
Short-Time Magnetron Sputtering for the Development of Carbon–Palladium Nanocomposites
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Activated Carbon Cloth Substrates
2.2. Synthesis of Carbon–Palladium Nanocomposites
2.3. Characterization Methods
3. Results & Discussion
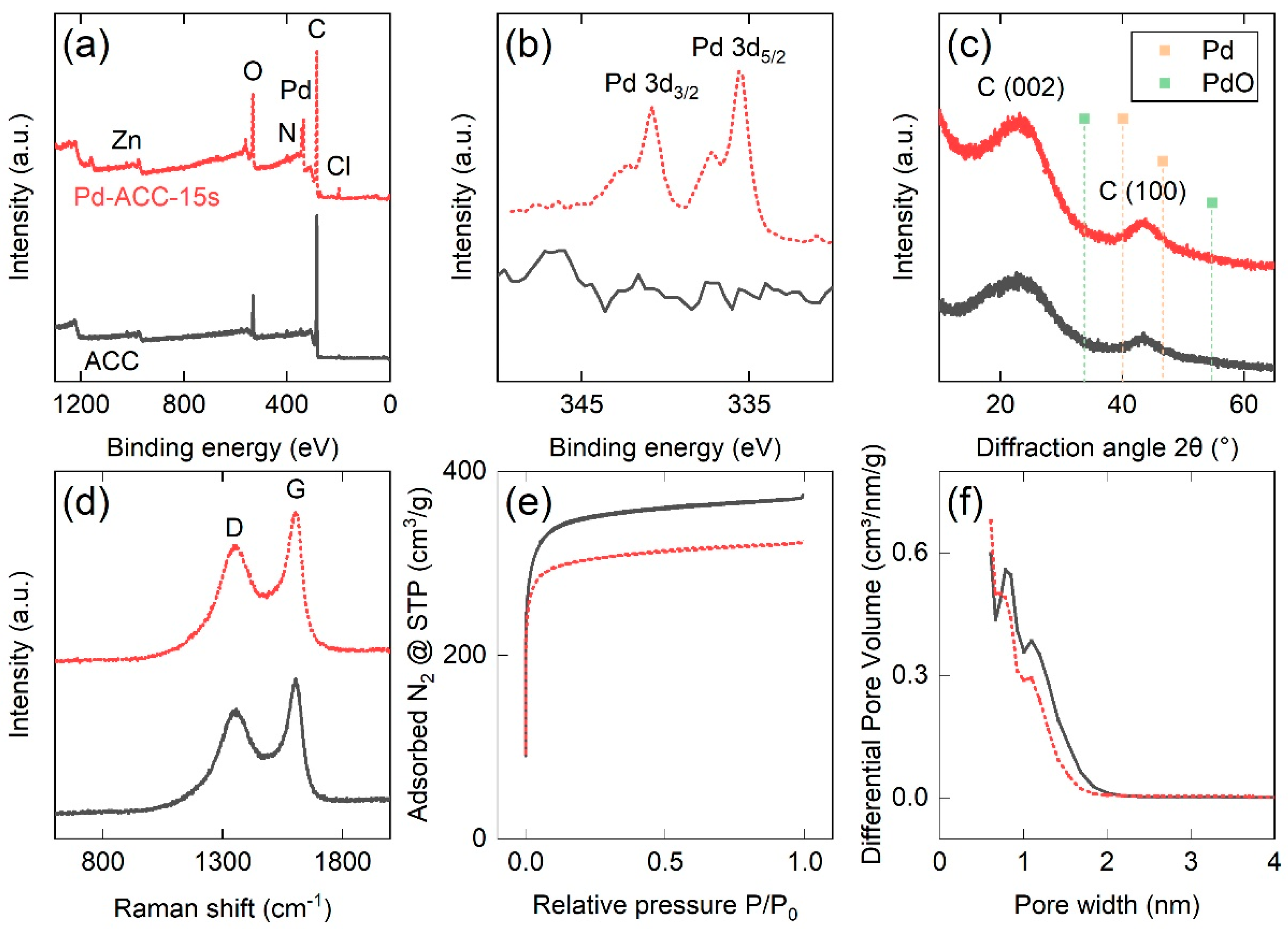
3.1. Surface Chemistry and Elemental Composition
3.2. Microstructural Characterization
3.3. Pore Structure Properties
3.4. Surface Morphology
3.5. Discussion on Growth Mechanisms
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, H.; Wen, G.; Nie, Y.; Zhang, G.; Duan, H. Flexible 3D carbon cloth as a high-performing electrode for energy storage and conversion. Nanoscale 2020, 12, 5261–5285. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, W.; Zheng, D.; Li, Z.; Zeng, Y.; Lu, X. Carbon cloth as an advanced electrode material for supercapacitors: Progress and challenges. J. Mater. Chem. A 2020, 8, 17938–17950. [Google Scholar] [CrossRef]
- Cukierman, A.L. Development and Environmental Applications of Activated Carbon Cloths. ISRN Chem. Eng. 2013, 2013, 261523. [Google Scholar] [CrossRef]
- Teradal, N.L.; Jelinek, R. Carbon Nanomaterials in Biological Studies and Biomedicine. Adv. Healthc. Mater. 2017, 6, 1700574. [Google Scholar] [CrossRef] [PubMed]
- Omanović-Mikličanin, E.; Badnjević, A.; Kazlagić, A.; Hajlovac, M. Nanocomposites: A brief review. Health Technol. 2020, 10, 51–59. [Google Scholar] [CrossRef]
- Phan, T.T.; Huynh, T.-C.; Manivasagan, P.; Mondal, S.; Oh, J. An Up-To-Date Review on Biomedical Applications of Palladium Nanoparticles. Nanomaterials 2020, 10, 66. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Parambhath, V.B.; Nagar, R.; Ramaprabhu, S. Effect of Nitrogen Doping on Hydrogen Storage Capacity of Palladium Decorated Graphene. Langmuir 2012, 28, 7826–7833. [Google Scholar] [CrossRef]
- Adams, B.D.; Chen, A. The role of palladium in a hydrogen economy. Mater. Today 2011, 14, 282–289. [Google Scholar] [CrossRef]
- Joudeh, N.; Saragliadis, A.; Koster, G.; Mikheenko, P.; Linke, D. Synthesis methods and applications of palladium nanoparticles: A review. Front. Nanotechnol. 2022, 4, 1062608. [Google Scholar] [CrossRef]
- Chamorro-Coral, W.; Caillard, A.; Brault, P.; Andreazza, P.; Coutanceau, C.; Baranton, S. The role of oxygen on the growth of palladium clusters synthesized by gas aggregation source. Plasma Process. Polym. 2019, 16, e1900006. [Google Scholar] [CrossRef]
- Pérez-Tijerina, E.; Pinilla, M.G.; Mejía-Rosales, S.; Ortiz-Méndez, U.; Torres, A.; José-Yacamán, M.; Perez-Tijerina, E.; Gracia Pinilla, M.; Mejıá-Rosales, S.; Ortiz-Mendez, U.; et al. Highly size-controlled synthesis of Au/Pd nanoparticles by inert-gas condensation. Faraday Discuss. 2008, 138, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Mattei, J.-G.; Grammatikopoulos, P.; Zhao, J.; Singh, V.; Vernieres, J.; Steinhauer, S.; Porkovich, A.; Danielson, E.; Nordlund, K.; Djurabekova, F.; et al. Gas-Phase Synthesis of Trimetallic Nanoparticles. Chem. Mater. 2019, 31, 2151–2163. [Google Scholar] [CrossRef]
- Cristoforetti, G.; Pitzalis, E.; Spiniello, R.; Ishak, R.; Giammanco, F.; Muniz-Miranda, M.; Caporali, S. Physico-chemical properties of Pd nanoparticles produced by Pulsed Laser Ablation in different organic solvents. Appl. Surf. Sci. 2012, 258, 3289–3297. [Google Scholar] [CrossRef]
- Singh, H.P.; Murr, L.E. Nucleation and growth characteristics of palladium and indium thin films. Metall. Trans. 1972, 3, 983–988. [Google Scholar] [CrossRef]
- Kelly, P.J.; Arnell, R.D. Magnetron sputtering: A review of recent developments and applications. Vacuum 2000, 56, 159–172. [Google Scholar] [CrossRef]
- Haas, V.; Birringer, R. The morphology and size of nanostructured Cu, Pd and W generated by sputtering. Nanostruct. Mater. 1992, 1, 491–504. [Google Scholar] [CrossRef]
- Tenny, K.M.; Forner-Cuenca, A.; Chiang, Y.-M.; Brushett, F.R. Comparing Physical and Electrochemical Properties of Different Weave Patterns for Carbon Cloth Electrodes in Redox Flow Batteries. J. Electrochem. Energy Convers. Storage 2020, 17, 041010. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chung, T.-Y.; Shen, C.-C.; Yu, M.-S.; Tsao, C.-S.; Shi, G.-N.; Huang, C.-C.; Ger, M.-D.; Lee, W.-L. Hydrogen storage performance in palladium-doped graphene/carbon composites. Int. J. Hydrog. Energy 2013, 38, 3681–3688. [Google Scholar] [CrossRef]
- Erdogan, F.O.; Celik, C.; Turkmen, A.C.; Sadak, A.E.; Cucu, E. Hydrogen sorption studies of palladium decorated graphene nanoplatelets and carbon samples. Int. J. Hydrog. Energy 2023, 48, 21476–21486. [Google Scholar] [CrossRef]
- Chen, A.; Ostrom, C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999–12044. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Y.; Hou, H.; You, T. Simultaneous electrochemical determination of dopamine, uric acid and ascorbic acid using palladium nanoparticle-loaded carbon nanofibers modified electrode. Biosens. Bioelectron. 2008, 24, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.K.; Krishnan, S.; Yoshimura, M.; Kumar, A. Pd nanoparticles and thin films for room temperature hydrogen sensor. Nanoscale Res. Lett. 2009, 4, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yu, Y.; Zhang, L.; Zhu, J.; Zhao, X.; Zhang, W. Flexible and Highly Sensitive Hydrogen Sensor Based on Organic Nanofibers Decorated by Pd Nanoparticles. Sensors 2019, 19, 1290. [Google Scholar] [CrossRef]
- Pócza, J.F.; Barna, A.; Barna, P.B.; Pozsgai, I.; Radnóczi, G. In situ electron microscopy of thin film growth. Jpn. J. Appl. Phys. 1974, 13, 525. [Google Scholar] [CrossRef]
- Kostoglou, N.; Koczwara, C.; Prehal, C.; Terziyska, V.; Babic, B.; Matovic, B.; Constantinides, G.; Tampaxis, C.; Charalambopoulou, G.; Steriotis, T.; et al. Nanoporous activated carbon cloth as a versatile material for hydrogen adsorption, selective gas separation and electrochemical energy storage. Nano Energy 2017, 40, 49–64. [Google Scholar] [CrossRef]
- Babić, B.M.; Milonjić, S.K.; Polovina, M.J.; Kaludierović, B. V Point of zero charge and intrinsic equilibrium constants of activated carbon cloth. Carbon 1999, 37, 477–481. [Google Scholar] [CrossRef]
- Polovina, M.; Babić, B.; Kaluderović, B.; Dekanski, A. Surface characterization of oxidized activated carbon cloth. Carbon 1997, 35, 1047–1052. [Google Scholar] [CrossRef]
- Kölbl, L.; Mitterer, C.; Franz, R. Synthesis of crystalline silver niobate thin films opening pathways for future process development. Vacuum 2023, 213, 112077. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific Inc. Palladium X-ray Photoelectron Spectra, Palladium Electron Configuration, and Other Elemental Information. Available online: https://www.thermofisher.com/uk/en/home/materials-science/learning-center/periodic-table/transition-metal/palladium.html (accessed on 24 November 2023).
- Briggs, D.; Seah, M.P. (Eds.) Practical Surface Analysis, Auger and X-ray Photoelectron Spectroscopy (Volume 1), 2nd ed.; Wiley: Hoboken, NJ, USA, 1996; ISBN 978-0471920816. [Google Scholar]
- Kibis, L.S.; Titkov, A.I.; Stadnichenko, A.I.; Koscheev, S.V.; Boronin, A.I. X-ray photoelectron spectroscopy study of Pd oxidation by RF discharge in oxygen. Appl. Surf. Sci. 2009, 255, 9248–9254. [Google Scholar] [CrossRef]
- Mason, M.G.; Gerenser, L.J.; Lee, S.-T. Electronic Structure of Catalytic Metal Clusters Studied by X-ray Photoemission Spectroscopy. Phys. Rev. Lett. 1977, 39, 288–291. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Kostoglou, N.; Liao, C.W.; Wang, C.Y.; Kondo, J.N.; Tampaxis, C.; Steriotis, T.; Giannakopoulos, K.; Kontos, A.G.; Hinder, S.; Baker, M.; et al. Effect of Pt nanoparticle decoration on the H2 storage performance of plasma-derived nanoporous graphene. Carbon 2021, 171, 294–305. [Google Scholar] [CrossRef]
- Holec, D.; Kostoglou, N.; Tampaxis, C.; Babic, B.; Mitterer, C.; Rebholz, C. Theory-guided metal-decoration of nanoporous carbon for hydrogen storage applications. Surf. Coat. Technol. 2018, 351, 42–49. [Google Scholar] [CrossRef]
- Arroyo-Ramírez, L.; Figueroa, Y.; Rodríguez, D.; Otaño, W.; Cabrera, C.R. Palladium Nanostructures Synthesis by Sputtering Deposition on HOPG Surfaces. ECS Trans. 2010, 28, 1. [Google Scholar] [CrossRef]
- Pantojas, V.M.; Rodríguez, D.; Morell, G.; Rivera, A.; Ortiz, C.; Santiago-Avilés, J.J.; Otaño, W. Synthesis of palladium with different nanoscale structures by sputtering deposition onto fiber templates. J. Nanophotonics 2008, 2, 021925. [Google Scholar] [CrossRef]
- Sitnikova, N.A.; Solovieva, A.O.; Permyakova, E.S.; Sheveyko, A.N.; Shtansky, D.V.; Manakhov, A.M. Silver Ions Incorporation into Nanofibers for Enhanced hMSC Viability. Chemistry 2022, 4, 931–939. [Google Scholar] [CrossRef]
- Wu, L.; Wu, H.; Wang, X.; Zhong, H.; Wang, Z.; Cai, G.; Jiang, C.; Ren, F. A general method for large-scale fabrication of metal nanoparticles embedded N-doped carbon fiber cloth with highly efficient hydrogen production in all pH range. Electrochim. Acta 2020, 353, 136475. [Google Scholar] [CrossRef]
- Mukherjee, S.; Gall, D. Structure zone model for extreme shadowing conditions. Thin Solid Films 2013, 527, 158–163. [Google Scholar] [CrossRef]
- Wang, J.; Huang, H.; Kesapragada, S.V.; Gall, D. Growth of Y-Shaped Nanorods through Physical Vapor Deposition. Nano Lett. 2005, 5, 2505–2508. [Google Scholar] [CrossRef]
- Petrov, I.; Barna, P.B.; Hultman, L.; Greene, J.E. Microstructural evolution during film growth. J. Vac. Sci. Technol. A 2003, 21, S117–S128. [Google Scholar] [CrossRef]
- Stephenson, A.W.; Baddeley, C.J.; Tikhov, M.S.; Lambert, R.M. Nucleation and growth of catalytically active Pd islands on Au(111)-22 × studied by scanning tunnelling microscopy. Surf. Sci. 1998, 398, 172–183. [Google Scholar] [CrossRef]
- Bräuer, G.; Szyszka, B.; Vergöhl, M.; Bandorf, R. Magnetron sputtering—Milestones of 30 years. Vacuum 2010, 84, 1354–1359. [Google Scholar] [CrossRef]
- Ludwig, R.; Kukla, R.; Josephson, E. Vacuum Web Coating—State of the Art and Potential for Electronics. Proc. IEEE 2005, 93, 1483–1490. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Material | SBET [m2/g] | Smicro [m2/g] | VGurvich [cm³/g] | Vmicro [cm³/g] | d50 [nm] |
|---|---|---|---|---|---|
| ACC | 1365 | 1279 | 0.57 | 0.50 | 0.77 |
| Pd-ACC-15s | 1202 | 1131 | 0.50 | 0.44 | 0.67 |
| Change | −11.9% | −11.6% | −12.3% | −12% | −12.1% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knabl, F.; Kostoglou, N.; Terziyska, V.; Hinder, S.; Baker, M.; Bousser, E.; Rebholz, C.; Mitterer, C. Short-Time Magnetron Sputtering for the Development of Carbon–Palladium Nanocomposites. Nanomaterials 2024, 14, 164. https://doi.org/10.3390/nano14020164
Knabl F, Kostoglou N, Terziyska V, Hinder S, Baker M, Bousser E, Rebholz C, Mitterer C. Short-Time Magnetron Sputtering for the Development of Carbon–Palladium Nanocomposites. Nanomaterials. 2024; 14(2):164. https://doi.org/10.3390/nano14020164
Chicago/Turabian StyleKnabl, Florian, Nikolaos Kostoglou, Velislava Terziyska, Steven Hinder, Mark Baker, Etienne Bousser, Claus Rebholz, and Christian Mitterer. 2024. "Short-Time Magnetron Sputtering for the Development of Carbon–Palladium Nanocomposites" Nanomaterials 14, no. 2: 164. https://doi.org/10.3390/nano14020164