
Selenium Nanoparticles in Protecting the Brain from Stroke: Possible Signaling and Metabolic Mechanisms


Abstract
:1. Introduction
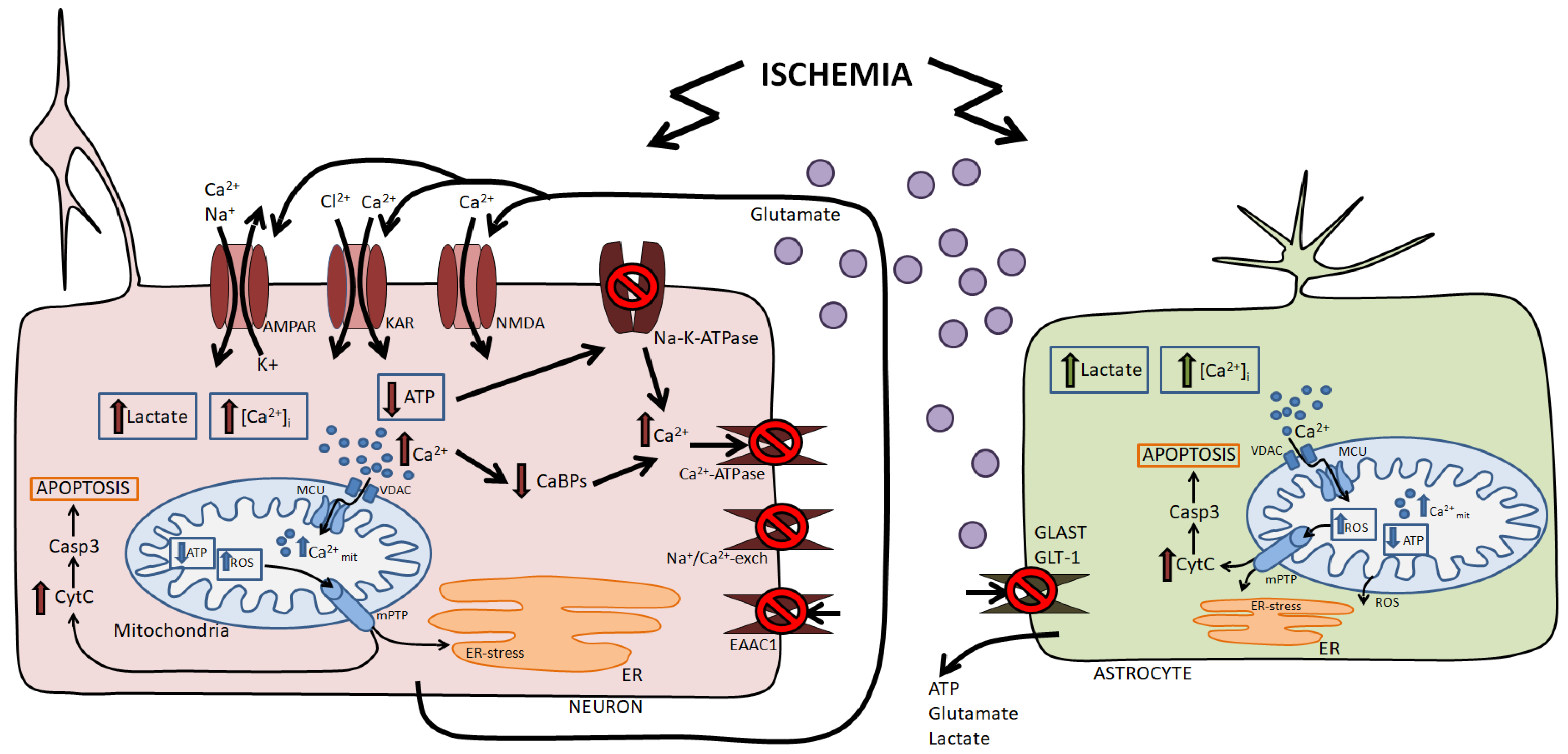
2. Damaging Factors of Ischemia
3. Nanoparticles for Brain Protection
3.1. Nanoparticles as Regulators of Cellular Redox Status
3.2. The Effect of the Shape and Diameter of Nanoparticles on Their Cytoprotective Properties
4. Role of Selenium and Selenoproteins on Neurodegeneration in the Brain

{kind=link}
{kind=link}
{kind=link}
| Physiological Action | Effects of Protein Disruption | Ref. | |
|---|---|---|---|
| GPX1 | Antioxidant action. Intracellular hydrogen peroxide utilization. Overexpression of GPX1 improves the differentiation of mouse embryonic stem cells into neural stem cells and dopaminergic neurons. | GPX1 gene knockout does not affect the normal development of mice. Knockout may exacerbate tissue damage if mice are subjected to brain damage using toxins or limiting cerebral blood flow. | [94,95,96] |
| GPX4 | Antioxidant action. GPx4 is the only enzyme that utilizes glutathione. GPx4 is the only GPX that can utilize membrane phospholipid hydroperoxides as its substrate, reducing phospholipids and cholesterol hydroperoxides. Protects neurons from death during ferroptosis through inhibition of lipid peroxidation. | The knockout causes embryonic lethality, and conditional GPX4 knockout mice exhibit cognitive disruption and hippocampal neurodegeneration. Mutations in the GPX4 gene cause spondylometaphyseal dysplasia sedagati type in children. Neuron-specific knockout causes astrocyte hyperproliferation and neuroinflammation. | [97,98,99,100,101,102] |
| TXNRD1 | Antioxidant action. Catalyzes electron flux from NADPH through TrxR to Trx, which then keeps cellular biomolecules (proteins, lipids, and DNA) in the reduced form. | Nervous system-specific inactivation leads to ataxia and tremors that are associated with cerebellar hypoplasia. Neuron-specific gene deletion leads to age-related neurodegeneration and impaired neuronal development. Conditional ablation of TXNRD1 in neuronal progenitors reveals only a mild cerebellar defect. | [49,87,103] |
| TXNRD2 | Antioxidant action. Participation in the regulation of proliferation. Inhibition of apoptosis. | A homozygous mutation in human TXNRD2 results in glucocorticoid deficiency without a cardiac phenotype. Nervous system-specific Txnrd2 knockout mice do not show any neurological abnormalities. Constitutive gene inactivation is embryonic-lethal. | [87,104,105] |
| Methionine sulfoxide reductase (MSRB1, SELENOR) | Responsible for the reduction in methionine sulfoxide. Involved in the regulation of synaptic plasticity by reducing oxidized CaMKIIα and CaMKIIβ in mice. | Does not cause neurodegeneration. Spatial memory and learning deficit, along with an upregulation of GFAP in MSRB1 deletion. Not directly shown; however, knockout of the methionine sulfoxide reductase A gene leads to neurodegenerative diseases, increased phosphorylation of the TAU protein (microtubule-associated protein), and loss of integrity of astrocytes and increased Aβ precipitation. It is likely that disruption of SEKENOR expression may lead to a similar effect. Spatial memory and learning deficit, along with an upregulation of GFAP in MSRB1 deletion. Not directly shown; however, knockout of the methionine sulfoxide reductase A gene leads to neurodegenerative diseases, increased phosphorylation of the TAU protein (microtubule-associated protein), loss of integrity of astrocytes, and increased Aβ precipitation. It is likely that disruption of SELENOR expression may lead to a similar effect. | [84,90,97,106] |
| SELENOW | Antioxidant action. | Knockout leads to increased H2O2-induced apoptosis of cortical neurons. | [49] |
| SELENOP | Transport of selenium into the brain. Antioxidant action. Modulatory effects in mesolimbic dopaminergic signaling. Exogenous SELENOP prevents the release of dopamine vesicles. Detoxification functions through the binding and inactivation of heavy (copper and cadmium) and transition metals (mercury and iron). | Ataxia. Epilepsy. Disruption of long-term potentiation. Loss of Parvalbumin interneurons. Reactive astrogliosis. Hippocampal neurogenesis is reduced. Depletion of SELENOP and its receptor ApoER2 results in spatial memory impairment in mice as well as defects in synaptic transmission. SELENOP-deficient mice exhibit selenium deficiency in the brain and myelin sheath abnormalities in the brainstem. Genetic deletion of SELENOP results in increased release of dopamine vesicles in response to methamphetamine. | [84,85,86,87,88,107,108,109,110,111,112] |
| SELENOI | Participation in myelin biosynthesis. Maintaining phospholipid homeostasis. | Inactivation of the gene in mice is embryonic-lethal. The SELENOI mutation causes atrophy of the cerebellum and brainstem, which can cause sensorineural deafness, blindness, and seizures. Homozygous missense mutations in SELENOI correlate with seizure activity in some individuals of a pedigree with hereditary spastic paraplegia. | [49,113,114] |
| Physiological Action | Effects of Protein Disruption | Ref. | |
|---|---|---|---|
| SELENOM | Participation in maintaining ER and cytosolic Ca2+ homeostasis. Overexpression of SELENOM in neurons reduces H2O2-induced [Ca2+]i increase. | Knockout leads to an increase in [Ca2+]i, probably due to its leakage from the ER, activation of oxidative stress, and apoptosis. In neurons overexpressing presenilin 2 (PS2), Ca2+ efflux from the ER was correlated with decreased SELENOM expression. | [49,121,122,123,124,125,126] |
| SELENOF | Control of N-glycosylated proteins folding through its interaction with UDP-glucose-glycoprotein glucosyltransferase. Participates in the secretion of some glycoproteins. Involved in adaptive ER stress. In response to moderate ER stress under the action of tunicamycin, SELENOF expression increases, and brain cells adapt. | A powerful stressor effect on the ER using DTT leads to a decrease in SELENOF expression and induction of apoptosis. Mice with SELENOF knockout were viable and fertile, with normal brain morphology and no activation of endoplasmic reticulum (ER) stress. Oxidative stress was elevated in the livers, and prominent nuclear cataracts developed at an early age. The expression of SELENOF mRNA was downregulated in the hippocampus and substantia nigra of a Parkinson’s mouse model. | [127,128,129,130] |
| SELENOT | Control of protein processing in the ER. Possessing oxidoreductase activity, it participates in the antioxidant protection of cells. Catalyzes redox reactions with thiol groups of thiol-disulfide oxidoreductases (ERp57 and protein disulfide isomerase) and various chaperones (BiP, calnexin, calreticulin, and glucose-regulated protein GRP94). Regulation of the protein N-glycosylation. Regulation of the Ca2+ ions pool in the ER. Regulation of dopaminergic neurotransmission (increased dopamine levels) through increased tyrosine hydrolase activity. | Mice with a neuron-specific knockout of SELENOT exhibit decreased volumes of the hippocampus, cerebral cortex, and cerebellum. Perturbation of SELENOT expression induces apoptosis in neurons during the postnatal period. Suppression of expression leads to increased ROS production and nitric oxide, depletion of the ER Ca2+ pool, disruption of hormone secretion, and activation of UPR signaling. | [49,88,120,131,132,133] |
| SELENOS | Anti-apoptotic effects. Participates in the folding and degradation of misfolded proteins associated with the ER (ERAD process). Overexpression increases the resistance of astrocytes to ER stress and inflammatory stimuli. | Suppression of expression correlates with astrocyte death. Gene knockout results in brain cell apoptosis mediated by ER stress. | [120,133,134] |
| SELENOK | Participation in the ERAD pathway of protein degradation in the ER. Participation in the restoration of the cell membrane bilipid layer. Regulation of brain cells Ca2+ homeostasis. Involvement in synaptic neurotransmission through functional interaction between SELENOK and NMDAR. Activation of IP3R and [Ca2+]i increase in microglia through the interaction of SELENOK with palmitoyltransferase (DHHC6). Activating microglial migration and phagocytosis to suppress brain neuroinflammation. | The knockout leads to the disruption of intracellular Ca2+ homeostasis and the functioning of synaptic glutamate receptors. Knockout results in an imbalance in the expression of NMDAR subunits in neurons and neurodegeneration. | [49,116,135] |
| SELENON | Protecting cells from oxidative stress. Regulation of Ca2+ homeostasis through interaction with the ryanodine receptor RYR1. Neutralizes the inhibitory effect of hydroperoxide on SERCA2b. | Not found. | [136,137,138] |
| DIO2 | Stabilizes brain thyroid hormones homeostasis. It is expressed predominantly in astrocytes, but through neuroglial interactions, it can regulate the neuronal network activity. | Impaired motor control. Leads to anxiety. | [139,140,141,142] |
5. Selenium Nanoparticles in Protecting Cells from Ischemic Factors
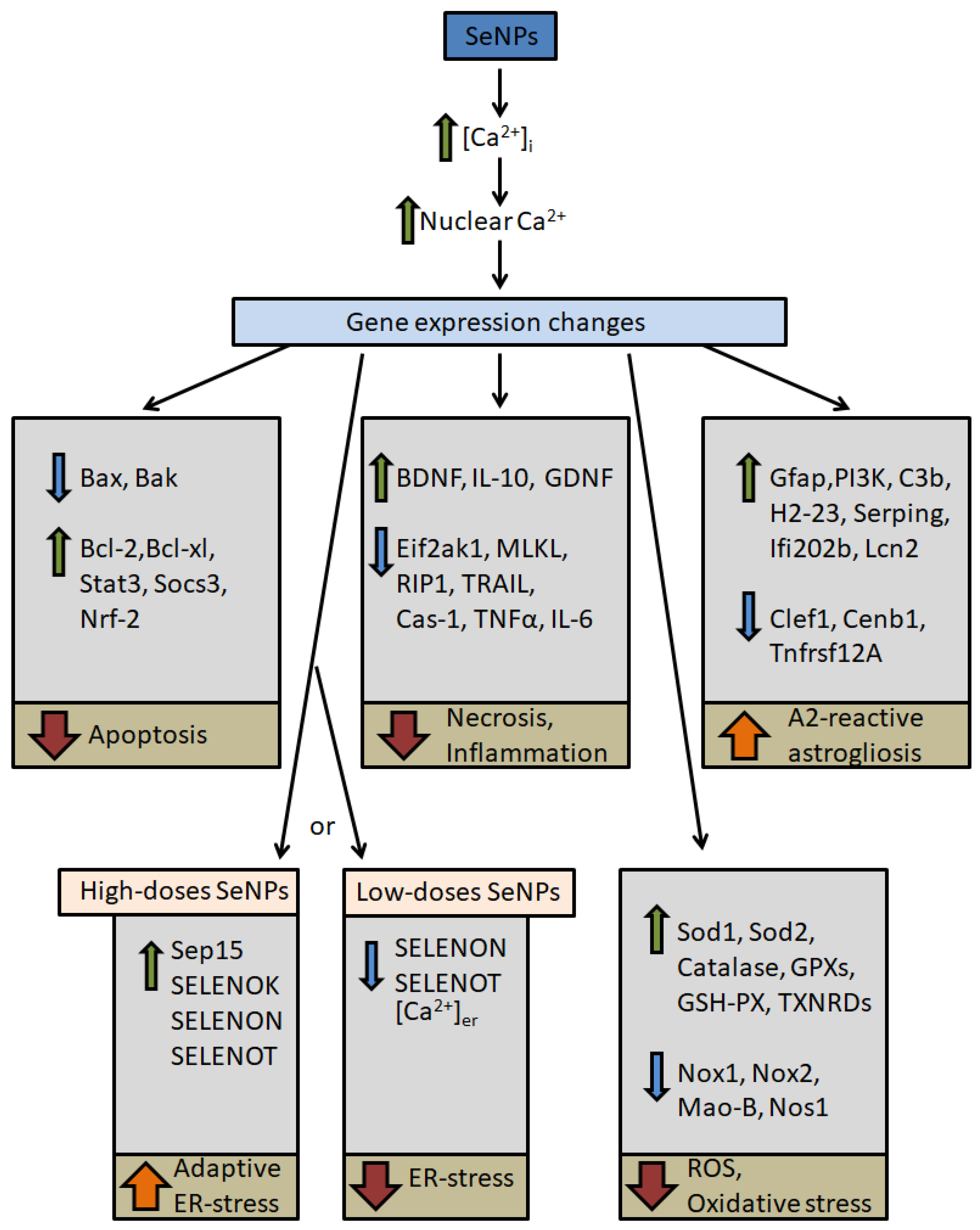
5.1. Possible Signaling Pathways for Cytoprotective Action of Selenium Nanoparticles
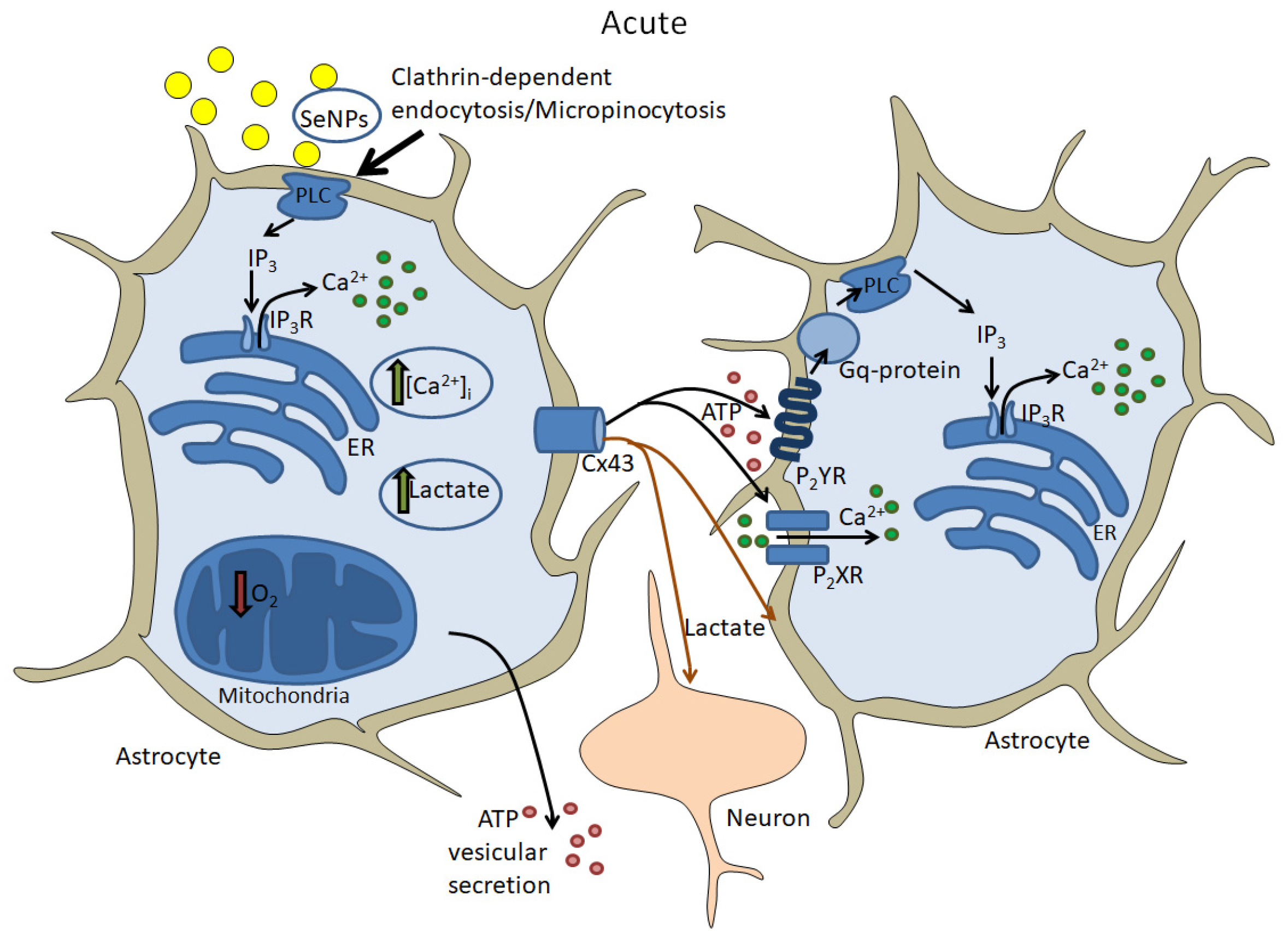
5.2. Acute Effects of Selenium Nanoparticles
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, 67–492. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Lifetime Risk of Stroke Collaborators; Feigin, V.L.; Nguyen, G.; Cercy, K.; Johnson, C.O.; Alam, T.; Parmar, P.G.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; et al. Global, Regional, and Country-Specific Lifetime Risks of Stroke, 1990 and 2016. N. Engl. J. Med. 2018, 379, 2429–2437. [Google Scholar] [PubMed]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. 2016, 5, 213. [Google Scholar]
- Duan, X.; Wen, Z.; Shen, H.; Shen, M.; Chen, G. Intracerebral Hemorrhage, Oxidative Stress, and Antioxidant Therapy. Oxidative Med. Cell Longev. 2016, 2016, 1203285. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients with Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, 344–418. [Google Scholar]
- Papanagiotou, P.; Ntaios, G. Endovascular Thrombectomy in Acute Ischemic Stroke. Circ. Cardiovasc. Interv. 2018, 11, 005362. [Google Scholar] [CrossRef] [PubMed]
- Tsivgoulis, G.; Katsanos, A.H.; Schellinger, P.D.; Köhrmann, M.; Varelas, P.; Magoufis, G.; Paciaroni, M.; Caso, V.; Alexandrov, A.W.; Gurol, E.; et al. Successful Reperfusion with Intravenous Thrombolysis Preceding Mechanical Thrombectomy in Large-Vessel Occlusions. Stroke 2018, 49, 232–235. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Zhang, T.T.; Li, W.; Meng, G.; Wang, P.; Liao, W. Strategies for transporting nanoparticles across the blood-brain barrier. Biomater. Sci. 2016, 4, 219–229. [Google Scholar] [CrossRef]
- Oh, J.; Lee, J.; Piao, C.; Jeong, J.H.; Lee, M. A self-assembled DNA-nanoparticle with a targeting peptide for hypoxia-inducible gene therapy of ischemic stroke. Biomater. Sci. 2019, 7, 2174–2190. [Google Scholar] [CrossRef]
- Kim, K.S.; Khang, G.; Lee, D. Application of nanomedicine in cardiovascular diseases and stroke. Curr. Pharm. Des. 2011, 17, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhao, M.; Chen, H.; Lenahan, C.; Zhou, X.; Ou, Y.; He, Y. The Role of Nanomaterials in Stroke Treatment: Targeting Oxidative Stress. Oxidative Med. Cell Longev. 2021, 2021, 8857486. [Google Scholar] [CrossRef] [PubMed]
- Moglianetti, M.; De Luca, E.; Pedone, D.; Marotta, R.; Catelani, T.; Sartori, B.; Amenitsch, H.; Retta, S.F.; Pompa, P.P. Platinum nanozymes recover cellular ROS homeostasis in an oxidative stress-mediated disease model. Nanoscale 2016, 8, 3739–3752. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Zhu, X.; Zhang, L.; Guo, F.; Zhang, M.; Tan, Y.; Gong, A.; Fang, Z.; Ju, H.; Wu, C.; et al. Biomineralization-Inspired Synthesis of Cerium-Doped Carbonaceous Nanoparticles for Highly Hydroxyl Radical Scavenging Activity. Nanoscale Res. Lett. 2018, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Turovsky, E.A.; Blinova, E.V. Therapeutic Potential and Main Methods of Obtaining Selenium Nanoparticles. Int. J. Mol. Sci. 2021, 22, 10808. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, R.; Shineh, G.; Mobaraki, M.; Doughty, S.; Tayebi, L. Structural parameters of nanoparticles affecting their toxicity for biomedical applications: A review. J. Nanopart. Res. 2023, 25, 43. [Google Scholar] [CrossRef] [PubMed]
- Yanar, F.; Carugo, D.; Zhang, X. Hybrid Nanoplatforms Comprising Organic Nanocompartments Encapsulating Inorganic Nanoparticles for Enhanced Drug Delivery and Bioimaging Applications. Molecules 2023, 28, 5694. [Google Scholar] [CrossRef]
- Jin, Q.; Cai, Y.; Li, S.; Liu, H.; Zhou, X.; Lu, C.; Gao, X.; Qian, J.; Zhang, J.; Ju, S.; et al. Edaravone-Encapsulated Agonistic Micelles Rescue Ischemic Brain Tissue by Tuning Blood-Brain Barrier Permeability. Theranostics 2017, 7, 884–898. [Google Scholar] [CrossRef]
- Turovskaya, M.V.; Zinchenko, V.P.; Babaev, A.A.; Epifanova, E.A.; Tarabykin, V.S.; Turovsky, E.A. Mutation in the Sip1 transcription factor leads to a disturbance of the preconditioning of AMPA receptors by episodes of hypoxia in neurons of the cerebral cortex due to changes in their activity and subunit composition. The protective effects of interleukin-10. Arch. Biochem. Biophys. 2018, 654, 126–135. [Google Scholar]
- Turovsky, E.A.; Turovskaya, M.V.; Gaidin, S.G.; Zinchenko, V.P. Cytokine IL-10, activators of PI3-kinase, agonists of α-2 adrenoreceptor and antioxidants prevent ischemia-induced cell death in rat hippocampal cultures. Arch. Biochem. Biophys. 2017, 615, 35–43. [Google Scholar] [CrossRef]
- Vergara, R.C.; Jaramillo-Riveri, S.; Luarte, A.; Moënne-Loccoz, C.; Fuentes, R.; Couve, A.; Maldonado, P.E. The Energy Homeostasis Principle: Neuronal Energy Regulation Drives Local Network Dynamics Generating Behavior. Front. Comput. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Farhat, E.; Devereaux, M.E.M.; Cheng, H.; Weber, J.M.; Pamenter, M.E. Na+/K+-ATPase activity is regionally regulated by acute hypoxia in naked mole-rat brain. Neurosci. Lett. 2021, 764, 136244. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Yu, L.; Xu, H. Calcium signaling in membrane repair. Semin. Cell Dev. Biol. 2015, 45, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xie, X.; Xing, X.; Sun, X. Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies. Int. J. Mol. Sci. 2022, 23, 9381. [Google Scholar] [CrossRef] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Najafov, A.; Py, B.F. Roles of Caspases in Necrotic Cell Death. Cell 2016, 167, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yuan, M.; Guo, Y.S.; Shen, X.Y.; Gao, Z.K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell. Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, Y.; Liu, Y.; Guo, Z.; Bai, T.; Zhou, P.; Wu, J.; Yang, Q.; Liu, Z.; Lu, X. Intrinsic Effects of Gold Nanoparticles on Oxygen-Glucose Deprivation/Reperfusion Injury in Rat Cortical Neurons. Neurochem. Res. 2019, 44, 1549–1566. [Google Scholar] [CrossRef]
- Ko, W.C.; Wang, S.J.; Hsiao, C.Y.; Hung, C.T.; Hsu, Y.J.; Chang, D.C.; Hung, C.F. Pharmacological Role of Functionalized Gold Nanoparticles in Disease Applications. Molecules 2022, 27, 1551. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Baryshev, A.S.; Gudkov, S.V.; Babenko, V.A.; Plotnikov, E.Y.; Turovsky, E.A. Cerium Oxide Nanoparticles Protect Cortical Astrocytes from Oxygen-Glucose Deprivation through Activation of the Ca2+ Signaling System. Int. J. Mol. Sci. 2023, 24, 14305. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, A.; Zhou, Z.; Connor, J.; Madhankumar, A.B.; Pamujula, S.; Sayes, C.M.; Kepley, C.L. Application of fullerenes in nanomedicine: An update. Nanomedicine 2013, 8, 1191–1208. [Google Scholar] [CrossRef]
- Baranes, K.; Shevach, M.; Shefi, O.; Dvir, T. Gold Nanoparticle-Decorated Scaffolds Promote Neuronal Differentiation and Maturation. Nano Lett. 2016, 16, 2916–2920. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kang, Y.; Liu, J.; Yin, S.; Huang, Z.; Shao, L. Nanomaterials alleviating redox stress in neurological diseases: Mechanisms and applications. J. Nanobiotechnol. 2022, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Lao, F.; Chen, L.; Li, W.; Ge, C.; Qu, Y.; Sun, Q.; Zhao, Y.; Han, D.; Chen, C. Fullerene nanoparticles selectively enter oxidation-damaged cerebral microvessel endothelial cells and inhibit JNK-related apoptosis. ACS Nano 2009, 3, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.; Shokr, A.; Willemen, N.; Bashir, S.M.; Shin, S.R.; Hassan, S. Microfluidic fabrication of lipid nanoparticles for the delivery of nucleic acids. Adv. Drug Deliv. Rev. 2022, 184, 114197. [Google Scholar] [CrossRef]
- El-Say, K.M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Song, G.L. Roles of Selenoproteins in Brain Function and the Potential Mechanism of Selenium in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 646518. [Google Scholar] [CrossRef]
- Jiang, X.; Kemal, L.; Yu, A. Silver-induced growth of selenium nanowires in aqueous solution. Mater. Lett. 2007, 61, 2584–2588. [Google Scholar] [CrossRef]
- Sarkar, J.; Saha, S.; Dey, P.; Acharya, K. Production of selenium nanorods by phytopathogen, Alternaria alternata. Adv. Sci. Lett. 2012, 10, 111–114. [Google Scholar] [CrossRef]
- Kumar, A.; Sevonkaev, I.; Goia, D.V. Synthesis of selenium particles with various morphologies. J. Colloid Interface Sci. 2014, 416, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Nayak, V.; Singh, K.R.B.; Singh, A.K.; Singh, R.P. Potentialities of selenium nanoparticles in biomedical science. New J. Chem. 2021, 45, 2849–2878. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.V.; Keliher, E.J.; Core, A.B.; Brown, D.; Weissleder, R. Characterizing the interactions of organic nanoparticles with renal epithelial cells in vivo. ACS Nano 2015, 9, 3641–3653. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Adewale, O.B.; Davids, H.; Cairncross, L.; Roux, S. Toxicological Behavior of Gold Nanoparticles on Various Models: Influence of Physicochemical Properties and Other Factors. Int. J. Toxicol. 2019, 38, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Gliga, A.R.; Skoglund, S.; Wallinder, I.O.; Fadeel, B.; Karlsson, H.L. Size-dependent cytotoxicity of silver nanoparticles in human lung cells: The role of cellular uptake, agglomeration and Ag release. Part. Fibre Toxicol. 2014, 11, 11. [Google Scholar] [CrossRef]
- Bhatia, D.; Mittal, A.; Malik, D.K. Antimicrobial potential and in vitro cytotoxicity study of polyvinyl pyrollidone-stabilised silver nanoparticles synthesised from Lysinibacillus boronitolerans. IET Nanobiotechnol. 2021, 15, 427–440. [Google Scholar] [CrossRef]
- Samberg, M.E.; Loboa, E.G.; Oldenburg, S.J.; Monteiro-Riviere, N.A. Silver nanoparticles do not influence stem cell differentiation but cause minimal toxicity. Nanomedicine 2012, 7, 1197–1209. [Google Scholar] [CrossRef]
- Sengstock, C.; Diendorf, J.; Epple, M.; Schildhauer, T.A.; Köller, M. Effect of silver nanoparticles on human mesenchymal stem cell differentiation. Beilstein J. Nanotechnol. 2014, 5, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Basu, A.; Bhattacharya, S. Selenium nanoparticles are less toxic than inorganic and organic selenium to mice in vivo. Nucleus 2019, 62, 259–268. [Google Scholar] [CrossRef]
- Sieber, F.; Daziano, J.P.; Günther, W.H.; Krieg, M.; Miyagi, K.; Sampson, R.W.; Ostrowski, M.D.; Anderson, G.S.; Tsujino, I.; Bula, R.J. Elemental selenium generated by the photobleaching of selenomerocyanine photosensitizers forms conjugates with serum macromolecules that are toxic to tumor cells. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Gudkov, S.V.; Plotnikov, E.Y.; Turovsky, E.A. Size-Dependent Cytoprotective Effects of Selenium Nanoparticles during Oxygen-Glucose Deprivation in Brain Cortical Cells. Int. J. Mol. Sci. 2022, 23, 7464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Ran, F.; Cui, Y.; Liu, C.; Zhao, Q.; Gao, Y.; Wang, D.; Wang, S. A comparison between sphere and rod nanoparticles regarding their in vivo biological behavior and pharmacokinetics. Sci. Rep. 2017, 7, 4131. [Google Scholar] [CrossRef] [PubMed]
- Niroumand, U.; Firouzabadi, N.; Goshtasbi, G.; Hassani, B.; Ghasemiyeh, P.; Mohammadi-Samani, S. The effect of size, morphology and surface properties of mesoporous silica nanoparticles on pharmacokinetic aspects and potential toxicity concerns. Front. Mater. 2023, 10, 1189463. [Google Scholar] [CrossRef]
- Xie, J.; MacEwan, M.R.; Li, X.; Sakiyama-Elbert, S.E.; Xia, Y. Neurite outgrowth on nanofiber scaffolds with different orders, structures, and surface properties. ACS Nano 2009, 26, 1151–1159. [Google Scholar] [CrossRef]
- Siddiqui, A.M.; Brunner, R.; Harris, G.M.; Miller, A.L., II; Waletzki, B.E.; Schmeichel, A.M.; Schwarzbauer, J.E.; Schwartz, J.; Yaszemski, M.J.; Windebank, A.J.; et al. Promoting Neuronal Outgrowth Using Ridged Scaffolds Coated with Extracellular Matrix Proteins. Biomedicines 2021, 9, 479. [Google Scholar] [CrossRef]
- Rauti, R.; Musto, M.; Bosi, S.; Prato, M.; Ballerini, L. Properties and behavior of carbon nanomaterials when interfacing neuronal cells: How far have we come? Carbon 2019, 143, 430–446. [Google Scholar] [CrossRef]
- Cellot, G.; Cilia, E.; Cipollone, S.; Rancic, V.; Sucapane, A.; Giordani, S.; Gambazzi, L.; Markram, H.; Grandolfo, M.; Scaini, D.; et al. Carbon nanotubes might improve neuronal performance by favouring electrical shortcuts. Nat. Nanotechnol. 2009, 4, 126–133. [Google Scholar] [CrossRef]
- Xiang, C.; Zhang, Y.; Guo, W.; Liang, X.J. Biomimetic carbon nanotubes for neurological disease therapeutics as inherent medication. Acta. Pharm. Sin. B 2020, 10, 239–248. [Google Scholar] [CrossRef]
- Gottipati, M.K.; Verkhratsky, A.; Parpura, V. Probing astroglia with carbon nanotubes: Modulation of form and function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130598. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Verkhratsky, A. Astrogliopathology: Could nanotechnology restore aberrant calcium signalling and pathological astroglial remodelling? Biochim. Biophys. Acta. 2013, 1833, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Turovsky, E.A.; Babenko, V.A.; Plotnikov, E.Y. The Mechanisms Underlying the Protective Action of Selenium Nanoparticles against Ischemia/Reoxygenation Are Mediated by the Activation of the Ca2+ Signaling System of Astrocytes and Reactive Astrogliosis. Int. J. Mol. Sci. 2021, 22, 12825. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Plotnikov, E.Y.; Baimler, I.V.; Gudkov, S.V.; Turovsky, E.A. Pilot Study of Cytoprotective Mechanisms of Selenium Nanorods (SeNrs) under Ischemia-like Conditions on Cortical Astrocytes. Int. J. Mol. Sci. 2023, 24, 12217. [Google Scholar] [CrossRef] [PubMed]
- Khandel, P.; Yadaw, R.K.; Soni, D.K.; Kanwar, L.; Shahi, S.K. Biogenesis of metal nanoparticles and their pharmacological applications: Present status and application prospects. J. Nanostruct. Chem. 2018, 8, 217–254. [Google Scholar] [CrossRef]
- Chintamani, R.B.; Salunkhe, K.S.; Chavan, M. Emerging use of green synthesis silver nanoparticle: An updated review. Int. J. Pharm. Sci. Res. 2018, 9, 4029–4055. [Google Scholar]
- Speckmann, B.; Grune, T. Epigenetic effects of selenium and their implications for health. Epigenetics 2015, 10, 179–190. [Google Scholar] [CrossRef]
- Sanguigno, L.; Guida, N.; Anzilotti, S.; Cuomo, O.; Mascolo, L.; Serani, A.; Brancaccio, P.; Pennacchio, G.; Licastro, E.; Pignataro, G.; et al. Stroke by inducing HDAC9-dependent deacetylation of HIF-1 and Sp1, promotes TfR1 transcription and GPX4 reduction, thus determining ferroptotic neuronal death. Int. J. Biol. Sci. 2023, 19, 2695–2710. [Google Scholar] [CrossRef]
- Dominiak, A.; Wilkaniec, A.; Wroczyński, P.; Adamczyk, A. Selenium in the Therapy of Neurological Diseases. Where is it Going? Curr. Neuropharmacol. 2016, 14, 282–299. [Google Scholar] [CrossRef]
- Yang, L.; Ma, Y.M.; Shen, X.L.; Fan, Y.C.; Zhang, J.Z.; Li, P.A.; Jing, L. The Involvement of Mitochondrial Biogenesis in Selenium Reduced Hyperglycemia-Aggravated Cerebral Ischemia Injury. Neurochem. Res. 2020, 45, 1888–1901. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Z.; Gong, P.; Yao, W.; Ba, Q.; Wang, H. Review on the health-promoting effect of adequate selenium status. Front. Nutr. 2023, 10, 1136458. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, N.; Drobyshev, E.; Blume, B.; Michalke, B. Selenium at the Neural Barriers: A Review. Front. Neurosci. 2021, 15, 630016. [Google Scholar] [CrossRef]
- Shi, T.; Song, J.; You, G.; Yang, Y.; Liu, Q.; Li, N. The Function of Selenium in Central Nervous System: Lessons from MsrB1 Knockout Mouse Models. Molecules 2021, 26, 1372. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.J.; Yorgason, J.T.; Mitchell, C.C.; Hagiwara, A.; Andres, M.A.; Kurokawa, S.; Steffensen, S.C.; Bellinger, F.P. Selenoprotein P Modulates Methamphetamine Enhancement of Vesicular Dopamine Release in Mouse Nucleus Accumbens Via Dopamine D2 Receptors. Front. Neurosci. 2021, 15, 631825. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef] [PubMed]
- Soerensen, J.; Jakupoglu, C.; Beck, H.; Förster, H.; Schmidt, J.; Schmahl, W.; Schweizer, U.; Conrad, M.; Brielmeier, M. The role of thioredoxin reductases in brain development. PLoS ONE 2008, 3, 1813. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci. 2021, 15, 652099. [Google Scholar] [CrossRef]
- Kim, H.Y.; Gladyshev, V.N. Methionine sulfoxide reductases: Selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem. J. 2007, 407, 321–329. [Google Scholar] [CrossRef]
- Shi, T.; Yang, Y.; Zhang, Z.; Zhang, L.; Song, J.; Ping, Y.; Du, X.; Song, G.; Liu, Q.; Li, N. Loss of MsrB1 perturbs spatial learning and long-term potentiation/long-term depression in mice. Neurobiol. Learn. Mem. 2019, 166, 107104. [Google Scholar] [CrossRef]
- Ding, W.; Wang, S.; Gu, J.; Yu, L. Selenium and human nervous system. Chin. Chem. Lett. 2023, 34, 108043. [Google Scholar] [CrossRef]
- Raman, A.V.; Pitts, M.W.; Seyedali, A.; Hashimoto, A.C.; Bellinger, F.P.; Berry, M.J. Selenoprotein W expression and regulation in mouse brain and neurons. Brain Behav. 2013, 3, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Selenoprotein P as an in vivo redox regulator: Disorders related to its deficiency and excess. J. Clin. Biochem. Nutr. 2020, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fontelles, C.C.; Ong, T.P. Selenium and Breast Cancer Risk: Focus on Cellular and Molecular Mechanisms. Adv. Cancer Res. 2017, 136, 173–192. [Google Scholar]
- Crack, P.J.; Taylor, J.M.; Flentjar, N.J.; de Haan, J.; Hertzog, P.; Iannello, R.C.; Kola, I. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J. Neurochem. 2001, 78, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Shimokawa, T.; Sekine-Suzuki, E.; Nyui, M.; Nakanishi, I.; Matsumoto, K.I. Preparation of an experimental mouse model lacking selenium-dependent glutathione peroxidase activities by feeding a selenium-deficient diet. J. Clin. Biochem. Nutr. 2021, 68, 123–130. [Google Scholar] [CrossRef]
- Pillai, R.; Uyehara-Lock, J.H.; Bellinger, F.P. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef]
- Cheff, D.M.; Muotri, A.R.; Stockwell, B.R.; Schmidt, E.E.; Ran, Q.; Kartha, R.V.; Johnson, S.C.; Mittal, P.; Arnér, E.S.J.; Wigby, K.M.; et al. Development of therapies for rare genetic disorders of GPX4: Roadmap and opportunities. Orphanet. J. Rare Dis. 2021, 16, 446. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Borchert, A.; Bräuer, A.U.; Kuhn, H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic. Biol. Med. 2007, 43, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Jabbar, S.; Mathews, P.; Kang, Y. Emerging Evidence of the Significance of Thioredoxin-1 in Hematopoietic Stem Cell Aging. Antioxidants 2022, 11, 1291. [Google Scholar] [CrossRef]
- Prasad, R.; Chan, L.F.; Hughes, C.R.; Kaski, J.P.; Kowalczyk, J.C.; Savage, M.O.; Peters, C.J.; Nathwani, N.; Clark, A.J.; Storr, H.L.; et al. Thioredoxin Reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J. Clin. Endocrinol. Metab. 2014, 99, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shi, Z.; Cheng, M.; Zhou, Z.; Chu, M.; Sun, L.; Zhou, J.C. Biology and Roles in Diseases of Selenoprotein I Characterized by Ethanolamine Phosphotransferase Activity and Antioxidant Potential. J. Nutr. 2023, 22, 3164–3172. [Google Scholar] [CrossRef]
- Pal, R.; Oien, D.B.; Ersen, F.Y.; Moskovitz, J. Elevated levels of brain-pathologies associated with neurodegenerative diseases in the methionine sulfoxide reductase A knockout mouse. Exp. Brain Res. 2007, 180, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, N.; Drobyshev, E.; Bjørklund, G.; Dubrovskii, Y.; Lysiuk, R.; Rayman, M.P. Selenium, selenoprotein P, and Alzheimer’s disease: Is there a link? Free Radic. Biol. Med. 2018, 127, 124–133. [Google Scholar] [CrossRef]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Burk, R.F. Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J. Nutr. 2004, 134, 157–161. [Google Scholar] [CrossRef]
- Byrns, C.N.; Pitts, M.W.; Gilman, C.A.; Hashimoto, A.C.; Berry, M.J. Mice lacking selenoprotein P and selenocysteine lyase exhibit severe neurological dysfunction, neurodegeneration, and audiogenic seizures. J. Biol. Chem. 2014, 289, 9662–9674. [Google Scholar] [CrossRef]
- Valentine, W.M.; Hill, K.E.; Austin, L.M.; Valentine, H.L.; Goldowitz, D.; Burk, R.F. Brainstem axonal degeneration in mice with deletion of selenoprotein p. Toxicol. Pathol. 2005, 33, 570–576. [Google Scholar] [CrossRef]
- Pitts, M.W.; Raman, A.V.; Hashimoto, A.C.; Todorovic, C.; Nichols, R.A.; Berry, M.J. Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 2012, 208, 58–68. [Google Scholar] [CrossRef]
- Leiter, O.; Zhuo, Z.; Rust, R.; Wasielewska, J.M.; Grönnert, L.; Kowal, S.; Overall, R.W.; Adusumilli, V.S.; Blackmore, D.G.; Southon, A.; et al. Selenium mediates exercise-induced adult neurogenesis and reverses learning deficits induced by hippocampal injury and aging. Cell Metab. 2022, 34, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.Y.; Al-Khayat, A.; Al-Murshedi, F.; Al-Futaisi, A.; Chioza, B.A.; Pedro Fernandez-Murray, J.; Self, J.E.; Salter, C.G.; Harlalka, G.V.; Rawlins, L.E.; et al. A mutation of EPT1 (SELENOI) underlies a new disorder of Kennedy pathway phospholipid biosynthesis. Brain 2017, 140, 547–554. [Google Scholar] [PubMed]
- Li, C.; Lai, H.; Cai, X.; Liu, Y.; Hong, L.; Huang, X.; Shao, L. Thioredoxin Reductase 2 Synergizes with Cytochrome c, Somatic to Alleviate Doxorubicin-Induced Oxidative Stress in Cardiomyocytes and Mouse Myocardium. Int. Heart J. 2023, 64, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Arnér, E.S.; Berry, M.J.; Brigelius-Flohé, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein Gene Nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef]
- Zhang, Y.; Roh, Y.J.; Han, S.-J.; Park, I.; Lee, H.M.; Ok, Y.S.; Lee, B.C.; Lee, S.-R. Role of Selenoproteins in Redox Regulation of Signaling and the Antioxidant System: A Review. Antioxidants 2020, 9, 383. [Google Scholar] [CrossRef]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Miyamoto, S.; Schulze, A. Ferroptosis: The Greasy Side of Cell Death. Chem. Res. Toxicol. 2019, 32, 362–369. [Google Scholar] [CrossRef]
- Solovyev, N.D. Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef]
- Jehan, C.; Cartier, D.; Bucharles, C.; Anouar, Y.; Lihrmann, I. Emerging roles of ER-resident selenoproteins in brain physiology and physiopathology. Redox Biol. 2022, 55, 102412. [Google Scholar] [CrossRef]
- Reeves, M.A.; Bellinger, F.P.; Berry, M.J. The neuroprotective functions of selenoprotein M and its role in cytosolic calcium regulation. Antioxid. Redox Signal. 2010, 12, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.Y.; Sin, J.S.; Kim, M.S.; Yim, S.Y.; Kim, Y.K.; Kim, C.K.; Kim, B.G.; Shim, S.B.; Jee, S.W.; Lee, S.H.; et al. Overexpression of human selenoprotein M differentially regulates the concentrations of antioxidants and H2O2, the activity of antioxidant enzymes, and the composition of white blood cells in a transgenic rat. Int. J. Mol. Med. 2008, 21, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Rogachev, V.V.; Novoselov, S.V.; Turovsky, E.A. SelenoM-knockdown enhances the protective effect of A-172 cancer cells against MSA-induced ER-stress and staurosporine-induced apoptosis. Opera Med. Physiol. 2021, 8, 23–32. [Google Scholar]
- Varlamova, E.G.; Goltyaev, M.V.; Turovsky, E.A. The Role of Selenoproteins SELENOM and SELENOT in the Regulation of Apoptosis, ER Stress and Calcium Homeostasis in the A-172 Human Glioblastoma Cell Line. Biology 2022, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Rogachev, V.V.; Goltyaev, M.V.; Varlamova, E.G.; Turovsky, E.A. Molecular Mechanisms of the Cytotoxic Effect of Recombinant Selenoprotein SELENOM on Human Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 6469. [Google Scholar] [CrossRef] [PubMed]
- Dogaru, C.B.; Duță, C.; Muscurel, C.; Stoian, I. “Alphabet” Selenoproteins: Implications in Pathology. Int. J. Mol. Sci. 2023, 24, 15344. [Google Scholar] [CrossRef]
- Tian, J.; Liu, J.; Li, J.; Zheng, J.; Chen, L.; Wang, Y.; Liu, Q.; Ni, J. The interaction of selenoprotein F (SELENOF) with retinol dehydrogenase 11 (RDH11) implied a role of SELENOF in vitamin A metabolism. Nutr. Metab. 2018, 15, 7. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Yoo, M.H.; Hatfield, D.L.; Gladyshev, V.N. Sep15, a thioredoxin-like selenoprotein, is involved in the unfolded protein response and differentially regulated by adaptive and acute ER stresses. Biochemistry 2009, 48, 8458–8465. [Google Scholar] [CrossRef]
- Kasaikina, M.V.; Fomenko, D.E.; Labunskyy, V.M.; Lachke, S.A.; Qiu, W.; Moncaster, J.A.; Zhang, J.; Wojnarowicz, M.W., Jr.; Natarajan, S.K.; Malinouski, M.; et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 2011, 286, 33203–33212. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, Y.L.; Zhu, H.; Sun, S.N.; Zheng, J.; Fan, H.H.; Wu, H.M.; Chen, S.F.; Cheng, W.H.; Zhu, J.H. Selenotranscriptomic analyses identify signature selenoproteins in brain regions in a mouse model of parkinson’s disease. PLoS ONE 2016, 11, 0163372. [Google Scholar] [CrossRef]
- Castex, M.T.; Arabo, A.; Bénard, M.; Roy, V.; Le Joncour, V.; Prévost, G.; Bonnet, J.J.; Anouar, Y.; Falluel-Morel, A. Selenoprotein T Deficiency Leads to Neurodevelopmental Abnormalities and Hyperactive Behavior in Mice. Mol. Neurobiol. 2016, 53, 5818–5832. [Google Scholar] [CrossRef] [PubMed]
- Pothion, H.; Jehan, C.; Tostivint, H.; Cartier, D.; Bucharles, C.; Falluel-Morel, A.; Boukhzar, L.; Anouar, Y.; Lihrmann, I. Selenoprotein T: An Essential Oxidoreductase Serving as a Guardian of Endoplasmic Reticulum Homeostasis. Antioxid. Redox Signal. 2020, 33, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Fradejas, N.; Serrano-Pérez Mdel, C.; Tranque, P.; Calvo, S. Selenoprotein S expression in reactive astrocytes following brain injury. Glia 2011, 59, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Lai, Q.; Han, Q.; Cai, N.; He, H.; Fang, W.; Yi, J.; Li, X.; Xu, H.; Li, X.; et al. Seleno-polymannuronate attenuates neuroinflammation by suppressing microglial and astrocytic activation. J. Funct. Foods 2018, 51, 113–120. [Google Scholar] [CrossRef]
- Meng, X.L.; Chen, C.L.; Liu, Y.Y.; Su, S.J.; Gou, J.M.; Huan, F.N.; Wang, D.; Liu, H.S.; Ben, S.B.; Lu, J. Selenoprotein SELENOK Enhances the Migration and Phagocytosis of Microglial Cells by Increasing the Cytosolic Free Ca2+ Level Resulted from the Up-Regulation of IP3R. Neuroscience 2019, 406, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Simmen, T. ER-luminal thiol/selenol-mediated regulation of Ca2+ signaling. Biochem. Soc. Trans. 2016, 44, 452–459. [Google Scholar] [CrossRef]
- Arbogast, S.; Beuvin, M.; Fraysse, B.; Zhou, H.; Muntoni, F.; Ferreiro, A. Oxidativestress in SEPN1-related myopathy: From pathophysiology to treatment. Ann. Neurol. 2009, 65, 677–686. [Google Scholar] [CrossRef]
- Marino, M.; Stoilova, T.; Giorgi, C.; Bachi, A.; Cattaneo, A.; Auricchio, A. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyper-oxidation by means of redox-regulating SERCA2 pump activity. Hum. Mol. Genet. 2015, 24, 1843–1855. [Google Scholar] [CrossRef]
- Bárez-López, S.; Grijota-Martínez, C.; Ausó, E.; Fernández-de Frutos, M.; Montero-Pedrazuela, A.; Guadaño-Ferraz, A. Adult Mice Lacking Mct8 and Dio2 Proteins Present Alterations in Peripheral Thyroid Hormone Levels and Severe Brain and Motor Skill Impairments. Thyroid 2019, 29, 1669–1682. [Google Scholar] [CrossRef]
- Bárez-López, S.; Bosch-García, D.; Gómez-Andrés, D.; Pulido-Valdeolivas, I.; Montero-Pedrazuela, A.; Obregon, M.J.; Guadaño-Ferraz, A. Abnormal motor phenotype at adult stages in mice lacking type 2 deiodinase. PLoS ONE 2014, 9, 103857. [Google Scholar] [CrossRef]
- Bocco, B.M.; Werneck-de-Castro, J.P.; Oliveira, K.C.; Fernandes, G.W.; Fonseca, T.L.; Nascimento, B.P.; McAninch, E.A.; Ricci, E.; Kvárta-Papp, Z.; Fekete, C.; et al. Type 2 Deiodinase Disruption in Astrocytes Results in Anxiety-Depressive-Like Behavior in Male Mice. Endocrinology 2016, 157, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Bárez-López, S.; Montero-Pedrazuela, A.; Bosch-García, D.; Venero, C.; Guadaño-Ferraz, A. Increased anxiety and fear memory in adult mice lacking type 2 deiodinase. Psychoneuroendocrinology 2017, 84, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Zhou, X.Y.; Li, C.S.; Liu, L.Q.; Huang, S.Y.; Zhou, S.N. Selenoprotein S expression in the rat brain following focal cerebral ischemia. Neurol. Sci. 2013, 34, 1671–1678. [Google Scholar] [CrossRef]
- Khurana, A.; Tekula, S.; Saifi, M.A.; Venkatesh, P.; Godugu, C. Therapeutic applications of selenium nanoparticles. Biomed. Pharmacother. 2019, 111, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Gangadoo, S.; Dinev, I.; Willson, N.L.; Moore, R.J.; Chapman, J.; Stanley, D. Nanoparticles of selenium as high bioavailable and non-toxic supplement alternatives for broiler chickens. Environ. Sci. Pollut. Res. Int. 2020, 27, 16159–16166. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhao, J.; Liu, P.; Ji, D.; Zhang, L.; Zhang, M.; Li, Y.; Xiao, Y. Preparation and in vitro evaluation of multi-target-directed selenium-chondroitin sulfate nanoparticles in protecting against the Alzheimer’s disease. Int. J. Biol. Macromol. 2020, 142, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Gholamigeravand, B.; Shahidi, S.; Amiri, I.; Samzadeh-kermani, A.; Abbasalipourkabir, R.; Soleimani, A.S. Administration of selenium nanoparticles reverses streptozotocin-induced neurotoxicity in the male rats. Metab. Brain Dis. 2021, 36, 1259–1266. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, Y.; Jin, X.; Li, Y.; Zhang, L. A novel synthesis of selenium nanoparticles encapsulated PLGA nanospheres with curcumin molecules for the inhibition of amyloid β aggregation in Alzheimer’s disease. J. Photochem. Photobiol. B 2019, 190, 98–102. [Google Scholar] [CrossRef]
- Yue, D.; Zeng, C.; Okyere, S.K.; Chen, Z.; Hu, Y. Glycine nano-selenium prevents brain oxidative stress and neurobehavioral abnormalities caused by MPTP in rats. J. Trace Elem. Med. Biol. 2021, 64, 126680. [Google Scholar] [CrossRef]
- Cong, W.; Bai, R.; Li, Y.F.; Wang, L.; Chen, C. Selenium nanoparticles as an efficient nanomedicine for the therapy of Huntington’s disease. ACS Appl. Mater. Interfaces 2019, 11, 34725–34735. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Khabatova, V.V.; Gudkov, S.V.; Plotnikov, E.Y.; Turovsky, E.A. Cytoprotective Properties of a New Nanocomplex of Selenium with Taxifolin in the Cells of the Cerebral Cortex Exposed to Ischemia/Reoxygenation. Pharmaceutics 2022, 14, 2477. [Google Scholar] [CrossRef]
- Deng, X.; Ouyang, P.; Xu, W.; Yang, E.; Bao, Z.; Wu, Y.; Gong, J.; Pan, J. Research progress of nano selenium in the treatment of oxidative stress injury during hepatic ischemia-reperfusion injury. Front. Pharmacol. 2023, 13, 1103483. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.M. Selenium Nanoparticles as Delivery System Against Various Diseases. Phyllanthaceae. Glob. J. Pharmaceu. Sci. 2023, 10, 555794. [Google Scholar]
- Abou Zaid, O.A.R.; El-Sonbaty, S.M.; Barakat, W.M. Ameliorative effect of selenium nanoparticles and ferulic acid on acrylamide-induced neurotoxicity in rats. Ann. Med. Biomed. Sci. 2017, 3, 35–45. [Google Scholar]
- Malik, A.; Ansari, J.A.; Ahmed, S.; Rani, A.; Ansari, S.Y.; Anwar, S. Emerging Selenium Nanoparticles for CNS Intervention. Biomedical Engineering. In Biotechnology—Biosensors, Biomaterials and Tissue Engineering Annual Volume 2023; IntechOpen: London, UK, 2023. [Google Scholar]
- Turovsky, E.A.; Mal’tseva, V.N.; Sarimov, R.M.; Simakin, A.V.; Gudkov, S.V.; Plotnikov, E.Y. Features of the cytoprotective effect of selenium nanoparticles on primary cortical neurons and astrocytes during oxygen-glucose deprivation and reoxygenation. Sci. Rep. 2022, 12, 1710. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Matsumoto, T.; Numakawa, Y.; Richards, M.; Yamawaki, S.; Kunugi, H. Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration. J. Toxicol. 2011, 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, M.V.; Gaidin, S.G.; Vedunova, M.V.; Babaev, A.A.; Turovsky, E.A. BDNF Overexpression Enhances the Preconditioning Effect of Brief Episodes of Hypoxia, Promoting Survival of GABAergic Neurons. Neurosci. Bull. 2020, 36, 733–760. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, S.; Dharmaraj, S. Selenium and selenoproteins: It’s role in regulation of inflammation. Inflammopharmacology 2020, 28, 667–695. [Google Scholar] [CrossRef]
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawski, K. Selenium Compounds as Novel Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 1009. [Google Scholar] [CrossRef]
- Mal’tseva, V.N.; Gudkov, S.V.; Turovsky, E.A. Modulation of the Functional State of Mouse Neutrophils by Selenium Nanoparticles In Vivo. Int. J. Mol. Sci. 2022, 23, 13651. [Google Scholar] [CrossRef]
- Alkhudhayri, A.; Al-Shaebi, E.M.; Qasem, M.A.A.; Murshed, M.; Mares, M.M.; Al-Quraishy, S.; Dkhil, M.A. Antioxidant and anti-apoptotic effects of selenium nanoparticles against murine eimeriosis. An. Acad. Bras. Ciências 2020, 92, 20191107. [Google Scholar] [CrossRef]
- Fakhri, S.; Abdian, S.; Zarneshan, S.N.; Moradi, S.Z.; Farzaei, M.H.; Abdollahi, M. Nanoparticles in Combating Neuronal Dysregulated Signaling Pathways: Recent Approaches to the Nanoformulations of Phytochemicals and Synthetic Drugs Against Neurodegenerative Diseases. Int. J. Nanomed. 2022, 17, 299–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, X.; Wei, Y. Selenium and Selenoproteins in Health. Biomolecules 2023, 13, 799. [Google Scholar] [CrossRef]
- Guo, L.; Xiao, J.; Liu, H.; Liu, H. Selenium nanoparticles alleviate hyperlipidemia and vascular injury in ApoE-deficient mice by regulating cholesterol metabolism and reducing oxidative stress. Metallomics 2020, 12, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A.; Lossi, L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. Int. J. Mol. Sci. 2022, 23, 15186. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; De Bartolo, A.; Granieri, M.C.; Rago, V.; Amelio, D.; Falbo, F.; Malivindi, R.; Mazza, R.; Cerra, M.C.; Boukhzar, L.; et al. The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress. Antioxidants 2022, 11, 571. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.R.; Fields, R.D. Activity-Dependent Gene Expression in Neurons. Neuroscientist 2021, 27, 355–366. [Google Scholar] [CrossRef]
- Park, D.J.; Koh, P.O. Diabetes aggravates decreases in hippocalcin and parvalbumin expression in focal cerebral ischemia. Neurosci. Lett. 2017, 662, 189–194. [Google Scholar] [CrossRef]
- Ebokaiwe, A.P.; Okori, S.; Nwankwo, J.O.; Ejike, C.E.C.C.; Osawe, S.O. Selenium nanoparticles and metformin ameliorate streptozotocin-instigated brain oxidative-inflammatory stress and neurobehavioral alterations in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 591–602. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Zinchenko, V.P.; Gaidin, S.G.; Turovskaya, M.V. Calcium-Binding Proteins Protect GABAergic Neurons of the Hippocampus from Hypoxia and Ischemia in vitro. Biochem. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 74–84. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Khabatova, V.V.; Gudkov, S.V.; Turovsky, E.A. Ca2+-Dependent Effects of the Selenium-Sorafenib Nanocomplex on Glioblastoma Cells and Astrocytes of the Cerebral Cortex: Anticancer Agent and Cytoprotector. Int. J. Mol. Sci. 2023, 24, 2411. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Zuidema, J.M.; Gilbert, R.J.; Gottipati, M.K. Biomaterial Approaches to Modulate Reactive Astroglial Response. Cells Tissues Organs 2018, 205, 372–395. [Google Scholar] [CrossRef] [PubMed]
- Moulson, A.J.; Squair, J.W.; Franklin, R.J.M.; Tetzlaff, W.; Assinck, P. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Front. Cell. Neurosci. 2021, 15, 703810. [Google Scholar] [CrossRef] [PubMed]
- Lira-Diaz, E.; Gonzalez-Pedroza, M.G.; Vasquez, C.; Morales-Luckie, R.A.; Gonzalez-Perez, O. Gold nanoparticles produce transient reactive gliosis in the adult brain. Neurosci. Res. 2021, 170, 76–86. [Google Scholar] [CrossRef]
- Lazic, A.; Balint, V.; Stanisavljevic Ninkovic, D.; Peric, M.; Stevanovic, M. Reactive and Senescent Astroglial Phenotypes as Hallmarks of Brain Pathologies. Int. J. Mol. Sci. 2022, 23, 4995. [Google Scholar] [CrossRef]
- Barzegar, A.; Javdani, M. Application of Chitosan Hydrogels in Traumatic Spinal Cord Injury; A Therapeutic Approach Based on the Anti-inflammatory and Antioxidant Properties of Selenium Nanoparticles. Front. Biomed. Technol. 2023, 10, 349–369. [Google Scholar]
- Varlamova, E.G.; Goltyaev, M.V.; Mal’tseva, V.N.; Turovsky, E.A.; Sarimov, R.M.; Simakin, A.V.; Gudkov, S.V. Mechanisms of the Cytotoxic Effect of Selenium Nanoparticles in Different Human Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 7798. [Google Scholar] [CrossRef]
- Khabatova, V.V.; Serov, D.A.; Tikhonova, I.V.; Astashev, M.E.; Nagaev, E.I.; Sarimov, R.M.; Matveyeva, T.A.; Simakin, A.V.; Gudkov, S.V. Selenium Nanoparticles Can Influence the Immune Response Due to Interactions with Antibodies and Modulation of the Physiological State of Granulocytes. Pharmaceutics 2022, 14, 2772. [Google Scholar] [CrossRef]
- Amani, H.; Habibey, R.; Shokri, F.; Hajmiresmail, S.J.; Akhavan, O.; Mashaghi, A.; Pazoki-Toroudi, H. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 2019, 9, 6044. [Google Scholar] [CrossRef]
- Shang, L.; Nienhaus, K.; Nienhaus, G.U. Engineered nanoparticles interacting with cells: Size matters. J. Nanobiotechnol. 2014, 12, 5. [Google Scholar] [CrossRef]
- Wang, T.; Bai, J.; Jiang, X.; Nienhaus, G.U. Cellular uptake of nanoparticles by membrane penetration: A study combining confocal microscopy with FTIR spectroelectrochemistry. ACS Nano 2012, 6, 1251–1259. [Google Scholar] [CrossRef]
- Smutná, T.; Dumková, J.; Kristeková, D.; Laštovičková, M.; Jedličková, A.; Vrlíková, L.; Dočekal, B.; Alexa, L.; Kotasová, H.; Pelková, V.; et al. Macrophage-mediated tissue response evoked by subchronic inhalation of lead oxide nanoparticles is associated with the alteration of phospholipases C and cholesterol transporters. Part. Fibre Toxicol. 2022, 19, 52. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhou, J.; Li, X.; Li, P.; Tian, G.; Zhang, C.; Zhou, D. Nano-selenium stablilized by Konjac Glucommannan and its biological activity in vitro. LWT 2022, 161, 113289. [Google Scholar] [CrossRef]
- Bajpai, S.; Mishra, M.; Kumar, H.; Tripathi, K.; Singh, S.K.; Pandey, H.P.; Singh, R.K. Effect of selenium on connexin expression, angiogenesis, and antioxidant status in diabetic wound healing. Biol. Trace Elem. Res. 2011, 144, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.R.; Talukder, M.; Li, C.X.; Zhao, Y.X.; Zhang, C.; Ge, J.; Li, J.L. Nano-selenium alleviates cadmium-induced neurotoxicity in cerebrum via inhibiting gap junction protein connexin 43 phosphorylation. Environ. Toxicol. 2023, 1–12. [Google Scholar] [CrossRef]
- Yamagata, K. Lactate Supply from Astrocytes to Neurons and its Role in Ischemic Stroke-induced Neurodegeneration. Neuroscience 2022, 481, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.S.; Frostig, R.D. Astrocyte-neuron lactate shuttle plays a pivotal role in sensory-based neuroprotection in a rat model of permanent middle cerebral artery occlusion. Sci. Rep. 2023, 13, 12799. [Google Scholar] [CrossRef]
- Gaidin, S.G.; Turovskaya, M.V.; Mal’tseva, V.N.; Zinchenko, V.P.; Blinova, E.V.; Turovsky, E.A. A complex neuroprotective effect of alpha-2-adrenergic receptor agonists in a model of cerebral ischemia–reoxygenation in vitro. Biochem. Suppl. Ser. A Membr. Cell Biol. 2019, 13, 319–333. [Google Scholar] [CrossRef]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Yang, Y.; Deng, G.; Wang, P.; Lv, G.; Mao, R.; Sun, Y.; Wang, B.; Liu, X.; Bian, L.; Zhou, D. A Selenium Nanocomposite Protects the Mouse Brain from Oxidative Injury Following Intracerebral Hemorrhage. Int. J. Nanomed. 2021, 16, 775–788. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Lin, F.; Liu, W.; Gu, R. Epigallocatechin-3-gallate selenium nanoparticles for neuroprotection by scavenging reactive oxygen species and reducing inflammation. Front. Bioeng. Biotechnol. 2022, 10, 989602. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turovsky, E.A.; Baryshev, A.S.; Plotnikov, E.Y. Selenium Nanoparticles in Protecting the Brain from Stroke: Possible Signaling and Metabolic Mechanisms. Nanomaterials 2024, 14, 160. https://doi.org/10.3390/nano14020160
Turovsky EA, Baryshev AS, Plotnikov EY. Selenium Nanoparticles in Protecting the Brain from Stroke: Possible Signaling and Metabolic Mechanisms. Nanomaterials. 2024; 14(2):160. https://doi.org/10.3390/nano14020160
Chicago/Turabian StyleTurovsky, Egor A., Alexey S. Baryshev, and Egor Y. Plotnikov. 2024. "Selenium Nanoparticles in Protecting the Brain from Stroke: Possible Signaling and Metabolic Mechanisms" Nanomaterials 14, no. 2: 160. https://doi.org/10.3390/nano14020160
APA StyleTurovsky, E. A., Baryshev, A. S., & Plotnikov, E. Y. (2024). Selenium Nanoparticles in Protecting the Brain from Stroke: Possible Signaling and Metabolic Mechanisms. Nanomaterials, 14(2), 160. https://doi.org/10.3390/nano14020160