Single-Drop Analysis of Epinephrine and Uric Acid on a Screen-Printed Carbon Electrode



Abstract
:1. Introduction
2. Experimental Methods
2.1. Solutions and Reagents
2.2. Screen-Printed Electrodes
2.3. Square-Wave Voltammetry
2.4. Single-Drop Analysis on an SPCE Sensor
2.5. Cyclic Voltammetry
2.6. Real Sample Analysis
3. Results and Discussion
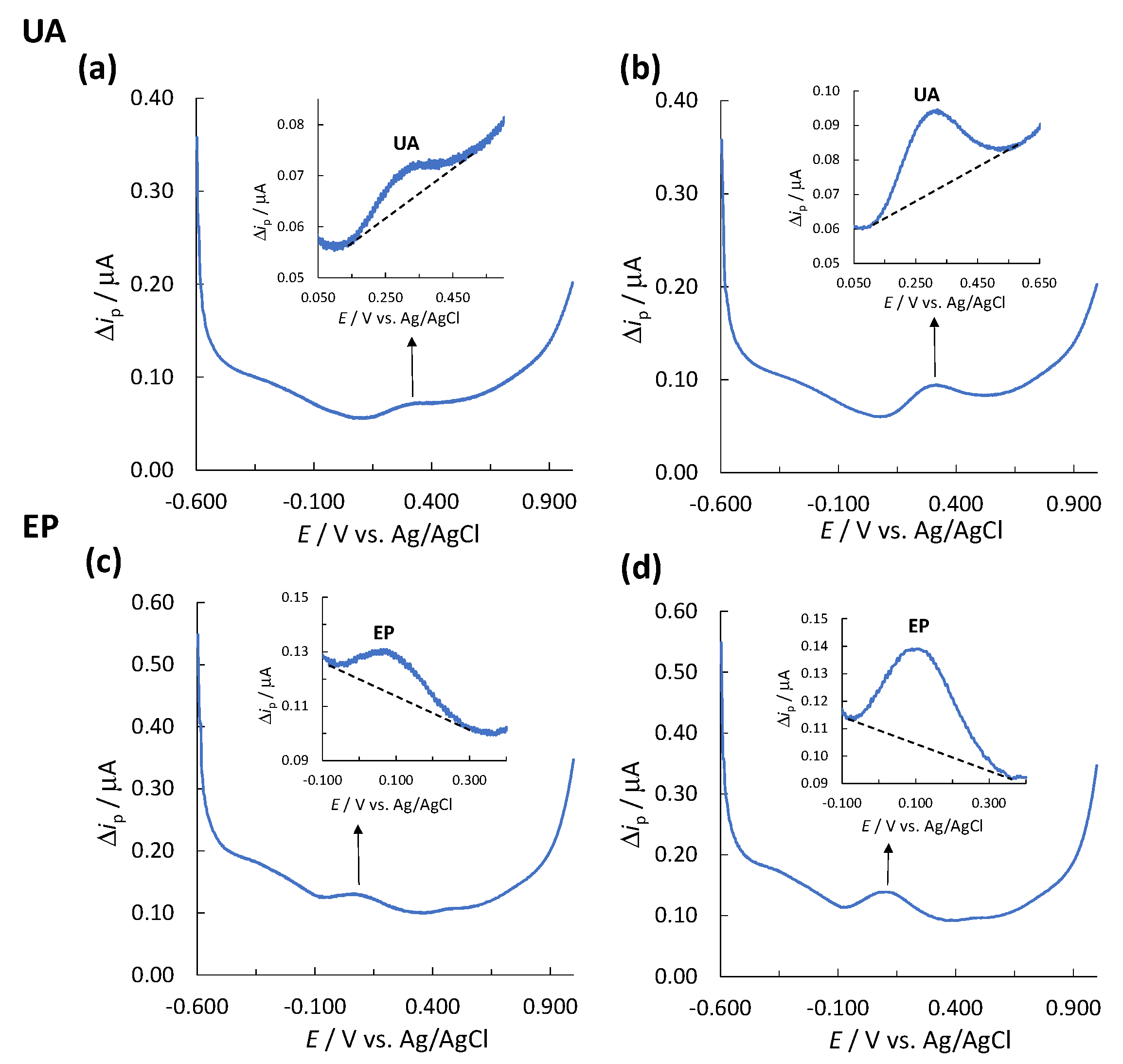
3.1. Limit of Detection and Limit of Quantification
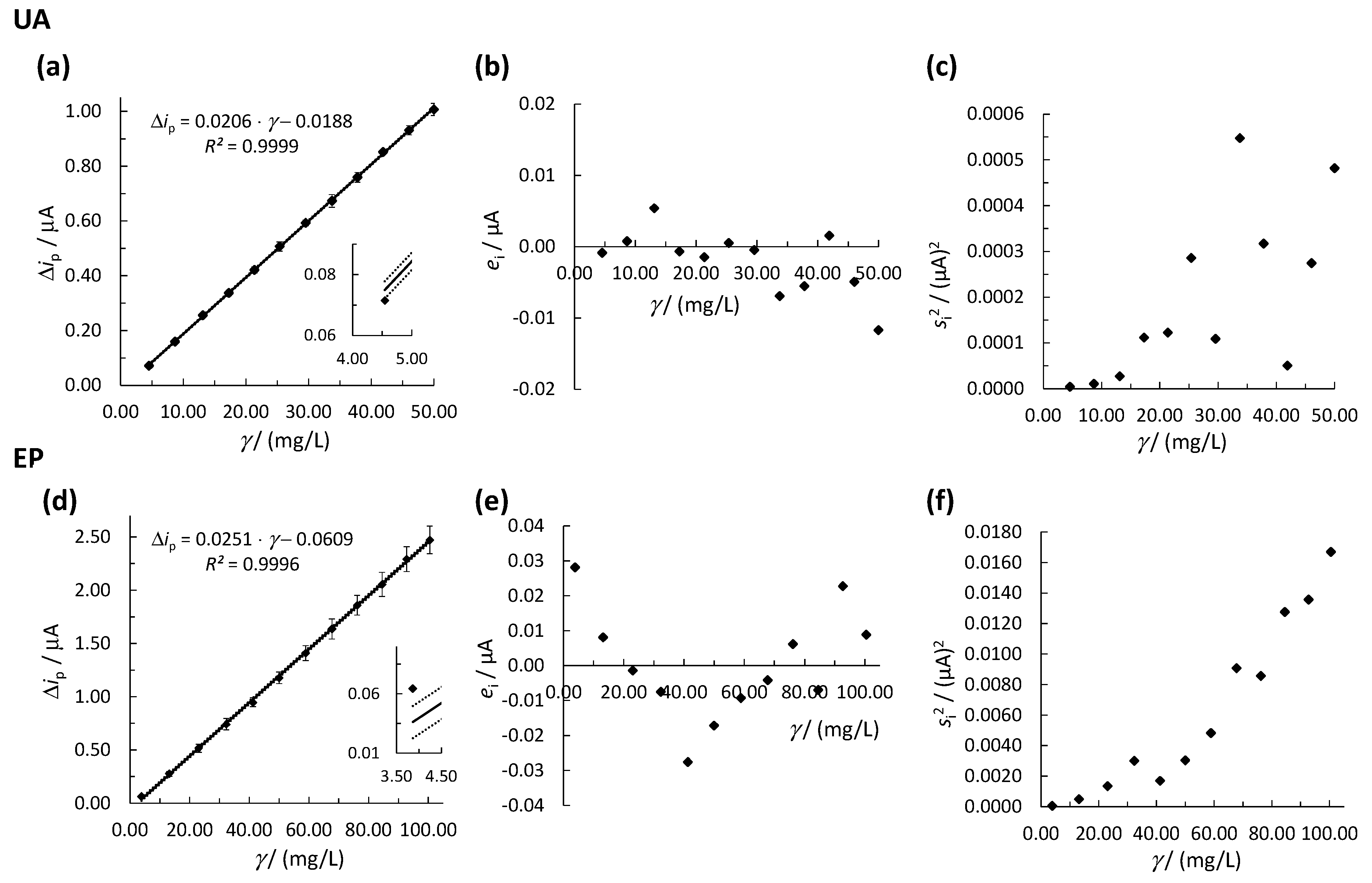
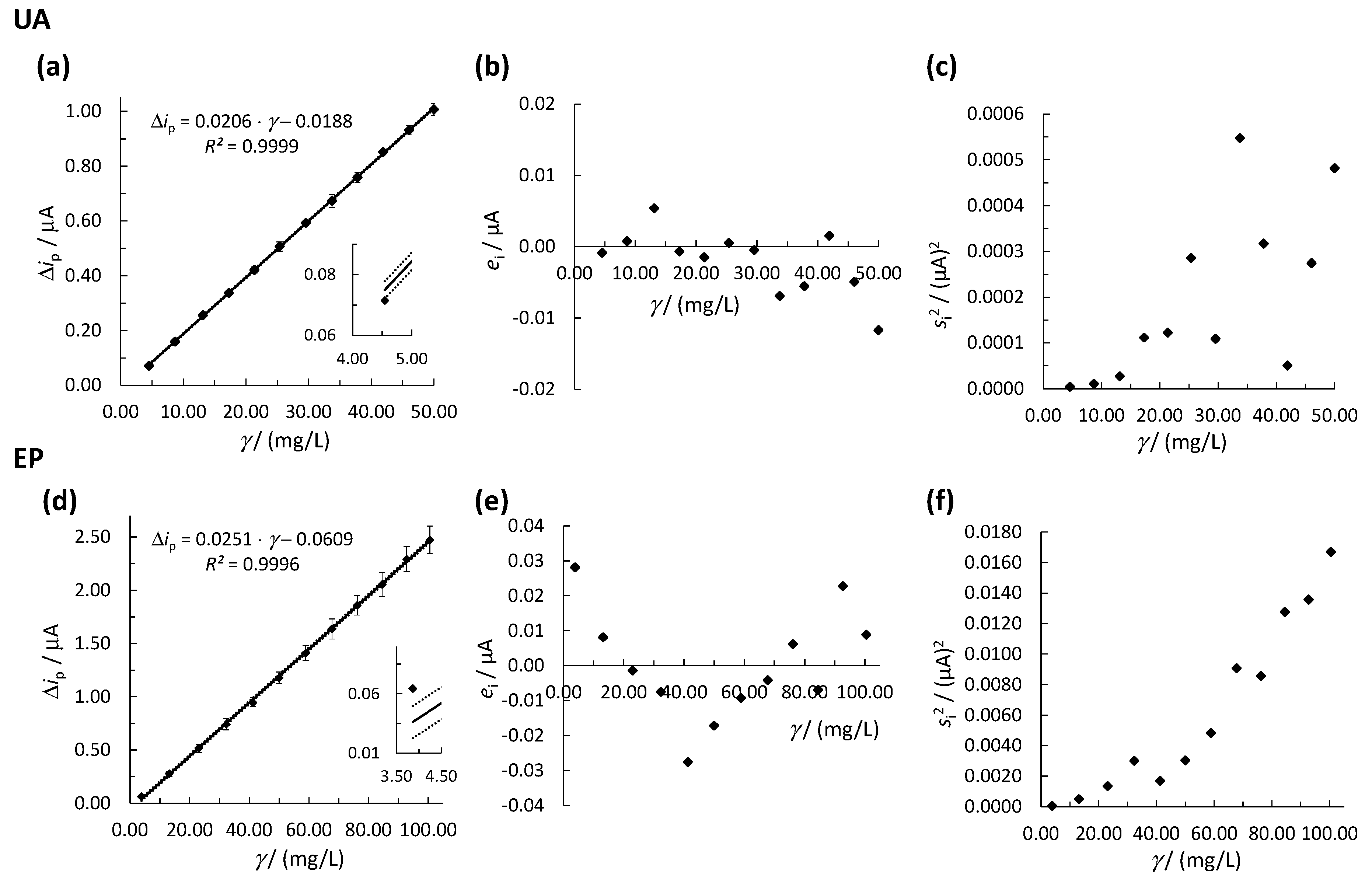
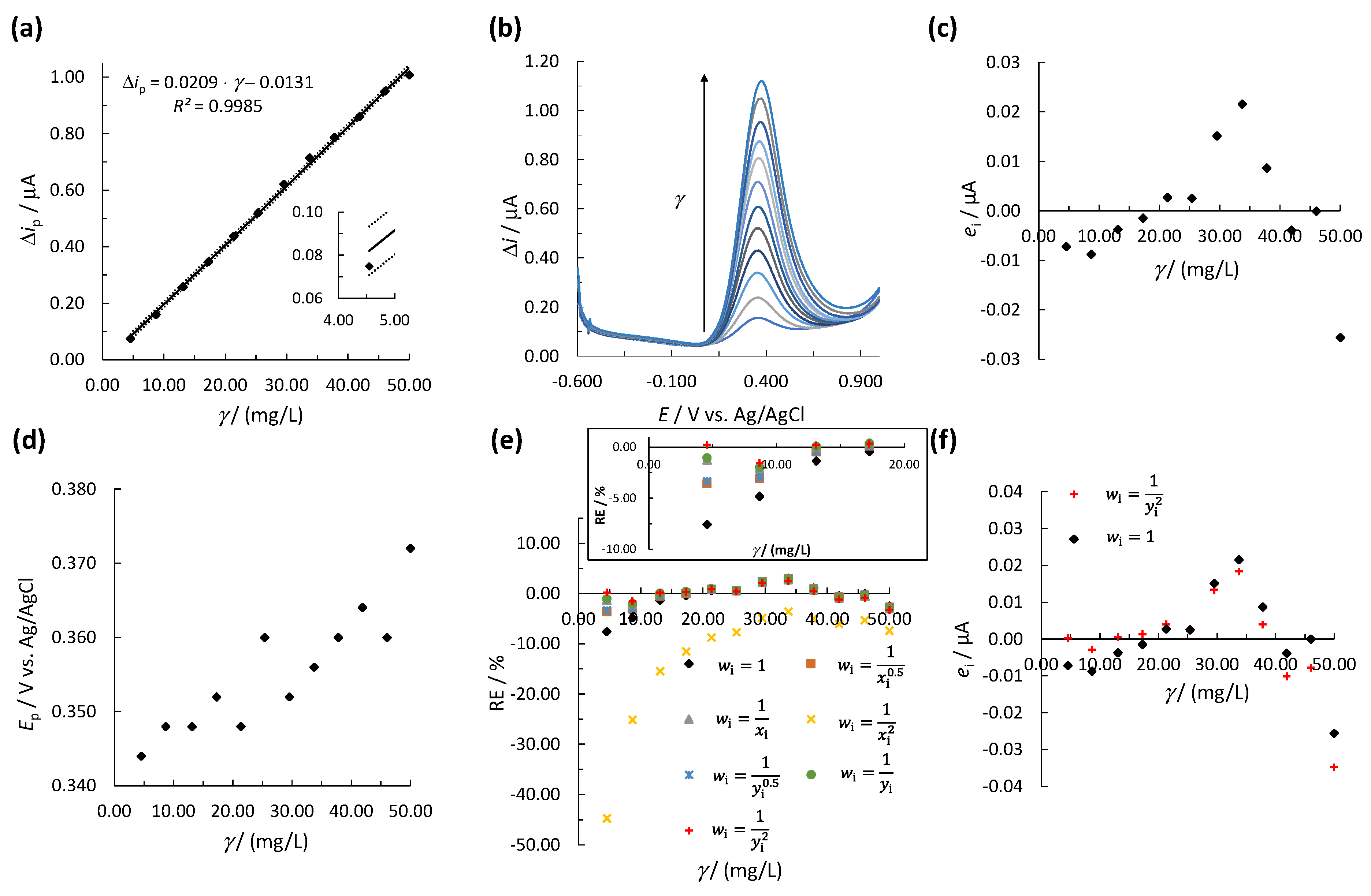
3.2. Determination of the Linear Concentration Range
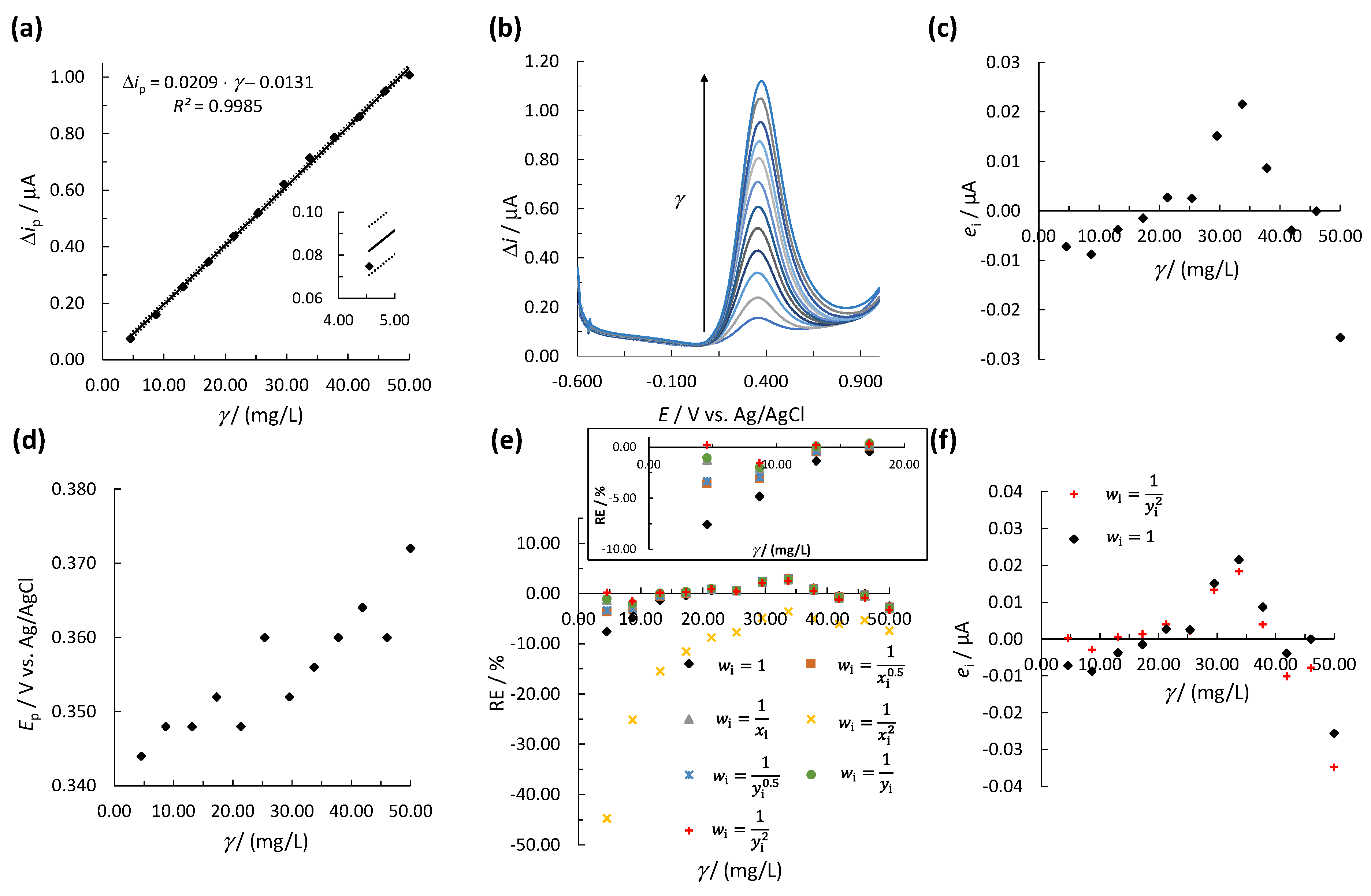
3.2.1. OLS Method
3.2.2. WLR Method
3.3. Accuracy and Precision
3.4. Robustness
4. Real Sample Analysis of EP and UA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ni, J.-A.; Ju, H.-X.; Chen, H.-Y.; Leech, D. Amperometric determination of epinephrine with an osmium complex and Nafion double-layer membrane modified electrode. Anal. Chim. Acta 1999, 378, 151–157. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Esler, M.D. Hypertension, Neurogenic. In Encyclopedia of Endocrine Diseases; Martini, L., Ed.; Elsevier: New York, NY, USA, 2004; pp. 603–608. [Google Scholar]
- Alpat, Ş.; Özdemir, K.; Kılınç Alpat, S. Voltammetric Determination of Epinephrine in Pharmaceutical Sample with a Tyrosinase Nanobiosensor. J. Sens. 2016, 2016, 5653975. [Google Scholar] [CrossRef] [Green Version]
- Ensafi, A.A.; Taei, M.; Khayamian, T. Simultaneous determination of ascorbic acid, epinephrine, and uric acid by differential pulse voltammetry using poly(p-xylenolsulfonephthalein) modified glassy carbon electrode. Colloids Surf. B Biointerfaces 2010, 79, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Luo, H.Q.; Li, N.B. Simultaneous voltammetric measurement of ascorbic acid, epinephrine and uric acid at a glassy carbon electrode modified with caffeic acid. Biosens. Bioelectron. 2006, 21, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Thivya, P.; Wilson, J. Electron rays irradiated polyaniline anchored over bovine serum albumin for simultaneous detection of epinephrine and uric acid. Microchem. J. 2019, 145, 883–891. [Google Scholar] [CrossRef]
- Mekassa, B.; Tessema, M.; Chandravanshi, B.S.; Baker, P.G.L.; Muya, F.N. Sensitive electrochemical determination of epinephrine at poly(L-aspartic acid)/electro-chemically reduced graphene oxide modified electrode by square wave voltammetry in pharmaceutics. J. Electroanal. Chem. 2017, 807, 145–153. [Google Scholar] [CrossRef]
- Mphuthi, N.G.; Adekunle, A.S.; Ebenso, E.E. Electrocatalytic oxidation of Epinephrine and Norepinephrine at metal oxide doped phthalocyanine/MWCNT composite sensor. Sci. Rep. 2016, 6, 26938. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Jin, Y.; Han, Q. Electrochemical Detection of Epinephrine Using an L-Glutamic Acid Functionalized Graphene Modified Electrode. Anal. Lett. 2014, 47, 1552–1563. [Google Scholar] [CrossRef]
- Al-Ameri, S.A.H. Spectrophotometric determination of adrenaline in pharmaceutical preparations. Arab. J. Chem. 2016, 9, S1000–S1004. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, K.M. Chapter 23—Adrenergic mechanisms and drugs. In Clinical Pharmacology, 11th ed.; Bennett, P.N., Brown, M.J., Sharma, P., Eds.; Churchill Livingstone: Oxford, UK, 2012; pp. 382–392. [Google Scholar]
- Ganti, A.; Pilkerton, R.; Falsgraf, E.; Vanwert, E.; Brouwer, N.; Beyer, A.; Byrne, B. Retrieval of Additional Epinephrine from Adrenaclick-Style Epinephrine Autoinjectors. Wilderness Environ. Med. 2021, 32, 55–58. [Google Scholar] [CrossRef]
- Shaker, M.S.; Wallace, D.V.; Golden, D.B.K.; Oppenheimer, J.; Bernstein, J.A.; Campbell, R.L.; Dinakar, C.; Ellis, A.; Greenhawt, M.; Khan, D.A.; et al. Anaphylaxis—A 2020 practice parameter update, systematic review, and Grading of Recommendations, Assessment, Development and Evaluation (GRADE) analysis. J. Allergy Clin. Immunol. 2020, 145, 1082–1123. [Google Scholar] [CrossRef] [Green Version]
- Motshakeri, M.; Phillips, A.R.J.; Kilmartin, P.A. Application of cyclic voltammetry to analyse uric acid and reducing agents in commercial milks. Food Chem. 2019, 293, 23–31. [Google Scholar] [CrossRef]
- Aryal, K.P.; Jeong, H.K. Simultaneous determination of ascorbic acid, dopamine, and uric acid with polyaniline/hemin/reduced graphite oxide composite. Chem. Phys. Lett. 2021, 768, 138405. [Google Scholar] [CrossRef]
- Du, J.; Yue, R.; Ren, F.; Yao, Z.; Jiang, F.; Yang, P.; Du, Y. Novel graphene flowers modified carbon fibers for simultaneous determination of ascorbic acid, dopamine and uric acid. Biosens. Bioelectron. 2014, 53, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, D.; Huang, J.; You, T. Simultaneous determination of dopamine, ascorbic acid and uric acid at electrochemically reduced graphene oxide modified electrode. Sens. Actuators B Chem. 2014, 193, 166–172. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, Z.; Liu, H.; Yang, M.; Lin, L.; Li, Q. Hemin-graphene oxide-pristine carbon nanotubes complexes with intrinsic peroxidase-like activity for the detection of H2O2 and simultaneous determination for Trp, AA, DA, and UA. Sens. Actuators B Chem. 2013, 188, 496–501. [Google Scholar] [CrossRef]
- Temoçin, Z. Modification of glassy carbon electrode in basic medium by electrochemical treatment for simultaneous determination of dopamine, ascorbic acid and uric acid. Sens. Actuators B Chem. 2013, 176, 796–802. [Google Scholar] [CrossRef]
- Hallaj, T.; Amjadi, M.; Mirbirang, F. S, N-doped carbon quantum dots enhanced Luminol-Mn(IV) chemiluminescence reaction for detection of uric acid in biological fluids. Microchem. J. 2020, 156, 104841. [Google Scholar] [CrossRef]
- Noroozifar, M.; Khorasani-Motlagh, M.; Akbari, R.; Bemanadi Parizi, M. Simultaneous and sensitive determination of a quaternary mixture of AA, DA, UA and Trp using a modified GCE by iron ion-doped natrolite zeolite-multiwall carbon nanotube. Biosens. Bioelectron. 2011, 28, 56–63. [Google Scholar] [CrossRef]
- Luo, Y.-C.; Do, J.-S.; Liu, C.-C. An amperometric uric acid biosensor based on modified Ir–C electrode. Biosens. Bioelectron. 2006, 22, 482–488. [Google Scholar] [CrossRef]
- Carrera, V.; Sabater, E.; Vilanova, E.; Sogorb, M.A. A simple and rapid HPLC–MS method for the simultaneous determination of epinephrine, norepinephrine, dopamine and 5-hydroxytryptamine: Application to the secretion of bovine chromaffin cell cultures. J. Chromatogr. B 2007, 847, 88–94. [Google Scholar] [CrossRef]
- Li, X.; Franke, A.A. Fast HPLC–ECD analysis of ascorbic acid, dehydroascorbic acid and uric acid. J. Chromatogr. B 2009, 877, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Gyllenhaal, O.; Johansson, L.; Vessman, J. Gas chromatography of epinephrine and norepinephrine after derivatization with chloroformates in aqueous media. J. Chromatogr. A 1980, 190, 347–357. [Google Scholar] [CrossRef]
- Chen, X.B.; Calder, A.G.; Prasitkusol, P.; Kyle, D.J.; Jayasuriya, M.C.N. Determination of 15N isotopic enrichment and concentrations of allantoin and uric acid in urine by gas chromatography/mass spectrometry. J. Mass Spectrom. 1998, 33, 130–137. [Google Scholar] [CrossRef]
- Bulatov, A.V.; Petrova, A.V.; Vishnikin, A.B.; Moskvin, A.L.; Moskvin, L.N. Stepwise injection spectrophotometric determination of epinephrine. Talanta 2012, 96, 62–67. [Google Scholar] [CrossRef]
- Moghadam, M.R.; Dadfarnia, S.; Shabani, A.M.H.; Shahbazikhah, P. Chemometric-assisted kinetic–spectrophotometric method for simultaneous determination of ascorbic acid, uric acid, and dopamine. Anal. Biochem. 2011, 410, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Song, G.; Lin, J.-M. Separation and determination of norepinephrine, epinephrine and isoprinaline enantiomers by capillary electrophoresis in pharmaceutical formulation and human serum. J. Chromatogr. A 2005, 1098, 166–171. [Google Scholar] [CrossRef]
- Pormsila, W.; Krähenbühl, S.; Hauser, P.C. Capillary electrophoresis with contactless conductivity detection for uric acid determination in biological fluids. Anal. Chim. Acta 2009, 636, 224–228. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, X.; Wang, J.; Wang, X.; Yang, N. Simultaneous determination of cysteine, ascorbic acid and uric acid by capillary electrophoresis with electrochemiluminescence. J. Electroanal. Chem. 2012, 674, 65–70. [Google Scholar] [CrossRef]
- Majer, D.; Mastnak, T.; Finšgar, M. An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination. Sensors 2020, 20, 7056. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Taei, M.; Khayamian, T.; Arabzadeh, A. Highly selective determination of ascorbic acid, dopamine, and uric acid by differential pulse voltammetry using poly(sulfonazo III) modified glassy carbon electrode. Sens. Actuators B Chem. 2010, 147, 213–221. [Google Scholar] [CrossRef]
- Chetankumar, K.; Kumara Swamy, B.E.; Sharma, S.C. Safranin amplified carbon paste electrode sensor for analysis of paracetamol and epinephrine in presence of folic acid and ascorbic acid. Microchem. J. 2021, 160, 105729. [Google Scholar] [CrossRef]
- Wu, C.; Li, J.; Liu, X.; Zhang, H.; Li, R.; Wang, G.; Wang, Z.; Li, Q.; Shangguan, E. Simultaneous voltammetric determination of epinephrine and acetaminophen using a highly sensitive CoAl-OOH/reduced graphene oxide sensor in pharmaceutical samples and biological fluids. Mater. Sci. Eng. C 2021, 119, 111557. [Google Scholar] [CrossRef]
- Immanuel, S.; Aparna, T.K.; Sivasubramanian, R. A facile preparation of Au—SiO2 nanocomposite for simultaneous electrochemical detection of dopamine and uric acid. Surf. Interfaces 2019, 14, 82–91. [Google Scholar] [CrossRef]
- Roy, P.R.; Okajima, T.; Ohsaka, T. Simultaneous electrochemical detection of uric acid and ascorbic acid at a poly(N,N-dimethylaniline) film-coated GC electrode. J. Electroanal. Chem. 2004, 561, 75–82. [Google Scholar] [CrossRef]
- Hayat, A.; Marty, J.L. Disposable screen printed electrochemical sensors: Tools for environmental monitoring. Sens. (Basel) 2014, 14, 10432–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, D.-W.; Xiu, G.; Long, Y.-T. Applications of screen-printed electrodes in current environmental analysis. Curr. Opin. Electrochem. 2017, 3, 137–143. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P. Screen-printed electrochemical sensors for monitoring metal pollutants. Trac Trends Anal. Chem. 2003, 22, 456–469. [Google Scholar] [CrossRef]
- González-Sánchez, M.I.; Gómez-Monedero, B.; Agrisuelas, J.; Iniesta, J.; Valero, E. Highly activated screen-printed carbon electrodes by electrochemical treatment with hydrogen peroxide. Electrochem. Commun. 2018, 91, 36–40. [Google Scholar] [CrossRef]
- Wang, W.; Fu, Y.; Lv, Q.; Bai, H.; Li, H.; Wang, Z.; Zhang, Q. Miniaturized device with a detachable three-electrode system and vibration motor for electrochemical analysis based on disposable electrodes. Sens. Actuators B Chem. 2019, 297, 126719. [Google Scholar] [CrossRef]
- Ji, D.; Liu, L.; Li, S.; Chen, C.; Lu, Y.; Wu, J.; Liu, Q. Smartphone-based cyclic voltammetry system with graphene modified screen printed electrodes for glucose detection. Biosens. Bioelectron. 2017, 98, 449–456. [Google Scholar] [CrossRef]
- Tseliou, F.; Pappas, P.; Spyrou, K.; Hrbac, J.; Prodromidis, M.I. Lab-on-a-screen-printed electrochemical cell for drop-volume voltammetric screening of flunitrazepam in untreated, undiluted alcoholic and soft drinks. Biosens. Bioelectron. 2019, 132, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Stanković, D.M.; Milanović, Z.; Švorc, Ľ.; Stanković, V.; Janković, D.; Mirković, M.; Đurić, S.V. Screen printed diamond electrode as efficient “point-of-care” platform for submicromolar determination of cytostatic drug in biological fluids and pharmaceutical product. Diam. Relat. Mater. 2021, 113, 108277. [Google Scholar] [CrossRef]
- Couto, R.A.S.; Costa, S.S.; Mounssef, B.; Pacheco, J.G.; Fernandes, E.; Carvalho, F.; Rodrigues, C.M.P.; Delerue-Matos, C.; Braga, A.A.C.; Moreira Gonçalves, L.; et al. Electrochemical sensing of ecstasy with electropolymerized molecularly imprinted poly(o-phenylenediamine) polymer on the surface of disposable screen-printed carbon electrodes. Sens. Actuators B Chem. 2019, 290, 378–386. [Google Scholar] [CrossRef]
- Raposo, F.; Ibelli-Bianco, C. Performance parameters for analytical method validation: Controversies and discrepancies among numerous guidelines. Trac Trends Anal. Chem. 2020, 129, 115913. [Google Scholar] [CrossRef]
- Rozet, E.; Ceccato, A.; Hubert, C.; Ziemons, E.; Oprean, R.; Rudaz, S.; Boulanger, B.; Hubert, P. Analysis of recent pharmaceutical regulatory documents on analytical method validation. J. Chromatogr. A 2007, 1158, 111–125. [Google Scholar] [CrossRef]
- da Silva, C.P.; Emídio, E.S.; de Marchi, M.R.R. Method validation using weighted linear regression models for quantification of UV filters in water samples. Talanta 2015, 131, 221–227. [Google Scholar] [CrossRef] [Green Version]
- De Beer, J.O.; De Beer, T.R.; Goeyens, L. Assessment of quality performance parameters for straight line calibration curves related to the spread of the abscissa values around their mean. Anal. Chim. Acta 2007, 584, 57–65. [Google Scholar] [CrossRef]
- Nascimento, R.S.; Froes, R.E.S.; e Silva, N.O.C.; Naveira, R.L.P.; Mendes, D.B.C.; Neto, W.B.; Silva, J.B.B. Comparison between ordinary least squares regression and weighted least squares regression in the calibration of metals present in human milk determined by ICP-OES. Talanta 2010, 80, 1102–1109. [Google Scholar] [CrossRef]
- Almeida, A.M.; Castel-Branco, M.M.; Falcão, A.C. Linear regression for calibration lines revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B 2002, 774, 215–222. [Google Scholar] [CrossRef]
- Otarola, J.J.; Cobo Solis, A.K.; Farias, M.E.; Garrido, M.; Mariano Correa, N.; Molina, P.G. Piroxicam-loaded nanostructured lipid carriers gel: Design and characterization by square wave voltammetry. Colloids Surf. A Physicochem. Eng. Asp. 2020, 606, 125396. [Google Scholar] [CrossRef]
- Finšgar, M.; Petovar, B.; Vodopivec, K. Bismuth-tin-film electrodes for Zn(II), Cd(II), and Pb(II) trace analysis. Microchem. J. 2019, 145, 676–685. [Google Scholar] [CrossRef]
- Finšgar, M.; Xhanari, K.; Otmačić Ćurković, H. Cyclic Voltammetry as an Electroanalytical Tool for Analysing the Reaction Mechanisms of Copper in Chloride Solution Containing Different Azole Compounds. Curr. Anal. Chem. 2020, 16, 465–474. [Google Scholar] [CrossRef]
- Borman, P.; Elder, D. Q2(R1) Validation of Analytical Procedures. In ICH Quality Guidelines; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 127–166. [Google Scholar]
- Komsta, L.; Vander Heyden, Y.; Sherma, J. Chemometrics in Chromatography; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Miller, J.N. Basic statistical methods for Analytical Chemistry. Part 2. Calibration and regression methods. A review. Analyst 1991, 116, 3–14. [Google Scholar] [CrossRef]
- Massart, D.L.; Buydens, L.M.; Vandeginste, B.G. Handbook of Chemometrics and Qualimetrics: Part A; Elsevier Science B.V.: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry; Pearson/Prentice Hall: Harlow, UK, 2010. [Google Scholar]
- Cardone, M.J.; Willavize, S.A.; Lacy, M.E. Method Validation Revisited: A Chemometric Approach. Pharm. Res. 1990, 7, 154–160. [Google Scholar] [CrossRef]
- Mansilha, C.; Melo, A.; Rebelo, H.; Ferreira, I.M.P.L.V.O.; Pinho, O.; Domingues, V.; Pinho, C.; Gameiro, P. Quantification of endocrine disruptors and pesticides in water by gas chromatography–tandem mass spectrometry. Method validation using weighted linear regression schemes. J. Chromatogr. A 2010, 1217, 6681–6691. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.L.; Reynolds, D.L.; Wright, D.S.; Pachla, L.A. Biological sample preparation and data reduction concepts in pharmaceutical analysis. J. Chromatogr. Sci. 1988, 26, 372–379. [Google Scholar] [CrossRef]
- Harvey, D. Modern Analytical Chemistry; McGraw-Hill: New York, NY, USA, 2000. [Google Scholar]
- Surve, D.H.; Jindal, A.B. Development and validation of reverse-phase high-performance liquid chromatographic (RP-HPLC) method for quantification of Efavirenz in Efavirenz-Enfuvirtide co-loaded polymer-lipid hybrid nanoparticles. J. Pharm. Biomed. Anal. 2019, 175, 112765. [Google Scholar] [CrossRef]
- Aszyk, J.; Kot, J.; Tkachenko, Y.; Woźniak, M.; Bogucka-Kocka, A.; Kot-Wasik, A. Novel liquid chromatography method based on linear weighted regression for the fast determination of isoprostane isomers in plasma samples using sensitive tandem mass spectrometry detection. J. Chromatogr. B 2017, 1051, 17–23. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime (UNODC). Guidance for the Validation of Analytical Methodology and Calibration of Equipment Used for Testing of Illicit Drugs in Seized Materials and Biological Specimens: A Commitment to Quality and Continuous Improvement; United Nations Publications: New York, NY, USA, 2009. [Google Scholar]
- Curran, J.M. The Frequentist Approach to Forensic Evidence Interpretation. In Encyclopedia of Forensic Sciences, 2nd ed.; Siegel, J.A., Saukko, P.J., Houck, M.M., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 286–291. [Google Scholar]
- Finšgar, M.; Perva-Uzunalić, A.; Behr, H.; Ledinek, N.; Knez, Ž.; Novak, Z. An Improved Reversed-Phase High-Performance Liquid Chromatography Method for the Analysis of Related Substances of Prednisolone in Active Ingredient. ACS Omega 2020, 5, 7987–8000. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Shiu, K.-K. Determination of Uric Acid at Electrochemically Activated Glassy Carbon Electrode. Electroanalysis 2001, 13, 1319–1325. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| SPCE | ||||||
|---|---|---|---|---|---|---|
| Analyte | Model No. | |||||
| UA | 1 (unweighted model) | 1 | 0.0209 | −0.0131 | 0.9985 | 24.74 |
| 2 | 0.0211 | −0.0176 | 0.9989 | 18.62 | ||
| 3 | 0.0212 | −0.0203 | 0.9992 | 15.43 | ||
| 4 | 0.0213 | −0.0223 | 0.9995 | 145.41 | ||
| 5 | 0.0211 | −0.0178 | 0.9990 | 18.32 | ||
| 6 | 0.0212 | −0.0205 | 0.9993 | 15.31 | ||
| 7 | 0.0213 | −0.0222 | 0.9995 | 14.02 |
| SPCE | |||||||
|---|---|---|---|---|---|---|---|
| OLS (Unweighted Model 1) | WLR (Weighted Model 7) | ||||||
| γ/(mg/L) (Theoretical) | γ/(mg/L) (Determined) | Average Recovery/% | RSD/% (n = 3) | γ/(mg/L) (Determined) | Average Recovery/% | RSD/% (n = 3) | |
| UA | 6.14 | 5.78 | 94.18 | 1.08 | 6.11 | 99.52 | 1.00 |
| 25.00 | 25.51 | 102.04 | 1.60 | 25.50 | 102.00 | 1.57 | |
| 47.62 | 47.79 | 100.37 | 0.80 | 47.40 | 99.54 | 0.79 | |
| EP | 5.69 | 6.69 | 117.64 | 0.91 | 5.70 | 100.20 | 1.10 |
| 50.25 | 49.84 | 99.20 | 0.46 | 50.22 | 99.95 | 0.47 | |
| 97.56 | 103.46 | 106.04 | 0.15 | 105.54 | 108.18 | 0.15 | |
| D-SPCE | ||||||
|---|---|---|---|---|---|---|
| Analyte | Model No. | |||||
| UA | 1 (unweighted model) | 1 | 0.4351 | 0.0250 | 1.0000 | 5.90 |
| 7 | 0.4369 | −0.0184 | 1.0000 | 4.03 | ||
| EP | 1 (unweighted model) | 1 | 0.2681 | 0.1295 | 0.9990 | 39.15 |
| 7 | 0.2727 | −0.0912 | 0.9994 | 20.65 |
| D-SPCE | |||||||
|---|---|---|---|---|---|---|---|
| OLS (Unweighted Model 1) | WLR (Weighted Model 7) | ||||||
| γ/(mg/L) (Theoretical) | γ/(mg/L) (Determined) | Recovery/% | RSD/% | γ/(mg/L) (Determined) | Recovery/% | RSD/% | |
| UA | 6.14 | 6.29 | 102.47 | 1.63 | 6.36 | 103.65 | 1.60 |
| 25.00 | 24.13 | 96.53 | 0.40 | 24.13 | 96.51 | 0.39 | |
| 47.62 | 44.81 | 94.10 | 1.10 | 44.72 | 93.91 | 1.09 | |
| EP | 5.69 | 3.99 | 70.23 | 6.71 | 4.74 | 83.27 | 5.57 |
| 50.25 | 53.22 | 105.93 | 3.15 | 53.13 | 105.75 | 3.11 | |
| 97.56 | 97.78 | 100.22 | 2.39 | 96.93 | 99.35 | 2.37 | |
| * γ/(mg/L) (Initially Determined) | γ/(mg/L) (Spiked) | ** γ/(mg/L) (Determined) | Average Recovery/% | RSD/% | *** γ/(mg/L) (Sample Content) | |
|---|---|---|---|---|---|---|
| UA in human urine | 6.33 | 25.00 | 29.90 | 94.35 | 5.65 | 316.29 |
| EP in an auto-injector | 23.92 | 50.52 | 74.95 | 101.01 | 2.75 | 478.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majer, D.; Finšgar, M. Single-Drop Analysis of Epinephrine and Uric Acid on a Screen-Printed Carbon Electrode. Biosensors 2021, 11, 285. https://doi.org/10.3390/bios11080285
Majer D, Finšgar M. Single-Drop Analysis of Epinephrine and Uric Acid on a Screen-Printed Carbon Electrode. Biosensors. 2021; 11(8):285. https://doi.org/10.3390/bios11080285
Chicago/Turabian StyleMajer, David, and Matjaž Finšgar. 2021. "Single-Drop Analysis of Epinephrine and Uric Acid on a Screen-Printed Carbon Electrode" Biosensors 11, no. 8: 285. https://doi.org/10.3390/bios11080285
APA StyleMajer, D., & Finšgar, M. (2021). Single-Drop Analysis of Epinephrine and Uric Acid on a Screen-Printed Carbon Electrode. Biosensors, 11(8), 285. https://doi.org/10.3390/bios11080285