
CO2 and SO2 Capture by Cryptophane-111 Porous Liquid: Insights from Molecular Dynamics Simulations and Computational Chemistry



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
3. Results and Discussion
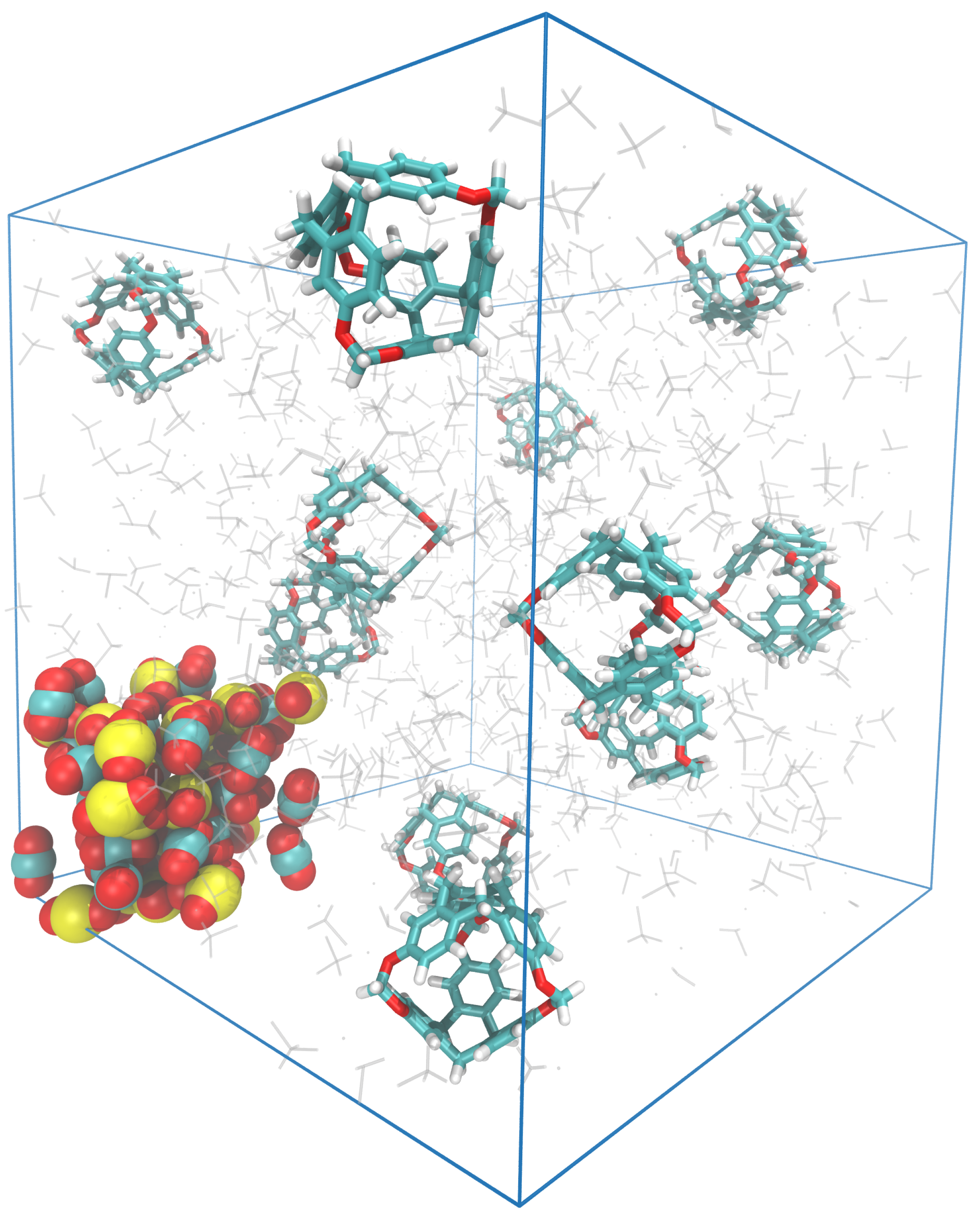
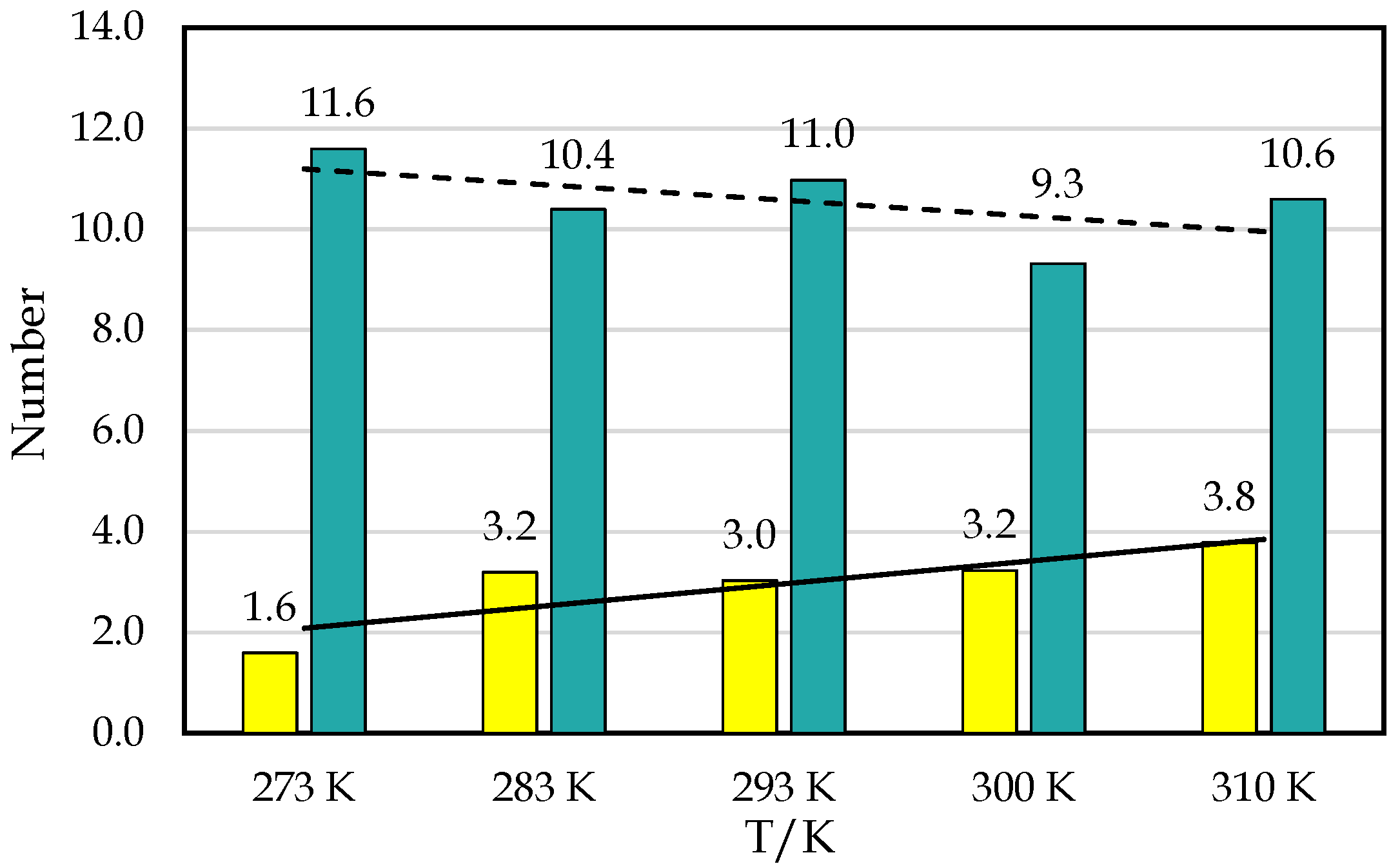
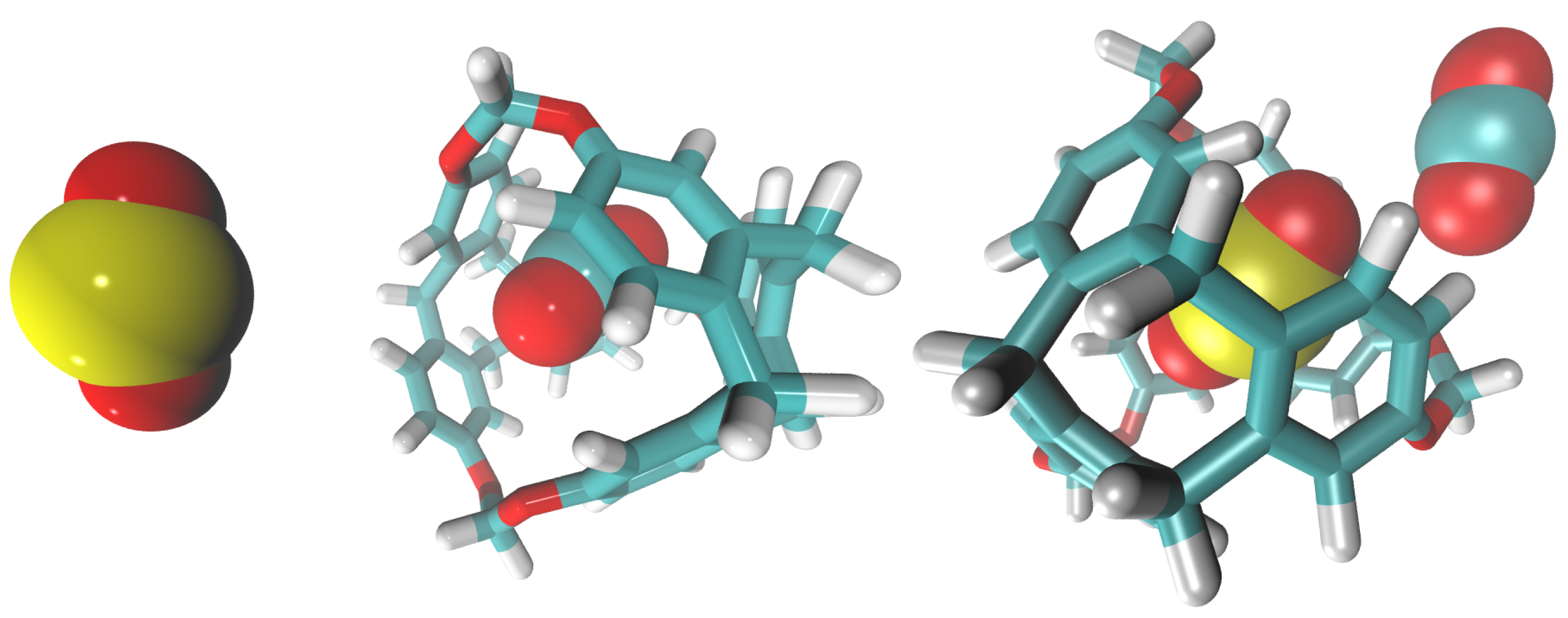
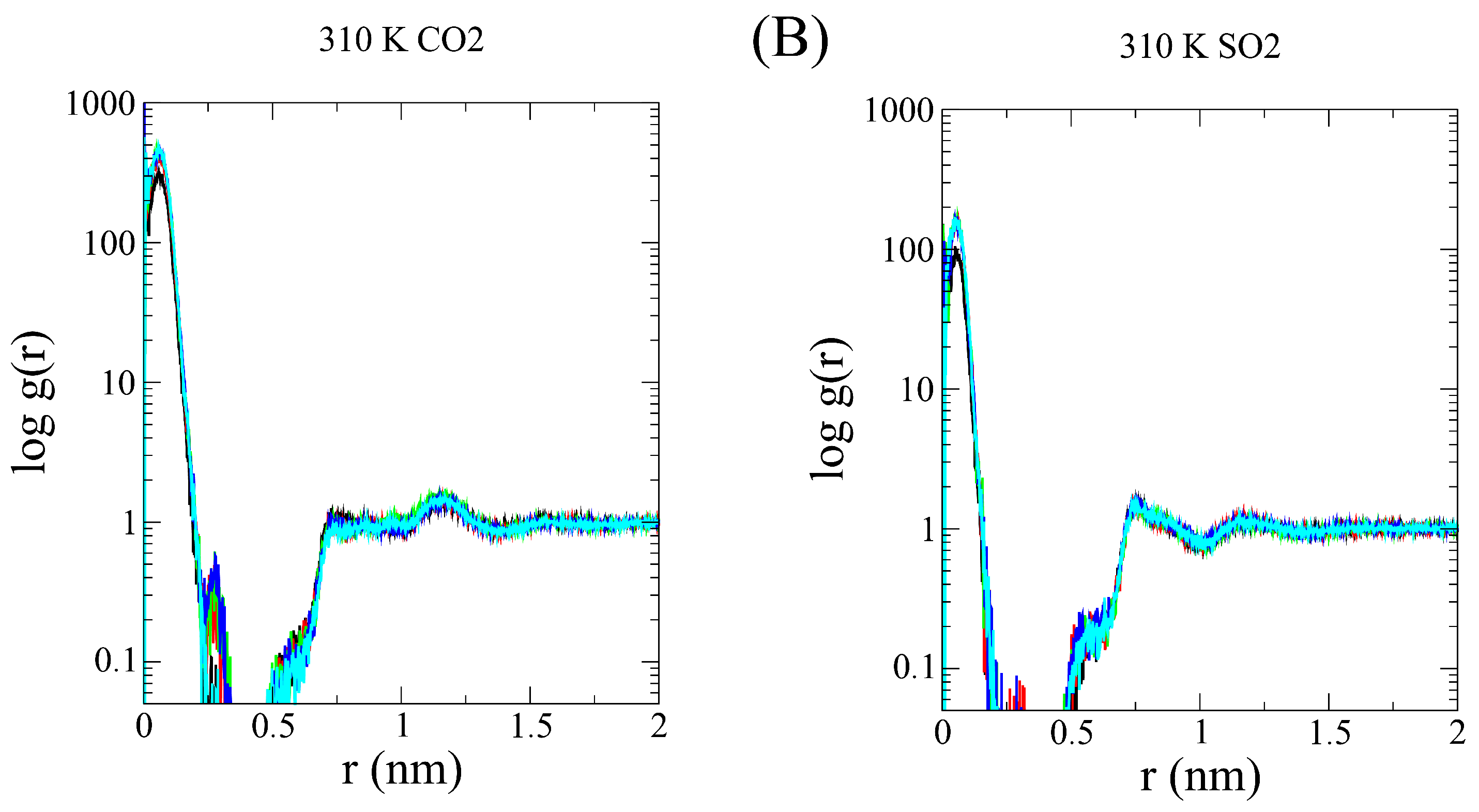
3.1. Simulation Analysis
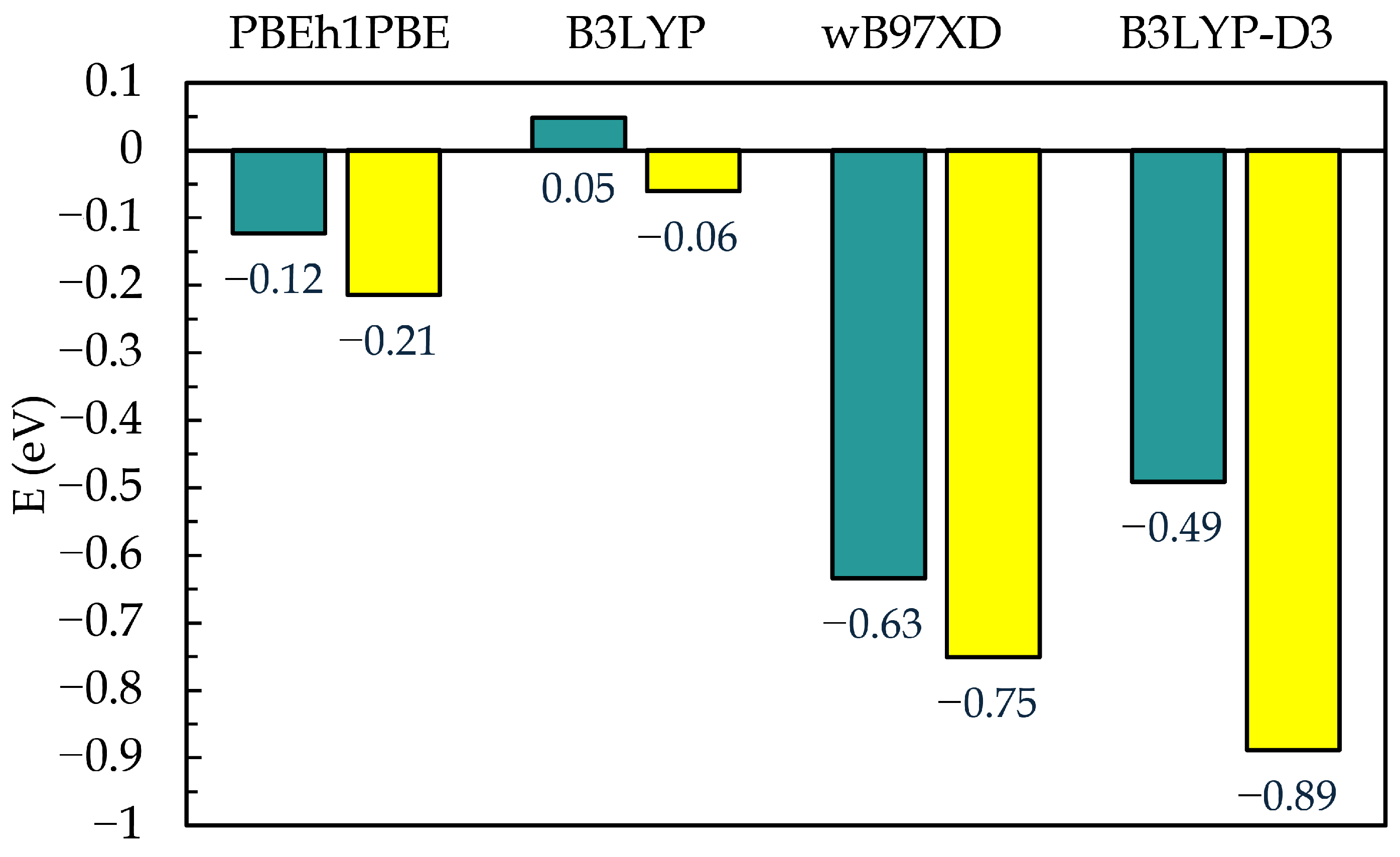
3.2. DFT Calculations and Stability
3.3. Future Approaches
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATB | Automatic Topology Builder |
| DCM | dichloromethane |
| C-111 | cryptophane-111 |
| MOF | Metal Organic Framework |
| PDB | Protein Data Bank |
| PL | porous liquid |
References
- Barnes, J.; Dove, M.; Lahsen, M.; Mathews, A.; McElwee, P.; McIntosh, R.; Moore, F.; O’Reilly, J.; Orlove, B.; Puri, R.; et al. Contribution of anthropology to the study of climate change. Nat. Clim. Change 2013, 3, 541–544. [Google Scholar] [CrossRef]
- Anukwonke, C.C.; Tambe, E.B.; Nwafor, D.C.; Malik, K.T. Climate Change and Interconnected Risks to Sustainable Development. In Climate Change: The Social and Scientific Construct; Bandh, S.A., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 71–86. [Google Scholar] [CrossRef]
- Lawrence, J.; Blackett, P.; Cradock-Henry, N.A. Cascading climate change impacts and implications. Clim. Risk Manag. 2020, 29, 100234. [Google Scholar] [CrossRef]
- Räsänen, A.; Juhola, S.; Nygren, A.; Käkönen, M.; Kallio, M.; Monge, A.; Kanninen, M. Climate change, multiple stressors and human vulnerability: A systematic review. Reg. Environ. Change 2016, 16, 2291–2302. [Google Scholar] [CrossRef]
- Shukla, P.; Skea, J.; Slade, R.; Khourdajie, A.A.; van Diemen, R.; McCollum, D.; Pathak, M.; Some, S.; Vyas, P.; Fradera, R.; et al. IPCC, 2022: Climate Change 2022: Mitigation of Climate Change. Contribution of Working Group III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2022. [Google Scholar]
- Wang, D.; Xin, Y.; Yao, D.; Li, X.; Ning, H.; Zhang, H.; Wang, Y.; Ju, X.; He, Z.; Yang, Z.; et al. Shining Light on Porous Liquids: From Fundamentals to Syntheses, Applications and Future Challenges. Adv. Funct. Mater. 2022, 32, 2104162. [Google Scholar] [CrossRef]
- O’Reilly, N.; Giri, N.; James, S.L. Porous liquids. Chem.—Eur. J. 2007, 13, 3020–3025. [Google Scholar] [CrossRef] [PubMed]
- James, S.L. The dam bursts for porous liquids. Adv. Mater. 2016, 28, 5712–5716. [Google Scholar] [CrossRef]
- Bennett, T.D.; Coudert, F.X.; James, S.L.; Cooper, A.I. The changing state of porous materials. Nat. Mater. 2021, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Jie, K.; Zhou, Y.; Ryan, H.P.; Dai, S.; Nitschke, J.R. Porous liquids: Engineering permanent porosity into liquids. Adv. Mater. 2021, 33, 2170136. [Google Scholar] [CrossRef]
- Melaugh, G.; Giri, N.; Davidson, C.E.; James, S.L.; Del Pópolo, M.G. Designing and understanding permanent microporosity in liquids. Phys. Chem. Chem. Phys. 2014, 16, 9422–9431. [Google Scholar] [CrossRef]
- Liu, H.; Liu, B.; Lin, L.C.; Chen, G.; Wu, Y.; Wang, J.; Gao, X.; Lv, Y.; Pan, Y.; Zhang, X.; et al. A hybrid absorption-adsorption method to efficiently capture carbon. Nat. Commun. 2014, 5, 5147. [Google Scholar] [CrossRef]
- Avila, J.; Červinka, C.; Dugas, P.Y.; Pádua, A.A.H.; Costa Gomes, M. Porous ionic liquids: Structure, stability, and gas absorption mechanisms. Adv. Mater. Interfaces 2021, 8, 2001982. [Google Scholar] [CrossRef]
- Li, P.; Chen, H.; Schott, J.A.; Li, B.; Zheng, Y.; Mahurin, S.M.; Jiang, D.E.; Cui, G.; Hu, X.; Wang, Y.; et al. Porous liquid zeolites: Hydrogen bonding-stabilized H-ZSM-5 in branched ionic liquids. Nanoscale 2019, 11, 1515–1519. [Google Scholar] [CrossRef]
- Avila, J.; Lepre, L.F.; Santini, C.; Tiano, M.; Denis-Quanquin, S.; Szeto, K.C.; Padua, A.; Costa Gomes, M. High-performance porous ionic liquids for low pressure CO2 capture. Angew. Chem. 2021, 60, 12876–12882. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Zhang, L.; Liu, C.; Wu, F.; Wang, Y.; Wang, Z.; Zhao, Z.; Wu, W.; Liang, Y.; et al. An in situ coupling strategy toward porous carbon liquid with permanent porosity. Small 2021, 17, e2006687. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.Z.; Fuoco, A. Porous liquids—Future for CO2 Capture and Separation? Curr. Res. Green Sustain. Chem. 2021, 4, 100070. [Google Scholar] [CrossRef]
- Brand, M.C.; Rankin, N.; Cooper, A.I.; Greenaway, R.L. Photoresponsive Type III Porous Liquids. Chem.—Eur. J. 2023, 29, e202202848. [Google Scholar] [CrossRef]
- Sarmad, S.; Nikjoo, D.; Mikkola, J.P. Innovative CO2 capture technologies: Exploring the potential of porous liquids containing deep eutectic solvents and hypercrosslinked polymers. Sep. Purif. Technol. 2025, 352, 128189. [Google Scholar] [CrossRef]
- Cheng, B.; Ma, J.; Ju, X.; Wei, F.; Cheng, W.; Li, P.; Liu, X. Metal-organic framework-based porous liquids via surface functionalization engineering for selective gas adsorption. Sep. Purif. Technol. 2024, 350, 127994. [Google Scholar] [CrossRef]
- Ji, D.; Xue, C.; Wen, Y.; Zhao, Y.; Gong, W.; Li, Y. Preparation and CO2 adsorption behavior of ZIF-67-based porous liquids: A molecular dynamics study. Sep. Purif. Technol. 2024, 347, 127564. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, S. The Role of the Nanoconfinement Effect in the Adsorption of Carbon Dioxide by Porous Liquids. ACS Sustain. Chem. Eng. 2024, 12, 10455–10465. [Google Scholar] [CrossRef]
- Oltean, M. Theoretical Study of Chemical Reactivity: From Nano-Objects to Follow the Entropy via Metadynamics Simulations. Ph.D. Dissertation, Université Joseph-Fourier-Grenoble I, Saint-Martin-d’Hères, France, 2010. [Google Scholar]
- Dou, S.; Liu, K.; Feng, Y.; Wang, X.; Zhang, B.; Yang, C.; Sun, M.; Han, J.; Duan, E. A new strategy to effectively capture and recover SO2 based on the functional porous liquids. J. Clean. Prod. 2024, 467, 143006. [Google Scholar] [CrossRef]
- Collado, P.; Piñeiro, M.M.; Pérez-Rodríguez, M. Molecular Simulation of CO2 and H2 Encapsulation in a Nanoscale Porous Liquid. Nanomaterials 2023, 13, 409. [Google Scholar] [CrossRef]
- Collado, P.; Piñeiro, M.M.; Pérez-Rodríguez, M. Molecular Simulation of SO2 Separation and Storage Using a Cryptophane-Based Porous Liquid. Int. J. Mol. Sci. 2024, 25, 2718. [Google Scholar] [CrossRef]
- Fogarty, H.A.; Berthault, P.; Brotin, T.; Huber, G.; Desvaux, H.; Dutasta, J.P. A cryptophane core optimized for xenon encapsulation. J. Am. Chem. Soc. 2007, 129, 10332–10333. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.I.; Lapidus, S.H.; Kane, C.M.; Holman, K.T. Extreme confinement of xenon by cryptophane-111 in the solid state. Angew. Chem. 2015, 127, 1491–1495. [Google Scholar] [CrossRef]
- Mecozzi, S.; Rebek, J., Jr. The 55 % solution: A formula for molecular recognition in the liquid state. Chemistry 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- El-Ayle, G.; Holman, K.T. Cryptophanes; Elsevier: Amsterdam, The Netherlands, 2017; pp. 199–249. [Google Scholar] [CrossRef]
- Buffeteau, T.; Pitrat, D.; Daugey, N.; Calin, N.; Jean, M.; Vanthuyne, N.; Ducasse, L.; Wien, F.; Brotin, T. Chiroptical properties of cryptophane-111. Phys. Chem. Chem. Phys. 2017, 19, 18303–18310. [Google Scholar] [CrossRef] [PubMed]
- Shirono, K.; Morimatsu, T.; Takemura, F. Gas Solubilities (CO2, O2, Ar, N2, H2, and He) in Liquid Chlorinated Methanes. J. Chem. Eng. Data 2008, 53, 1867–1871. [Google Scholar] [CrossRef]
- Vonderheiden, F.H.; Eldridge, J.W. The System Carbon Dioxide-Methylene Chloride. Solubility, Vapor Pressure, Liquid Density, and Activity Coefficients. J. Chem. Eng. Data 1963, 8, 20–21. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; Otero-Fernández, J.; Comesaña, A.; Fernández-Fernández, A.M.; Piñeiro, M.M. Simulation of Capture and Release Processes of Hydrogen by β-Hydroquinone Clathrate. ACS Omega 2018, 3, 18771–18782. [Google Scholar] [CrossRef]
- Torré, J.P.; Gornitzka, H.; Coupan, R.; Dicharry, C.; Pérez-Rodríguez, M.; Comesaña, A.; Piñeiro, M.M. Insights into the Crystal Structure and Clathration Selectivity of Organic Clathrates Formed with Hydroquinone and (CO2 + CH4) Gas Mixtures. J. Phys. Chem. C 2019, 123, 14582–14590. [Google Scholar] [CrossRef]
- Fernández-Fernández, A.M.; Conde, M.M.; Pérez-Sánchez, G.; Pérez-Rodríguez, M.; Piñeiro, M.M. Molecular simulation of methane hydrate growth confined into a silica pore. J. Mol. Liq. 2022, 362, 119698. [Google Scholar] [CrossRef]
- Méndez-Morales, T.; Montes-Campos, H.; Pérez-Rodríguez, M.; Piñeiro, M.M. Evaluation of hydrogen storage ability of hydroquinone clathrates using molecular simulations. J. Mol. Liq. 2022, 360, 119487. [Google Scholar] [CrossRef]
- Rodríguez-García, B.; Piñeiro, M.M.; Pérez-Rodríguez, M. Diffusion and dynamics of noble gases in hydroquinone clathrate channels. J. Chem. Phys. 2023, 158, 044503. [Google Scholar] [CrossRef]
- Rodríguez García, B.; Piñeiro, M.M.; Pérez-Rodríguez, M. Influence of Lennard-Jones Parameters in the Temperature Dependence of Real Gases Diffusion through Nanochannels. Nanomaterials 2023, 13, 1534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, Q.; Tao, H.; Wang, S.; Lin, W.; Wang, X.; Li, H.; Shi, G.; Wang, C. Highly Selective Absorption of Low-Concentration SO2 over CO2 in Flue Gas through Cation-Tunable Protic Ionic Liquids. Energy Fuels 2023, 37, 1180–1186. [Google Scholar] [CrossRef]
- Fan, Y.L.; Zhang, H.P.; Yin, M.J.; Krishna, R.; Feng, X.F.; Wang, L.; Luo, M.B.; Luo, F. High Adsorption Capacity and Selectivity of SO2 over CO2 in a Metal–Organic Framework. Inorg. Chem. 2021, 60, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.F.R.; McClung, M.; McReynolds, C.; Daly, H.; Greer, A.J.; Jacquemin, J.; Hardacre, C. Understanding the Competitive Gas Absorption of CO2 and SO2 in Superbase Ionic Liquids. Ind. Eng. Chem. Res. 2018, 57, 17033–17042. [Google Scholar] [CrossRef]
- Choi, W.Y.; Lee, D.; Jang, K.; Yoo, Y.; Park, J. A novel process for selective absorption of CO2/SO2 mixture gas with a single absorbent derived from seawater-based industrial wastewater. Desalination 2023, 560, 116661. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, X.; Li, H.; Peng, J.; Wu, Y.; Hu, X. Cyano-Containing Protic Ionic Liquids for Highly Selective Absorption of SO2 from CO2: Experimental Study and Theoretical Analysis. Ind. Eng. Chem. Res. 2016, 55, 11012–11021. [Google Scholar] [CrossRef]
- Qinlan, L.; Feng, B.; Zhuhan, L.; Zhou, Q.; Zhang, Y.; Li, N. Experimental study on simultaneous absorption and desorption of CO2/SO2/NOx using aqueous MDEA + DMSO solutions. Energy Fuels 2018, 32, 3647–3659. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated force field Topology Builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated topology builder version 3.0: Prediction of solvation free enthalpies in water and hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput.-Aided Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef]
- Potoff, J.J.; Siepmann, J.I. Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Hamani, A.W.S.; Bazile, J.P.; Hoang, H.; Luc, H.T.; Daridon, J.L.; Galliero, G. Thermophysical properties of simple molecular liquid mixtures: On the limitations of some force fields. J. Mol. Liq. 2020, 303, 112663. [Google Scholar] [CrossRef]
- Pérez-Sánchez, G.; González-Salgado, D.; Piñeiro, M.M.; Vega, C. Fluid-solid equilibrium of carbon dioxide as obtained from computer simulations of several popular potential models: The role of the quadrupole. J. Chem. Phys. 2013, 138, 084506. [Google Scholar] [CrossRef]
- Míguez, J.M.; Conde, M.M.; Torré, J.P.; Blas, F.J.; Piñeiro, M.M.; Vega, C. Molecular dynamics simulation of CO2 hydrates: Prediction of three phase coexistence line. J. Chem. Phys. 2015, 142, 124505. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M. Gromacs: A parallel computer for molecular dynamics simulations. In Physics Computing 92; DeGroot, R., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale: International Conference on Exascale Applications and Software; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J.; Gullingsrud, J.; Grayson, P.; Schulten, K. A System for Interactive Molecular Dynamics Simulation. In Proceedings of the 2001 ACM Symposium on Interactive 3D Graphics; Hughes, J.F., Séquin, C.H., Eds.; ACM: New York, NY, USA, 2001; pp. 191–194. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ernzerhof, M.; Perdew, J.P. Generalized gradient approximation to the angle- and system-averaged exchange hole. J. Chem. Phys. 1998, 109, 3313–3320. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Bálint, D.; Jäntschi, L. Comparison of Molecular Geometry Optimization Methods Based on Molecular Descriptors. Mathematics 2021, 9, 2855. [Google Scholar] [CrossRef]
- Cukras, J.; Sadlej, J. The influence of the dispersion corrections on the performance of DFT method in modeling HNgY noble gas molecules and their complexes. Chem. Phys. Lett. 2018, 691, 319–324. [Google Scholar] [CrossRef]
- Patel, D.; Margolese, D.; Dykea, T.R. Electric dipole moment of SO2 in ground and excited vibrational states. J. Chem. Phys. 1979, 70, 2740–2747. [Google Scholar] [CrossRef]

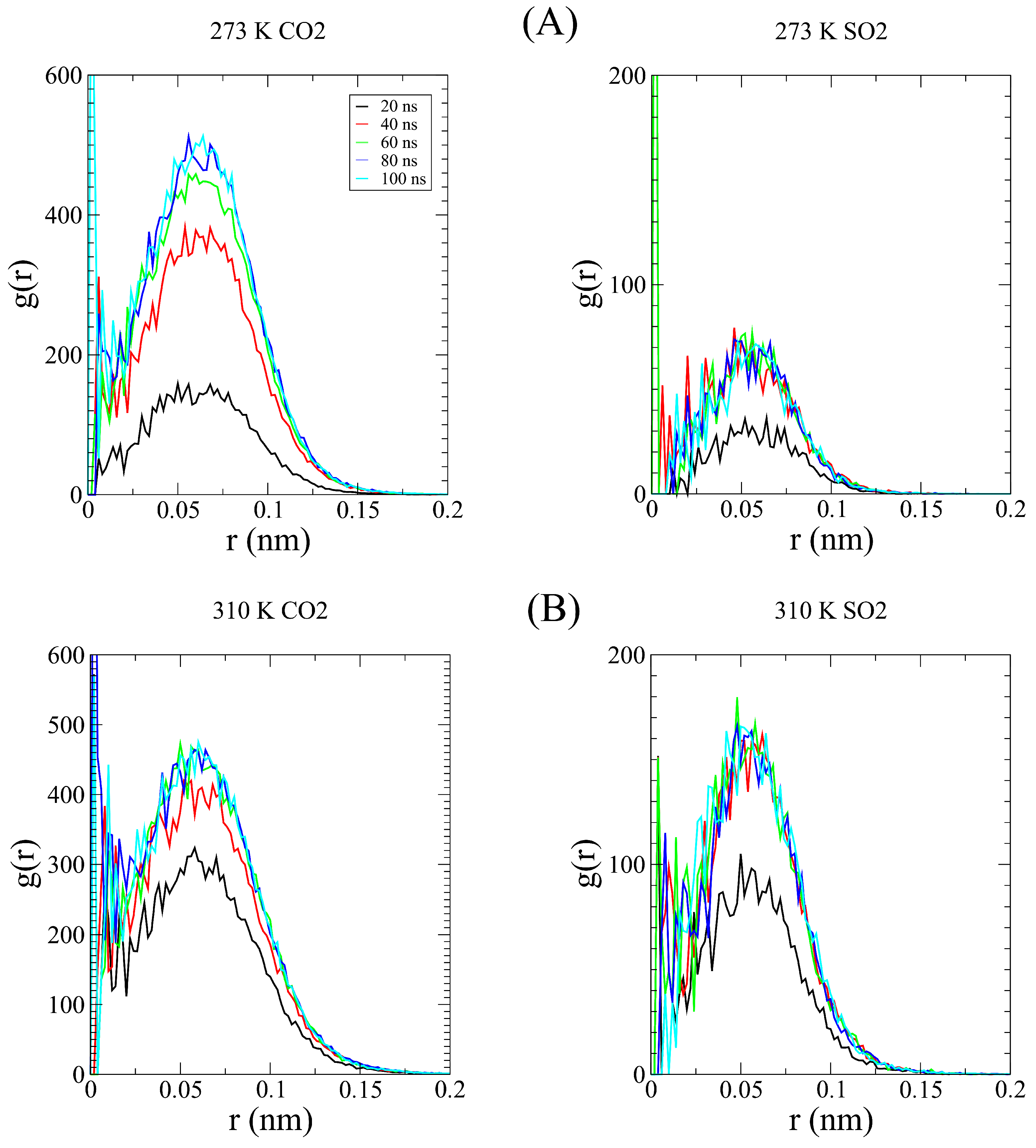
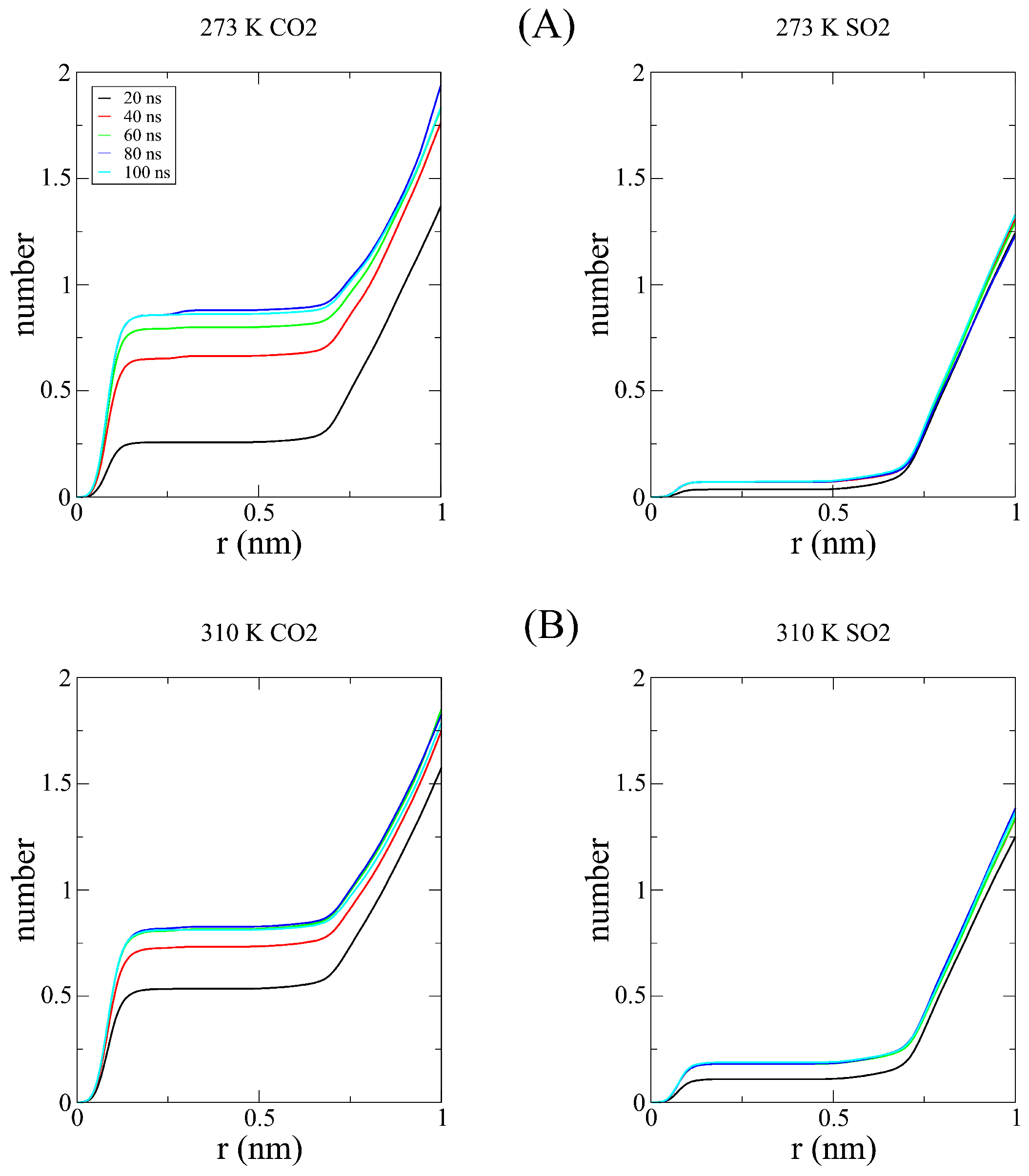
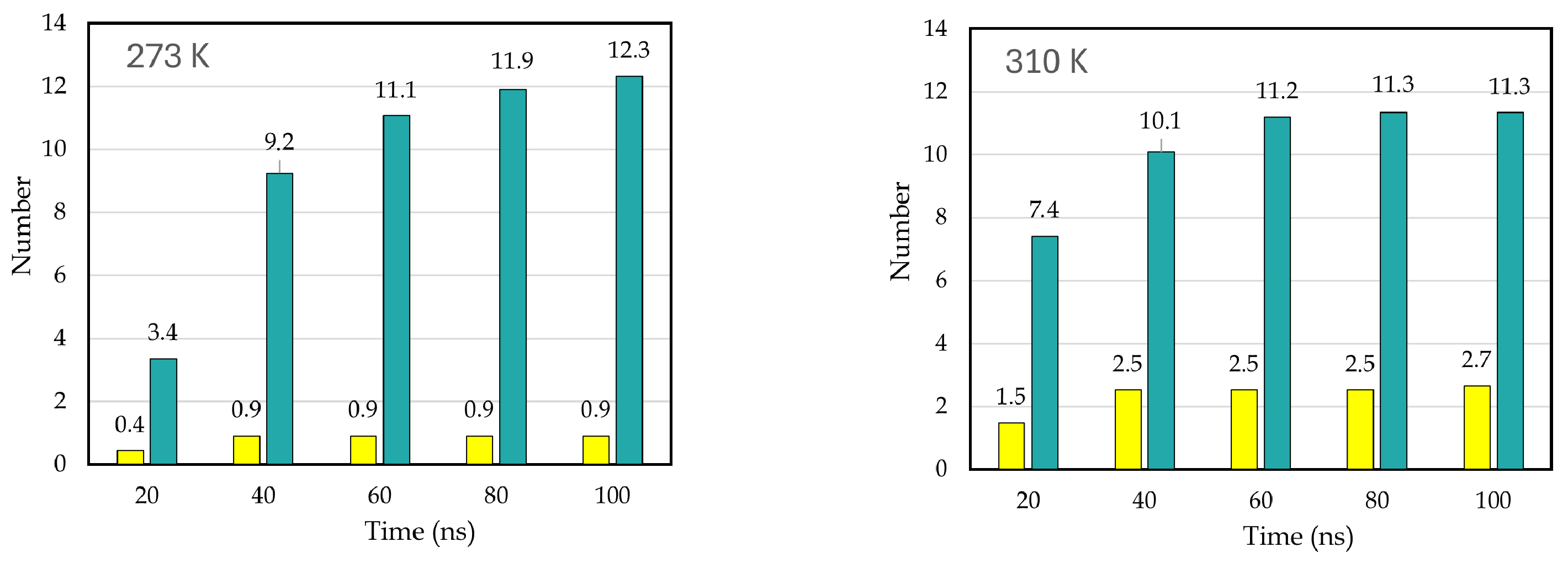
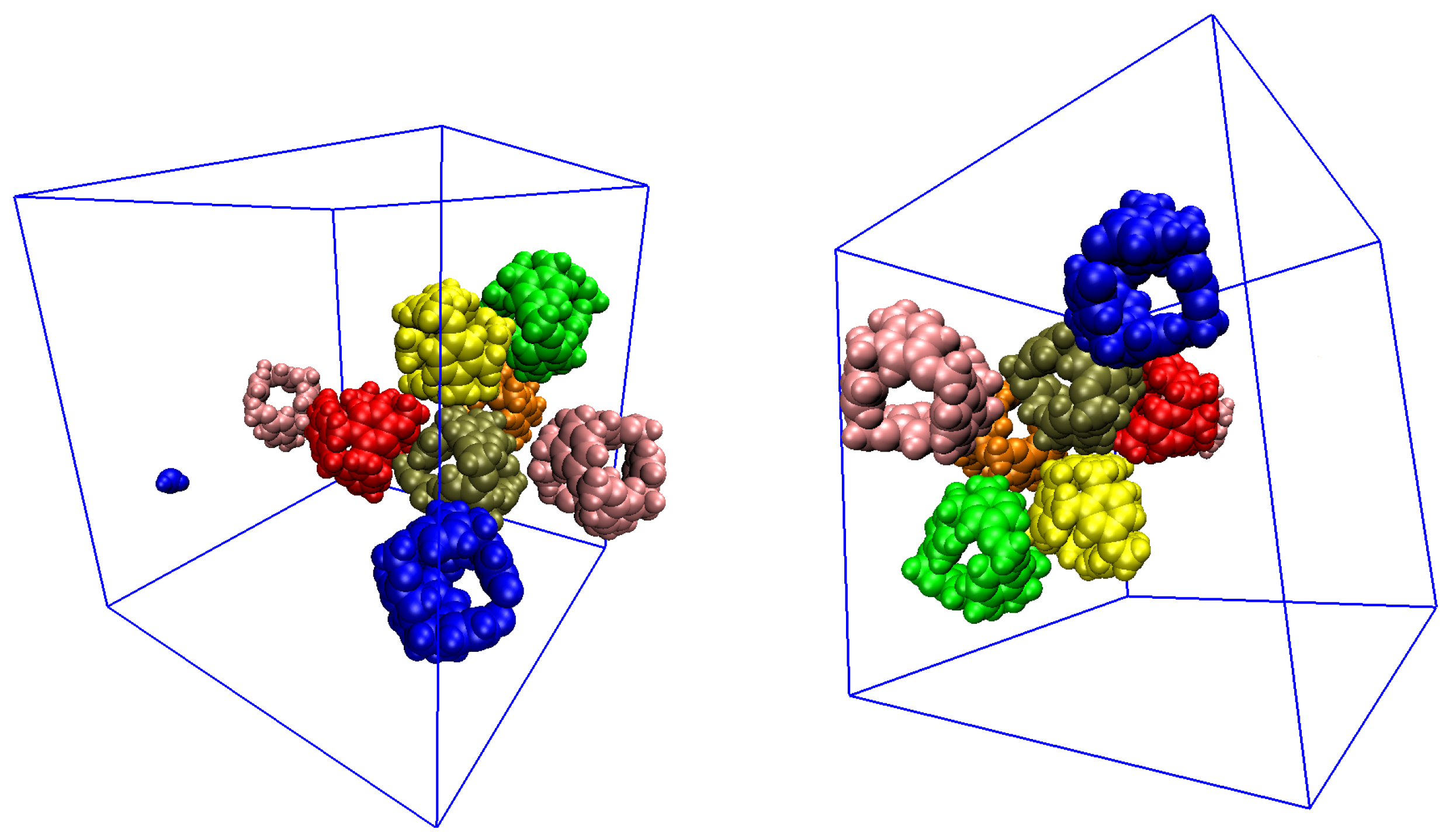
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collado, P.; Piñeiro, M.M.; Pérez-Rodríguez, M. CO2 and SO2 Capture by Cryptophane-111 Porous Liquid: Insights from Molecular Dynamics Simulations and Computational Chemistry. Nanomaterials 2025, 15, 616. https://doi.org/10.3390/nano15080616
Collado P, Piñeiro MM, Pérez-Rodríguez M. CO2 and SO2 Capture by Cryptophane-111 Porous Liquid: Insights from Molecular Dynamics Simulations and Computational Chemistry. Nanomaterials. 2025; 15(8):616. https://doi.org/10.3390/nano15080616
Chicago/Turabian StyleCollado, Pablo, Manuel M. Piñeiro, and Martín Pérez-Rodríguez. 2025. "CO2 and SO2 Capture by Cryptophane-111 Porous Liquid: Insights from Molecular Dynamics Simulations and Computational Chemistry" Nanomaterials 15, no. 8: 616. https://doi.org/10.3390/nano15080616
APA StyleCollado, P., Piñeiro, M. M., & Pérez-Rodríguez, M. (2025). CO2 and SO2 Capture by Cryptophane-111 Porous Liquid: Insights from Molecular Dynamics Simulations and Computational Chemistry. Nanomaterials, 15(8), 616. https://doi.org/10.3390/nano15080616