

Enhancement of Triple-Negative Breast Cancer-Specific Induction of Cell Death by Silver Nanoparticles by Combined Treatment with Proteotoxic Stress Response Inhibitors


Abstract
:1. Introduction
2. Materials and Methods
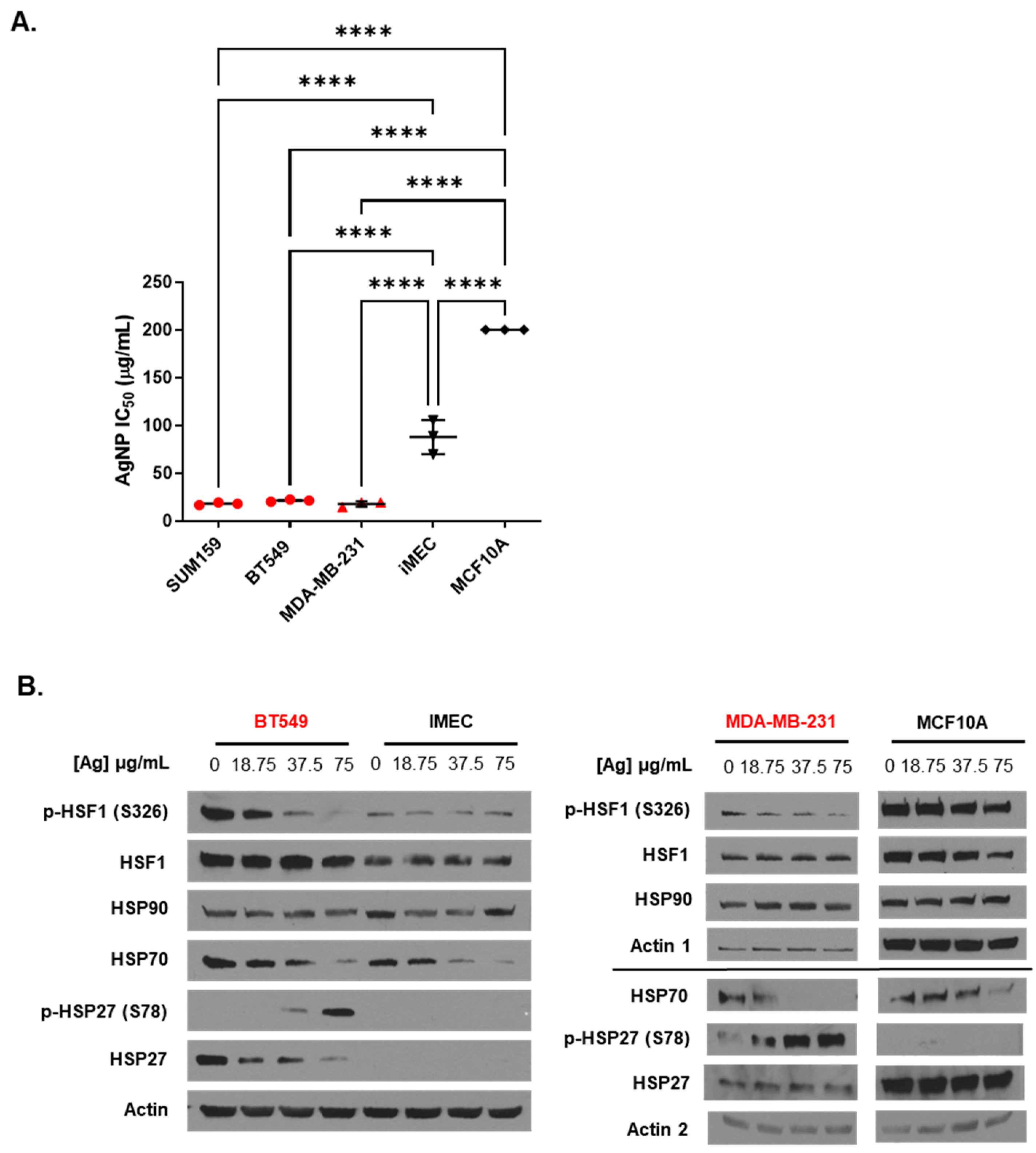
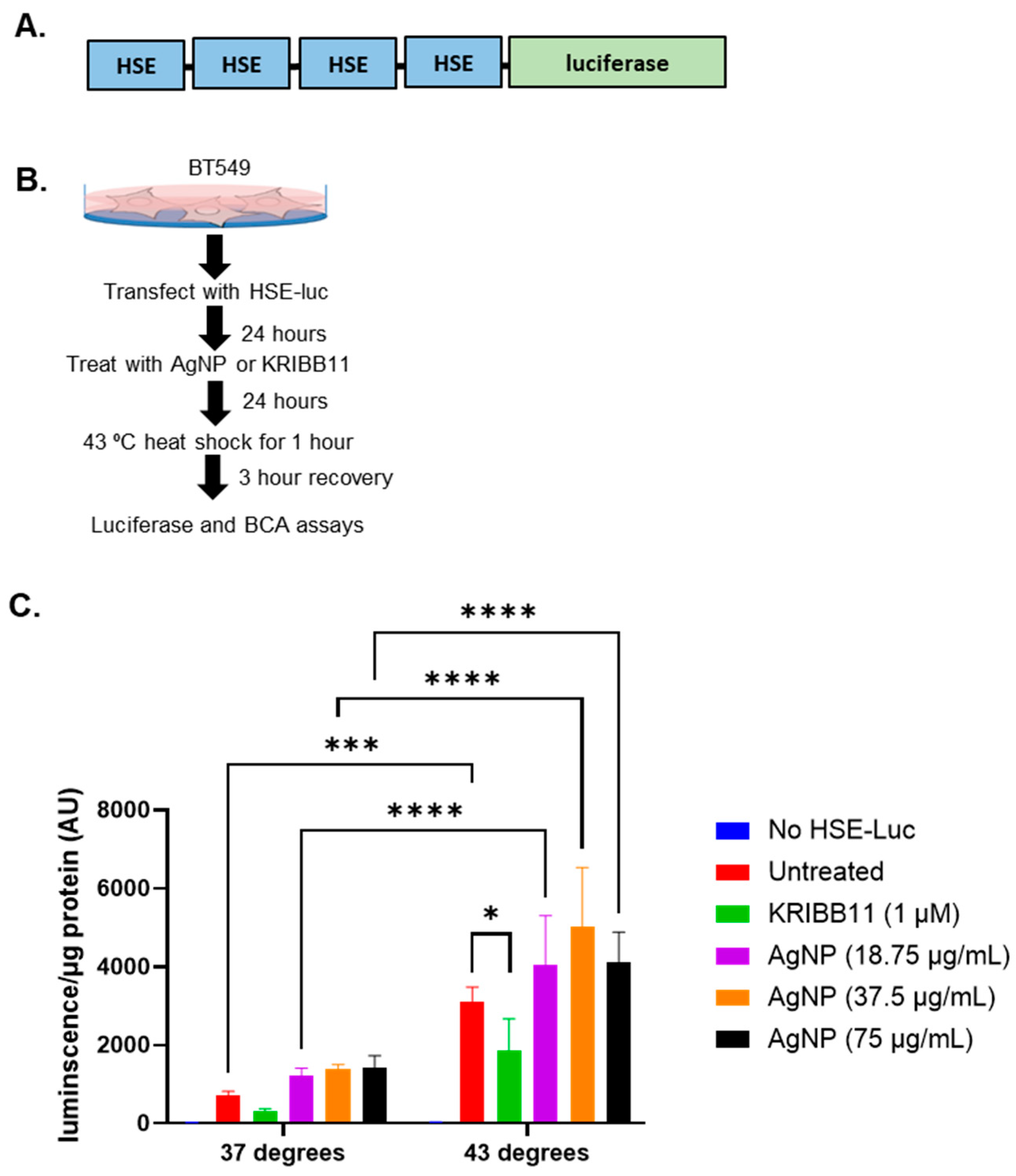
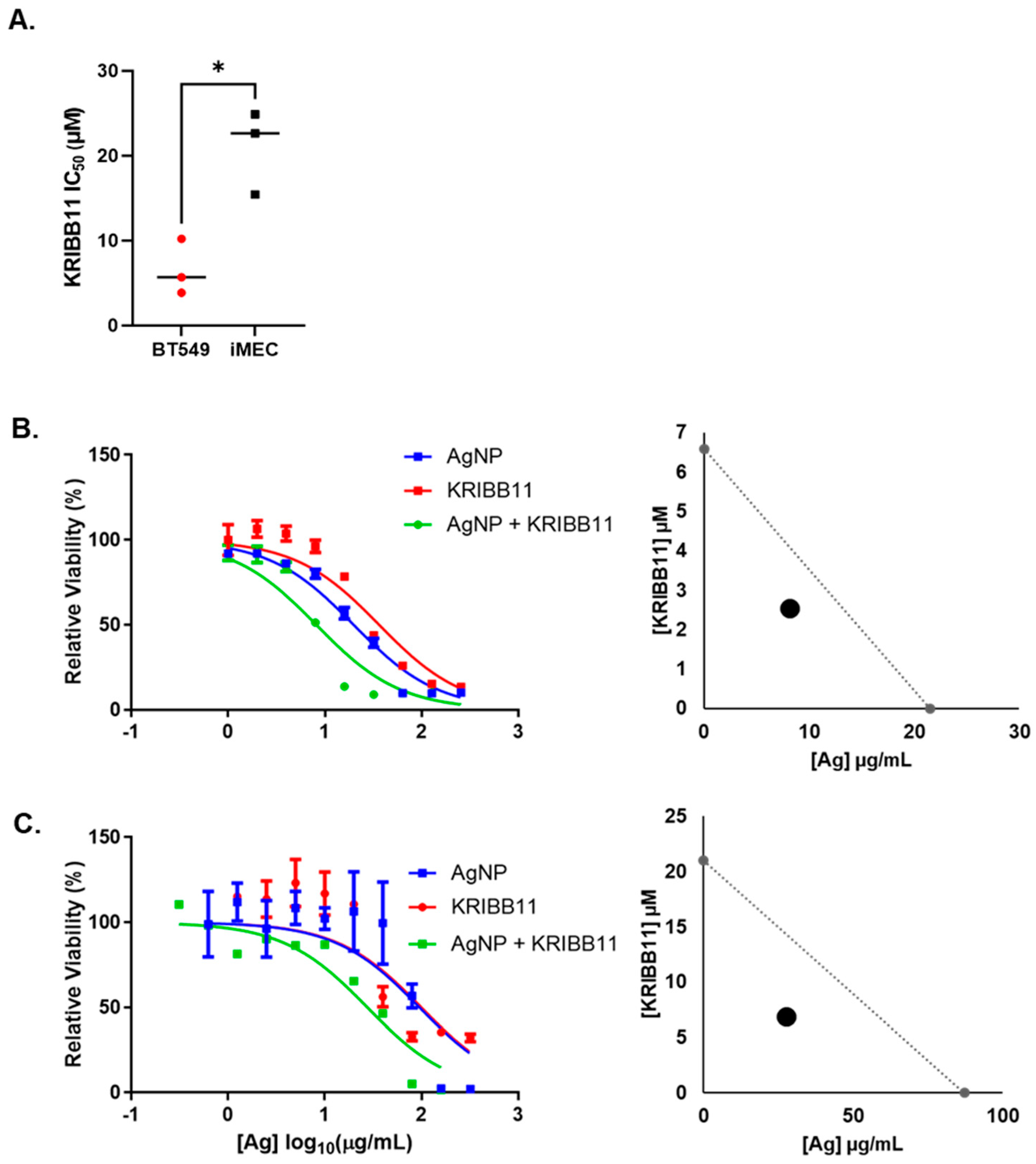
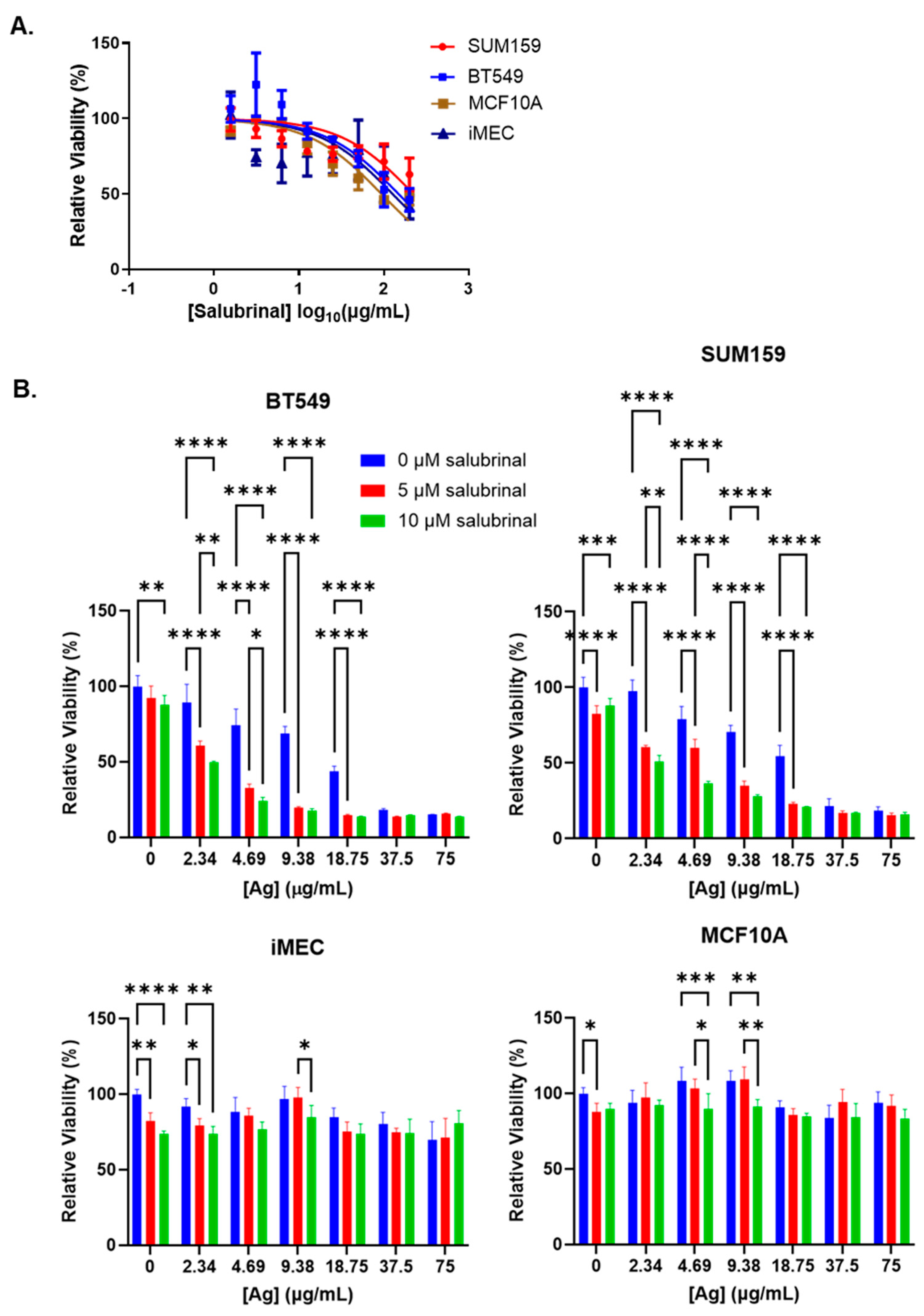
3. Results
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. [Google Scholar] [CrossRef] [PubMed]
- Ho Zhi Guang, M.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Sannino, S.; Brodsky, J.L. Targeting protein quality control pathways in breast cancer. BMC Biol. 2017, 15, 109. [Google Scholar] [CrossRef]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-Mesenchymal Transition Activates PERK-eIF2 alpha and Sensitizes Cells to Endoplasmic Reticulum Stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- Banerjee, S.; Ansari, A.A.; Upadhyay, S.P.; Mettman, D.J.; Hibdon, J.R.; Quadir, M.; Ghosh, P.; Kambhampati, A.; Banerjee, S.K. Benefits and Pitfalls of a Glycosylation Inhibitor Tunicamycin in the Therapeutic Implication of Cancers. Cells 2024, 13, 395. [Google Scholar] [CrossRef]
- Shen, S.; Du, X.J.; Liu, J.; Sun, R.; Zhu, Y.H.; Wang, J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J. Control. Release 2015, 208, 14–24. [Google Scholar] [CrossRef]
- Anchordoquy, T.; Artzi, N.; Balyasnikova, I.V.; Barenholz, Y.; La-Beck, N.M.; Brenner, J.S.; Chan, W.C.W.; Decuzzi, P.; Exner, A.A.; Gabizon, A.; et al. Mechanisms and Barriers in Nanomedicine: Progress in the Field and Future Directions. ACS Nano 2024, 18, 13983–13999. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Gawel, A.M.; Singh, R.; Debinski, W. Metal-Based Nanostructured Therapeutic Strategies for Glioblastoma Treatment—An Update. Biomedicines 2022, 10, 1598. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Saha, M.; Ghosh, S.S. Nanoparticle mediated alteration of EMT dynamics: An approach to modulate cancer therapeutics. Mater. Adv. 2020, 1, 2614–2630. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Saha, S.; Wang, E.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Inhibition of tumor growth and metastasis by a self-therapeutic nanoparticle. Proc. Natl. Acad. Sci. USA 2013, 110, 6700–6705. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Song, L.; Hu, X.; Liu, C.; Shi, J.; Wang, H.; Zhan, L.; Song, H. Inhibition of Epithelial-Mesenchymal Transition and Tissue Regeneration by Waterborne Titanium Dioxide Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 3449–3458. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Qian, P.; Lu, X.; Sun, B.; Zhang, X.; Wang, L.; Gao, X.; Li, H.; Chen, Z. Gd-metallofullerenol nanomaterial as non-toxic breast cancer stem cell-specific inhibitor. Nat. Commun. 2015, 6, 5988. [Google Scholar] [CrossRef]
- Seaberg, J.; Clegg, J.R.; Bhattacharya, R.; Mukherjee, P. Self-Therapeutic Nanomaterials: Applications in Biology and Medicine. Mater. Today 2023, 62, 190–224. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Rohde, M.M.; Fahrenholtz, C.D.; Swanner, J.; Sloop, J.; Donati, G.L.; Furdui, C.M.; Singh, R. Low Doses of Silver Nanoparticles Selectively Induce Lipid Peroxidation and Proteotoxic Stress in Mesenchymal Subtypes of Triple-Negative Breast Cancer. Cancers 2021, 13, 4217. [Google Scholar] [CrossRef]
- Fageria, L.; Bambroo, V.; Mathew, A.; Mukherjee, S.; Chowdhury, R.; Pande, S. Functional Autophagic Flux Regulates AgNP Uptake and the Internalized Nanoparticles Determine Tumor Cell Fate by Temporally Regulating Flux. Int. J. Nanomed. 2019, 14, 9063–9076. [Google Scholar] [CrossRef]
- Gopisetty, M.K.; Kovács, D.; Igaz, N.; Rónavári, A.; Bélteky, P.; Rázga, Z.; Venglovecz, V.; Csoboz, B.; Boros, I.M.; Kónya, Z.; et al. Endoplasmic reticulum stress: Major player in size-dependent inhibition of P-glycoprotein by silver nanoparticles in multidrug-resistant breast cancer cells. J. Nanobiotechnol. 2019, 17, 9. [Google Scholar] [CrossRef]
- Simard, J.C.; Durocher, I.; Girard, D. Silver nanoparticles induce irremediable endoplasmic reticulum stress leading to unfolded protein response dependent apoptosis in breast cancer cells. Apoptosis 2016, 21, 1279–1290. [Google Scholar] [CrossRef]
- Simard, J.C.; Vallieres, F.; de Liz, R.; Lavastre, V.; Girard, D. Silver nanoparticles induce degradation of the endoplasmic reticulum stress sensor activating transcription factor-6 leading to activation of the NLRP-3 inflammasome. J. Biol. Chem. 2015, 290, 5926–5939. [Google Scholar] [CrossRef] [PubMed]
- Swanner, J.; Fahrenholtz, C.D.; Tenvooren, I.; Bernish, B.W.; Sears, J.J.; Hooker, A.; Furdui, C.M.; Alli, E.; Li, W.; Donati, G.L.; et al. Silver nanoparticles selectively treat triple-negative breast cancer cells without affecting non-malignant breast epithelial cells in vitro and in vivo. FASEB Bioadv. 2019, 1, 639–660. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Bhattacharyya, S.; Kudgus, R.A.; Giri, K.; Bhattacharya, R.; Mukherjee, P. Intrinsic therapeutic applications of noble metal nanoparticles: Past, present and future. Chem. Soc. Rev. 2012, 41, 2943–2970. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Balogi, Z.; Khachatryan, W.; Gao, H.; Vígh, L.; Multhoff, G. Membrane-Associated Heat Shock Proteins in Oncology: From Basic Research to New Theranostic Targets. Cells 2020, 9, 1263. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef]
- Green, M.; Schuetz, T.J.; Sullivan, E.K.; Kingston, R.E. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol. Cell. Biol. 1995, 15, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Bharadwaj, S.; O’Carroll, R.; Ovsenek, N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol. Cell. Biol. 1998, 18, 4949–4960. [Google Scholar] [CrossRef]
- Krakowiak, J.; Zheng, X.; Patel, N.; Feder, Z.A.; Anandhakumar, J.; Valerius, K.; Gross, D.S.; Khalil, A.S.; Pincus, D. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 2018, 7, e31668. [Google Scholar] [CrossRef]
- Brunet Simioni, M.; De Thonel, A.; Hammann, A.; Joly, A.L.; Bossis, G.; Fourmaux, E.; Bouchot, A.; Landry, J.; Piechaczyk, M.; Garrido, C. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 2009, 28, 3332–3344. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, X.Y.; Han, R.; Zhang, T.T.; Chen, C.; Qin, Z.H.; Sheng, R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol. Sin. 2013, 34, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; He, K.L.; Li, X.; Wang, L.L.; Liu, C.L.; He, Y.Y. Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2α. Mol. Med. Rep. 2015, 12, 1043–1049. [Google Scholar] [CrossRef]
- Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef]
- Wu, C.T.; Sheu, M.L.; Tsai, K.S.; Chiang, C.K.; Liu, S.H. Salubrinal, an eIF2α dephosphorylation inhibitor, enhances cisplatin-induced oxidative stress and nephrotoxicity in a mouse model. Free Radic. Biol. Med. 2011, 51, 671–680. [Google Scholar] [CrossRef]
- Rohde, M.M.; Snyder, C.M.; Sloop, J.; Solst, S.R.; Donati, G.L.; Spitz, D.R.; Furdui, C.M.; Singh, R. The mechanism of cell death induced by silver nanoparticles is distinct from silver cations. Part. Fibre Toxicol. 2021, 18, 37. [Google Scholar] [CrossRef]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Swanner, J.; Mims, J.; Carroll, D.L.; Akman, S.A.; Furdui, C.M.; Torti, S.V.; Singh, R.N. Differential cytotoxic and radiosensitizing effects of silver nanoparticles on triple-negative breast cancer and non-triple-negative breast cells. Int. J. Nanomed. 2015, 10, 3937–3953. [Google Scholar]
- Tsaytler, P.; Bertolotti, A. Exploiting the selectivity of protein phosphatase 1 for pharmacological intervention. FEBS J. 2013, 280, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Beer, C.; Foldbjerg, R.; Hayashi, Y.; Sutherland, D.S.; Autrup, H. Toxicity of silver nanoparticles—Nanoparticle or silver ion? Toxicol. Lett. 2012, 208, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Falconer, J.L.; Grainger, D.W. In vivo comparisons of silver nanoparticle and silver ion transport after intranasal delivery in mice. J. Control. Release 2018, 269, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Topping, V.D.; Keltner, Z.; Sprando, R.L.; Yourick, J.J. Toxicity of nano- and ionic silver to embryonic stem cells: A comparative toxicogenomic study. J. Nanobiotechnol. 2017, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-C.; Huang, L.-J.; Yang, R.-C. Silver nanoparticles induces heat shock response and provides an anti-inflammatory effect in Clone 9 cells. FASEB J. 2013, 27, lb708. [Google Scholar] [CrossRef]
- Holmila, R.; Wu, H.; Lee, J.; Tsang, A.W.; Singh, R.; Furdui, C.M. Integrated Redox Proteomic Analysis Highlights New Mechanisms of Sensitivity to Silver Nanoparticles. Molelular Cell. Proteom. 2021, 100073. [Google Scholar] [CrossRef]
- Holmila, R.J.; Vance, S.A.; King, S.B.; Tsang, A.W.; Singh, R.; Furdui, C.M. Silver Nanoparticles Induce Mitochondrial Protein Oxidation in Lung Cells Impacting Cell Cycle and Proliferation. Antioxidants 2019, 8, 552. [Google Scholar] [CrossRef]
- Smith, J.N.; Thomas, D.G.; Jolley, H.; Kodali, V.K.; Littke, M.H.; Munusamy, P.; Baer, D.R.; Gaffrey, M.J.; Thrall, B.D.; Teeguarden, J.G. All that is silver is not toxic: Silver ion and particle kinetics reveals the role of silver ion aging and dosimetry on the toxicity of silver nanoparticles. Part. Fibre Toxicol. 2018, 15, 47. [Google Scholar] [CrossRef]
- Ahamed, M.; Posgai, R.; Gorey, T.J.; Nielsen, M.; Hussain, S.M.; Rowe, J.J. Silver nanoparticles induced heat shock protein 70, oxidative stress and apoptosis in Drosophila melanogaster. Toxicol. Appl. Pharmacol. 2010, 242, 263–269. [Google Scholar] [CrossRef]
- Tsai, T.N.; Lee, T.Y.; Liu, M.S.; Ho, J.J.; Huang, L.J.; Liu, C.J.; Chen, T.J.; Yang, R.C. Nonlethal dose of silver nanoparticles attenuates TNF-alpha-induced hepatic epithelial cell death through HSP70 overexpression. Am. J. Physiol.-Cell Physiol. 2015, 308, C959–C963. [Google Scholar] [CrossRef]
- Xin, L.; Wang, J.; Wu, Y.; Guo, S.; Tong, J. Increased oxidative stress and activated heat shock proteins in human cell lines by silver nanoparticles. Hum. Exp. Toxicol. 2015, 34, 315–323. [Google Scholar] [CrossRef]
- Alagar Boopathy, L.R.; Jacob-Tomas, S.; Alecki, C.; Vera, M. Mechanisms tailoring the expression of heat shock proteins to proteostasis challenges. J. Biol. Chem. 2022, 298, 101796. [Google Scholar] [CrossRef] [PubMed]
- Parcellier, A.; Brunet, M.; Schmitt, E.; Col, E.; Didelot, C.; Hammann, A.; Nakayama, K.; Nakayama, K.I.; Khochbin, S.; Solary, E.; et al. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 2006, 20, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, A.L.; Kurchashova, S.Y.; Golyshev, S.A.; Polyakov, V.Y.; Wunderink, H.F.; Kanon, B.; Budagova, K.R.; Kabakov, A.E.; Kampinga, H.H. Regulation of stress-induced intracellular sorting and chaperone function of Hsp27 (HspB1) in mammalian cells. Biochem. J. 2007, 407, 407–417. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Quan, J.H.; Gao, F.F.; Lee, M.; Yuk, J.M.; Cha, G.H.; Chu, J.Q.; Wang, H.; Lee, Y.H. Involvement of endoplasmic reticulum stress response and IRE1-mediated ASK1/JNK/Mcl-1 pathways in silver nanoparticle-induced apoptosis of human retinal pigment epithelial cells. Toxicology 2020, 442, 152540. [Google Scholar] [CrossRef]
- Zhang, R.; Piao, M.J.; Kim, K.C.; Kim, A.D.; Choi, J.Y.; Choi, J.; Hyun, J.W. Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int. J. Biochem. Cell Biol. 2012, 44, 224–232. [Google Scholar] [CrossRef]
- Dey, S.; Fageria, L.; Sharma, A.; Mukherjee, S.; Pande, S.; Chowdhury, R.; Chowdhury, S. Silver nanoparticle-induced alteration of mitochondrial and ER homeostasis affects human breast cancer cell fate. Toxicol. Rep. 2022, 9, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.X. Tracking the Cellular Degradation of Silver Nanoparticles: Development of a Generic Kinetic Model. ACS Nano 2024, 18, 13308–13321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Goia, D.V. Comparative Analysis of Commercial Colloidal Silver Products. Int. J. Nanomed. 2020, 15, 10425–10434. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Ansar, M.; Speshock, J.; Ivanciuc, T.; Qu, Y.; Casola, A.; Garofalo, R.P. Antiviral and Immunomodulatory Activity of Silver Nanoparticles in Experimental RSV Infection. Viruses 2019, 11, 732. [Google Scholar] [CrossRef]
- Weaver, J.L.; Tobin, G.A.; Ingle, T.; Bancos, S.; Stevens, D.; Rouse, R.; Howard, K.E.; Goodwin, D.; Knapton, A.; Li, X.; et al. Evaluating the potential of gold, silver, and silica nanoparticles to saturate mononuclear phagocytic system tissues under repeat dosing conditions. Part. Fibre Toxicol. 2017, 14, 25. [Google Scholar] [CrossRef]
- De Jong, W.H.; Van Der Ven, L.T.; Sleijffers, A.; Park, M.V.; Jansen, E.H.; Van Loveren, H.; Vandebriel, R.J. Systemic and immunotoxicity of silver nanoparticles in an intravenous 28 days repeated dose toxicity study in rats. Biomaterials 2013, 34, 8333–8343. [Google Scholar] [CrossRef]
- Jarak, I.; Carrola, J.; Barros, A.S.; Gil, A.M.; Pereira, M.L.; Corvo, M.L.; Duarte, I.F. From the Cover: Metabolism Modulation in Different Organs by Silver Nanoparticles: An NMR Metabolomics Study of a Mouse Model. Toxicol. Sci. 2017, 159, 422–435. [Google Scholar] [CrossRef]
- Wieler, L.; Vittos, O.; Mukherjee, N.; Sarkar, S. Reduction in the COVID-19 pneumonia case fatality rate by silver nanoparticles: A randomized case study. Heliyon 2023, 9, e14419. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Media Formulation |
|---|---|
| SUM-159 | HAM’s F12 supplemented with penicillin (250 units/mL), streptomycin (250 μg/mL), 2 mM L-glutamine, 5 μg/mL insulin, 1 μg/mL hydrocortisone, 10 μM HEPES, and 5% fetal bovine serum |
| BT-549 | RPMI supplemented with penicillin (250 units/mL), streptomycin (250 μg/mL), and 10% fetal bovine serum |
| MDA-MB-231 | DMEM/F12 supplemented with penicillin (250 units/mL), streptomycin (250 μg/mL), 2 mmol/L L-glutamine, and 10% fetal bovine serum |
| MCF-10A | DMEM/F12 supplemented with penicillin (250 units/mL), streptomycin (250 μg/mL), 2 mM L-glutamine, 10 μg/mL insulin, 20 ng/mL EGF, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, and 5% heat-inactivated horse serum |
| iMEC | DMEM/F12 supplemented with 10 µg/mL insulin, 20 ng/mL hEGF, 0.5 μg/mL hydrocortisone, and 10% fetal bovine serum |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snyder, C.M.; Mateo, B.; Patel, K.; Fahrenholtz, C.D.; Rohde, M.M.; Carpenter, R.; Singh, R.N. Enhancement of Triple-Negative Breast Cancer-Specific Induction of Cell Death by Silver Nanoparticles by Combined Treatment with Proteotoxic Stress Response Inhibitors. Nanomaterials 2024, 14, 1564. https://doi.org/10.3390/nano14191564
Snyder CM, Mateo B, Patel K, Fahrenholtz CD, Rohde MM, Carpenter R, Singh RN. Enhancement of Triple-Negative Breast Cancer-Specific Induction of Cell Death by Silver Nanoparticles by Combined Treatment with Proteotoxic Stress Response Inhibitors. Nanomaterials. 2024; 14(19):1564. https://doi.org/10.3390/nano14191564
Chicago/Turabian StyleSnyder, Christina M., Beatriz Mateo, Khushbu Patel, Cale D. Fahrenholtz, Monica M. Rohde, Richard Carpenter, and Ravi N. Singh. 2024. "Enhancement of Triple-Negative Breast Cancer-Specific Induction of Cell Death by Silver Nanoparticles by Combined Treatment with Proteotoxic Stress Response Inhibitors" Nanomaterials 14, no. 19: 1564. https://doi.org/10.3390/nano14191564