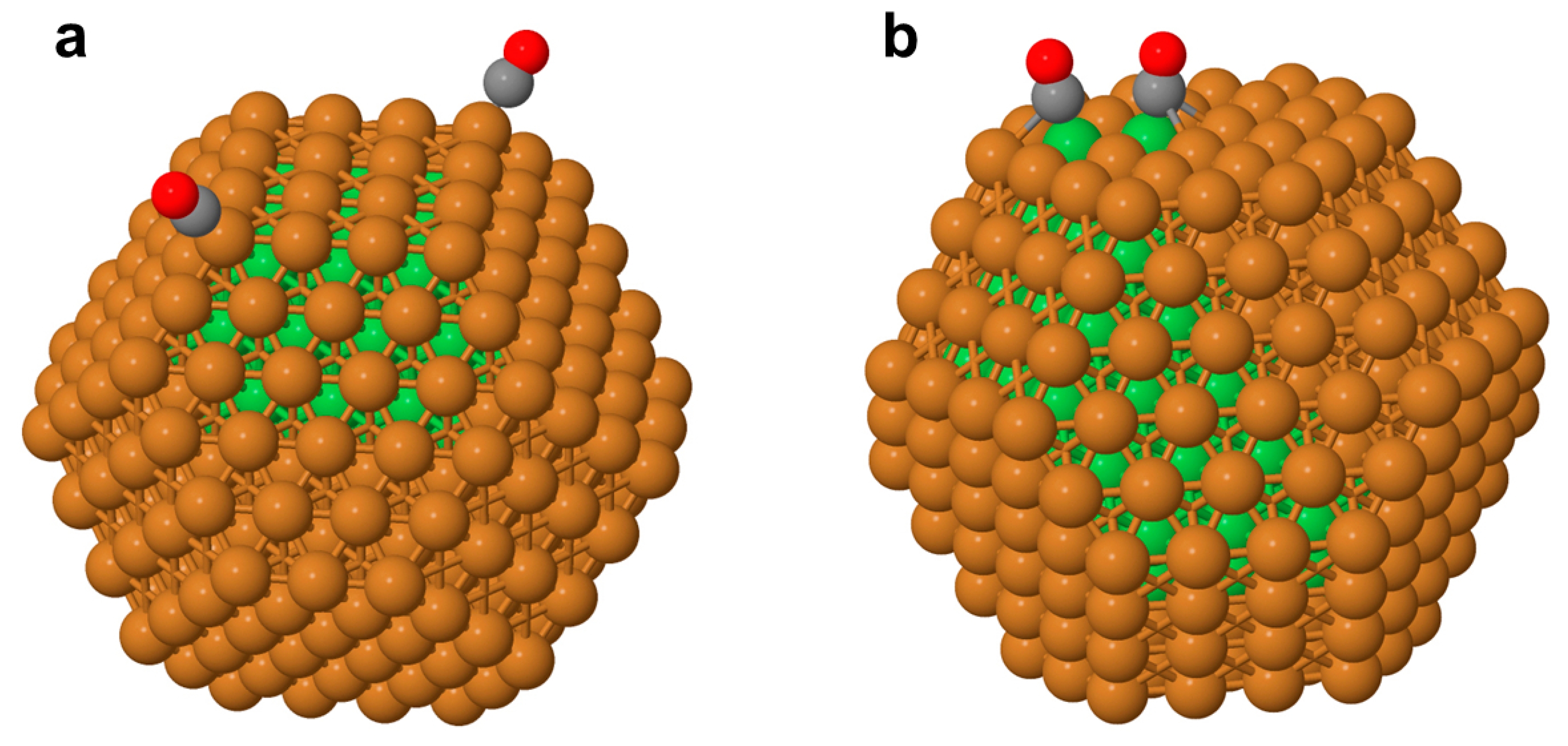
3.3. Surface Segregation of Co in the Cu303Co102 Nanoparticle Induced by CO Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From theory to applications of alloy clusters and nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef]
- Zhao, J.-Z.; Ahmed, T.; Jiang, H.-X.; He, J.; Sun, Q. Solidification of Immiscible Alloys: A Review. Acta Metall. Sin. 2017, 30, 1–28. [Google Scholar] [CrossRef]
- Ahn, J.P.; Jung, Y.K.; Park, J.K.; Huh, M.Y. Direct observation of cobalt precipitates in nanocrystalline Cu–Co powder synthesised by laser ablation. Powder Metall. 2005, 48, 338–342. [Google Scholar] [CrossRef]
- Zhao, T.; Liu, Y.; Wan, H.; Li, Z.; Wu, Y.; Yan, H. Electromagnetic wave absorption properties of carbon-encapsulated Co–Cu alloy nanoparticles prepared by gas explosion. Mater. Chem. Phys. 2023, 310, 128486. [Google Scholar] [CrossRef]
- Wang, J.; Chernavskii, P.A.; Khodakov, A.Y.; Wang, Y. Structure and catalytic performance of alumina-supported copper-cobalt catalysts for carbon monoxide hydrogenation. J. Catal. 2012, 286, 51–61. [Google Scholar] [CrossRef]
- Mo, X.; Tsai, Y.-T.; Gao, J.; Mao, D.; Goodwin, J.G. Effect of component interaction on the activity of Co/CuZnO for CO hydrogenation. J. Catal. 2012, 285, 208–215. [Google Scholar] [CrossRef]
- Xiang, Y.; Chitry, V.; Liddicoat, P.; Felfer, P.; Cairney, J.; Ringer, S.; Kruse, N. Long-Chain Terminal Alcohols through Catalytic CO Hydrogenation. J. Am. Chem. Soc. 2013, 135, 7114–7117. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.; Beijer, S.; Smith, M.L.; He, M.; Au, Y.; Wang, Z.; Bruce, D.A.; de Jong, K.P.; Spivey, J.J.; de Jongh, P.E. Design and synthesis of copper-cobalt catalysts for the selective conversion of synthesis gas to ethanol and higher alcohols. Angew. Chem. Int. Ed. 2014, 53, 6397–6401. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Barbosa, R.; Kruse, N. Higher Alcohols through CO Hydrogenation over CoCu Catalysts: Influence of Precursor Activation. ACS Catal. 2014, 4, 2792–2800. [Google Scholar] [CrossRef]
- Su, J.; Zhang, Z.; Fu, D.; Liu, D.; Xu, X.-C.; Shi, B.; Wang, X.; Si, R.; Jiang, Z.; Xu, J.; et al. Higher alcohols synthesis from syngas over CoCu/SiO2 catalysts: Dynamic structure and the role of Cu. J. Catal. 2016, 336, 94–106. [Google Scholar] [CrossRef]
- Mendes, L.V.P.; Snider, J.L.; Fleischman, S.D.; Kibsgaard, J.; McEnaney, J.M.; Aranda, D.A.G.; Jaramillo, T.F. Polyol synthesis of cobalt-copper alloy catalysts for higher alcohol synthesis from syngas. Catal. Lett. 2017, 147, 2352–2359. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Li, X.; Zhong, L.; Wang, H.; Liu, H.; Wei, W.; Sun, Y. Direct conversion of CO2 to long-chain hydrocarbon fuels over K-promoted CoCu/TiO2 catalysts. Catal. Today 2018, 311, 65–73. [Google Scholar] [CrossRef]
- Ge, X.; Sun, H.; Dong, K.; Tao, Y.Q.; Wang, Q.; Chen, Y.Z.; Zhang, G.L.; Cui, P.; Wang, Y.; Zhang, Q.H. Copper-cobalt catalysts supported on mechanically mixed HZSM-5 and -Al2O3 for higher alcohols synthesis via carbon monoxide hydrogenation. RSC Adv. 2019, 9, 14592–14598. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Z.-J.; Yang, C.; Zha, S.; Neyman, K.M.; Studt, F.; Gong, J. Adsorption preference determines segregation direction: A shortcut to more realistic surface models of alloy catalysts. ACS Catal. 2019, 9, 5011–5018. [Google Scholar] [CrossRef]
- Eren, B.; Torres, D.; Karslıoğlu, O.; Liu, Z.; Wu, C.H.; Stacchiola, D.; Bluhm, H.; Somorjai, G.A.; Salmeron, M. Structure of copper-cobalt surface alloys in equilibrium with carbon monoxide gas. J. Am. Chem. Soc. 2018, 140, 6575–6581. [Google Scholar] [CrossRef] [PubMed]
- Sykes, E.C.H.; Christopher, P. Recent advances in single-atom catalysts and single-atom alloys: Opportunities for exploring the uncharted phase space in-between. Curr. Opin. Chem. Eng. 2020, 29, 67–73. [Google Scholar] [CrossRef]
- Hannagan, R.T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-atom alloy catalysis. Chem. Rev. 2020, 120, 12044–12088. [Google Scholar] [CrossRef] [PubMed]
- Mashkovsky, I.S.; Markov, P.V.; Rassolov, A.V.; Patil, E.D.; Stakheev, A.Y. Progress in single-atom methodology in modern catalysis. Russ. Chem. Revs. 2023, 92, RCR5087. [Google Scholar] [CrossRef]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.-K.; Liu, L.-M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef]
- Jirkovsky, J.S.; Panas, I.; Ahlberg, E.; Halasa, M.; Romani, S.; Schiffrin, D.J. Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H2O2 production. J. Am. Chem. Soc. 2011, 133, 19432–19441. [Google Scholar] [CrossRef]
- Sun, G.; Zhao, Z.J.; Mu, R.; Zha, S.; Li, L.; Chen, S.; Gong, J. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat. Commun. 2018, 9, 4454. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.M.; Kovács, G.; Ferrando, R.; Neyman, K.M. How to determine accurate chemical ordering in several nanometer large bimetallic crystallites from electronic structure calculations. Chem. Sci. 2015, 6, 3868–3880. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.; Kozlov, S.M.; Neyman, K.M. Versatile optimization of chemical ordering in bimetallic nanoparticles. J. Phys. Chem. C 2017, 121, 10803–10808. [Google Scholar] [CrossRef]
- Danielis, N.; Vega, L.; Fronzoni, G.; Stener, M.; Bruix, A.; Neyman, K.M. AgPd, AuPd, and AuPt nanoalloys with Ag- or Au-rich compositions: Modeling chemical ordering and optical properties. J. Phys. Chem. C 2021, 125, 17372–17384. [Google Scholar] [CrossRef] [PubMed]
- Mamatkulov, M.; Yudanov, I.V.; Bukhtiyarov, A.V.; Prosvirin, I.P.; Bukhtiyarov, V.I.; Neyman, K.M. Pd segregation on the surface of bimetallic PdAu nanoparticles induced by low coverage of adsorbed CO. J. Phys. Chem. C 2019, 123, 8037–8046. [Google Scholar] [CrossRef]
- Khalakhan, I.; Vega, L.; Vorokhta, M.; Skála, T.; Viñes, F.; Yakovlev, Y.V.; Neyman, K.M.; Matolínová, I. Irreversible structural dynamics on the surface of bimetallic PtNi alloy catalyst under alternating oxidizing and reducing environments. Appl. Catal. B Environ. 2020, 264, 118476. [Google Scholar] [CrossRef]
- Vega, L.; Aleksandrov, H.A.; Farris, R.; Bruix, A.; Viñes, F.; Neyman, K.M. Chemical ordering in Pt-Au, Pt-Ag and Pt-Cu nanoparticles from density functional calculations using a topological approach. Mater. Adv. 2021, 2, 6589–6602. [Google Scholar] [CrossRef]
- Xie, X.; Briega-Martos, V.; Farris, R.; Dopita, M.; Vorokhta, M.; Skála, T.; Matolínová, I.; Neyman, K.M.; Cherevko, S.; Khalakhan, I. Optimal Pt-Au alloying for efficient and stable oxygen reduction reaction catalyst. ACS Appl. Mater. Interfaces 2023, 15, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Farris, R.; Merinov, B.V.; Bruix, A.; Neyman, K.M. Effects of Zr dopants on properties of PtNi nanoparticles for ORR catalysis: A DFT modeling. J. Chem. Phys. 2024, 160, 124706. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous–semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the Projector Augmented-Wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Viñes, F.; Illas, F.; Neyman, K.M. On the mechanism of formation of metal nanowires by self-assembly. Angew. Chem. Int. Ed. 2007, 46, 7094–7097. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, C.; Zha, S.; Sharapa, D.; Studt, F.; Zhao, Z.-J.; Gong, J. Moderate Surface Segregation Promotes Selective Ethanol Production in CO2 Hydrogenation Reaction over CoCu Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202109027. [Google Scholar] [CrossRef]
- Collinge, G.; Kruse, N.; McEwen, J.-S. Role of carbon monoxide in catalyst reconstruction for CO hydrogenation: First-principles study of the composition, structure, and stability of Cu/Co(10-12) as a function of CO pressure. J. Phys. Chem. C 2017, 121, 2181–2191. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L.; Nørskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990. [Google Scholar] [CrossRef]
- Zha, S.; Sharapa, D.I.; Liu, S.; Zhao, Z.-J.; Studt, F. Modeling CoCu Nanoparticles Using Neural Network-Accelerated Monte Carlo Simulations. J. Phys. Chem. A 2022, 126, 9440–9446. [Google Scholar] [CrossRef]
- Ferrando, R.; Fortunelli, A.; Rossi, G. Quantum effects on the structure of pure and binary metallic nanoclusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 085449. [Google Scholar] [CrossRef]
- Billas, I.M.L.; Châtelain, A.; de Heer, W.A. Magnetism from the atom to the bulk in iron, cobalt, and nickel clusters. Science 1994, 265, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Aleksandrov, H.A.; Neyman, K.M.; Hadjiivanov, K.I.; Vayssilov, G.N. Can the state of platinum species be unambiguously determined by the stretching frequency of adsorbed CO probe molecule? Phys. Chem. Chem. Phys. 2016, 18, 22108–22121. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | |||||
|---|---|---|---|---|---|
| Cu50Co151 | 29 | −953 | −948 | −829 | −762 |
| Cu101Co100 | 58 | −283 | −615 | −267 | −102 |
| Cu151Co50 | 60 | −542 | −695 | −480 | −316 |
| Cu303Co102 | 55 | −734 | −385 | −503 | −255 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neyman, K.M.; Alemany, P. Chemical Orderings in CuCo Nanoparticles: Topological Modeling Using DFT Calculations. Nanomaterials 2024, 14, 1242. https://doi.org/10.3390/nano14151242
Neyman KM, Alemany P. Chemical Orderings in CuCo Nanoparticles: Topological Modeling Using DFT Calculations. Nanomaterials. 2024; 14(15):1242. https://doi.org/10.3390/nano14151242
Chicago/Turabian StyleNeyman, Konstantin M., and Pere Alemany. 2024. "Chemical Orderings in CuCo Nanoparticles: Topological Modeling Using DFT Calculations" Nanomaterials 14, no. 15: 1242. https://doi.org/10.3390/nano14151242
APA StyleNeyman, K. M., & Alemany, P. (2024). Chemical Orderings in CuCo Nanoparticles: Topological Modeling Using DFT Calculations. Nanomaterials, 14(15), 1242. https://doi.org/10.3390/nano14151242