Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons

 , , and
, , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Air-Liquid Interface Organoid Cultures
2.2. Statistical Analysis
2.3. String Network Analysis
3. Results
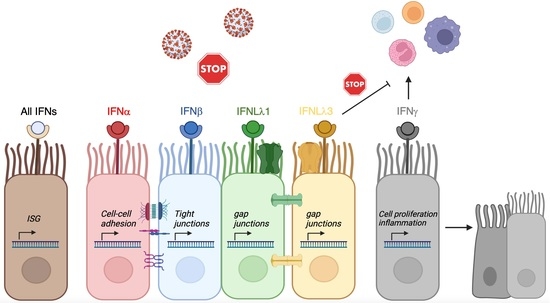
3.1. Differential IFN-Type-Specific Secreted Factors despite Overlapping Pan-IFN-Triggered ISG Expression
3.2. IFN-Type-Specific Induction of Distinct Transcription Factor Patterns in Airway Epithelia
3.3. Induction of Pro-Inflammatory Rather than Homeostatic Genes by All IFNs Except IFNλ3
3.4. Enhancement of Cell-Cell Adhesion by Type-I, of Intercellular Signaling and Ion Channels by Type-III and of Cell Growth by Type-II IFNs
3.5. IFN-Induced Hub DEGs Connect Signaling Pathways in the Airway Epithelium
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med. Virol. 2022, 94, 869–877. [Google Scholar] [CrossRef]
- Gao, S.; von der Malsburg, A.; Paeschke, S.; Behlke, J.; Haller, O.; Kochs, G.; Daumke, O. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 2010, 465, 502–506. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, A.; Barnes, B.J.; Amrute, S.; Yeow, W.S.; Megjugorac, N.; Dai, J.; Feng, D.; Chung, E.; Pitha, P.M.; Fitzgerald-Bocarsly, P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 2003, 74, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e876. [Google Scholar] [CrossRef] [PubMed]
- Henden, A.S.; Koyama, M.; Robb, R.J.; Forero, A.; Kuns, R.D.; Chang, K.; Ensbey, K.S.; Varelias, A.; Kazakoff, S.H.; Waddell, N.; et al. IFN-lambda therapy prevents severe gastrointestinal graft-versus-host disease. Blood 2021, 138, 722–737. [Google Scholar] [CrossRef]
- Wheelock, E.F. Interferon-Like Virus-Inhibitor Induced in Human Leukocytes by Phytohemagglutinin. Science 1965, 149, 310–311. [Google Scholar] [CrossRef]
- Zissler, U.M.; Chaker, A.M.; Effner, R.; Ulrich, M.; Guerth, F.; Piontek, G.; Dietz, K.; Regn, M.; Knapp, B.; Theis, F.J.; et al. Interleukin-4 and interferon-gamma orchestrate an epithelial polarization in the airways. Mucosal. Immunol. 2016, 9, 917–926. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Bou Ghanem, E.N.; Nelson, C.C.; D’Orazio, S.E. T cell-intrinsic factors contribute to the differential ability of CD8+ T cells to rapidly secrete IFN-gamma in the absence of antigen. J. Immunol. 2011, 186, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, B.; Molina, S.A.; Koval, M. Claudins: Gatekeepers of lung epithelial function. Semin. Cell Dev. Biol. 2015, 42, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.; Provost, K.; Niu, N.; Homer, R.; Cohn, L. IFN-gamma acts on the airway epithelium to inhibit local and systemic pathology in allergic airway disease. J. Immunol. 2011, 187, 3815–3820. [Google Scholar] [CrossRef]
- Lee, P.H.; Park, S.; Lee, Y.G.; Choi, S.M.; An, M.H.; Jang, A.S. The Impact of Environmental Pollutants on Barrier Dysfunction in Respiratory Disease. Allergy Asthma Immunol. Res. 2021, 13, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.F.; Thompson, L.J.; Ziegler, S.F. TSLP drives acute TH2-cell differentiation in lungs. J. Allergy Clin. Immunol. 2020, 146, 1406–1418.e1407. [Google Scholar] [CrossRef] [PubMed]
- Jakwerth, C.A.; Ordovas-Montanes, J.; Blank, S.; Schmidt-Weber, C.B.; Zissler, U.M. Role of Respiratory Epithelial Cells in Allergic Diseases. Cells 2022, 11, 1387. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanah, F.; Hamblin, M.R.; Rezaei, N. The immune system and COVID-19: Friend or foe? Life Sci. 2020, 256, 117900. [Google Scholar] [CrossRef]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef]
- Chemudupati, M.; Kenney, A.D.; Bonifati, S.; Zani, A.; McMichael, T.M.; Wu, L.; Yount, J.S. From APOBEC to ZAP: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 382–394. [Google Scholar] [CrossRef]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Jakwerth, C.A.; Feuerherd, M.; Guerth, F.M.; Oelsner, M.; Schellhammer, L.; Giglberger, J.; Pechtold, L.; Jerin, C.; Kugler, L.; Mogler, C.; et al. Early reduction of SARS-CoV-2-replication in bronchial epithelium by kinin B2 receptor antagonism. J. Mol. Med. 2022, 100, 613–627. [Google Scholar] [CrossRef]
- Li, X.; Hawkins, G.A.; Moore, W.C.; Hastie, A.T.; Ampleford, E.J.; Milosevic, J.; Li, H.; Busse, W.W.; Erzurum, S.C.; Kaminski, N.; et al. Expression of asthma susceptibility genes in bronchial epithelial cells and bronchial alveolar lavage in the Severe Asthma Research Program (SARP) cohort. J. Asthma 2016, 53, 775–782. [Google Scholar] [CrossRef]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890.e882. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, K.G.; Wang, J.; Luo, L.; Fujiwara, Y.; Orkin, S.H.; Beier, D.R. Gata4 is necessary for normal pulmonary lobar development. Am. J. Respir. Cell Mol. Biol. 2007, 36, 391–397. [Google Scholar] [CrossRef]
- Hebert, K.D.; McLaughlin, N.; Galeas-Pena, M.; Zhang, Z.; Eddens, T.; Govero, A.; Pilewski, J.M.; Kolls, J.K.; Pociask, D.A. Targeting the IL-22/IL-22BP axis enhances tight junctions and reduces inflammation during influenza infection. Mucosal Immunol. 2020, 13, 64–74. [Google Scholar] [CrossRef]
- Lazear, H.M.; Daniels, B.P.; Pinto, A.K.; Huang, A.C.; Vick, S.C.; Doyle, S.E.; Gale, M., Jr.; Klein, R.S.; Diamond, M.S. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 2015, 7, 284ra259. [Google Scholar] [CrossRef]
- Odendall, C.; Voak, A.A.; Kagan, J.C. Type III IFNs Are Commonly Induced by Bacteria-Sensing TLRs and Reinforce Epithelial Barriers during Infection. J. Immunol. 2017, 199, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Steelant, B.; Farre, R.; Wawrzyniak, P.; Belmans, J.; Dekimpe, E.; Vanheel, H.; Van Gerven, L.; Kortekaas Krohn, I.; Bullens, D.M.A.; Ceuppens, J.L.; et al. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J. Allergy. Clin. Immunol. 2016, 137, 1043–1053.e1045. [Google Scholar] [CrossRef] [PubMed]
- D’Agnillo, F.; Walters, K.A.; Xiao, Y.; Sheng, Z.M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Oldstone, M.B. Infected CD8alpha- dendritic cells are the predominant source of IL-10 during establishment of persistent viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Lu, M.M.; Zhang, Y.; Tucker, P.W.; Zhou, D.; Morrisey, E.E. Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development 2007, 134, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Ewart, S.L.; Kuperman, D.; Schadt, E.; Tankersley, C.; Grupe, A.; Shubitowski, D.M.; Peltz, G.; Wills-Karp, M. Quantitative trait loci controlling allergen-induced airway hyperresponsiveness in inbred mice. Am. J. Respir. Cell Mol. Biol. 2000, 23, 537–545. [Google Scholar] [CrossRef]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef]
- Stifter, S.A.; Bhattacharyya, N.; Pillay, R.; Florido, M.; Triccas, J.A.; Britton, W.J.; Feng, C.G. Functional Interplay between Type I and II Interferons Is Essential to Limit Influenza A Virus-Induced Tissue Inflammation. PLoS Pathog. 2016, 12, e1005378. [Google Scholar] [CrossRef]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [CrossRef]
- Mohseni Afshar, Z.; Barary, M.; Babazadeh, A.; Tavakoli Pirzaman, A.; Hosseinzadeh, R.; Alijanpour, A.; Allahgholipour, A.; Miri, S.R.; Sio, T.T.; Sullman, M.J.M.; et al. The role of cytokines and their antagonists in the treatment of COVID-19 patients. Rev. Med. Virol. 2022, e2372. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.T.; Hagner, S.; Ruchti, F.; Radzikowska, U.; Tan, G.; Altunbulakli, C.; Eljaszewicz, A.; Moniuszko, M.; Akdis, M.; Akdis, C.A.; et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy 2019, 74, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; McCrudden, C.M.; Kwok, H.F. Prognostic significance of combining VEGFA, FLT1 and KDR mRNA expression in lung cancer. Oncol. Lett. 2015, 10, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Onodera, Y.; Kuroiwa, M.; Nomimura, S.; Kubo, Y.; Nam, J.M.; Kajiwara, K.; Nada, S.; Oneyama, C.; Sabe, H.; et al. The Rho guanine nucleotide exchange factor ARHGEF5 promotes tumor malignancy via epithelial-mesenchymal transition. Oncogenesis 2016, 5, e258. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erb, A.; Zissler, U.M.; Oelsner, M.; Chaker, A.M.; Schmidt-Weber, C.B.; Jakwerth, C.A. Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons. Biosensors 2022, 12, 929. https://doi.org/10.3390/bios12110929
Erb A, Zissler UM, Oelsner M, Chaker AM, Schmidt-Weber CB, Jakwerth CA. Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons. Biosensors. 2022; 12(11):929. https://doi.org/10.3390/bios12110929
Chicago/Turabian StyleErb, Anna, Ulrich M. Zissler, Madlen Oelsner, Adam M. Chaker, Carsten B. Schmidt-Weber, and Constanze A. Jakwerth. 2022. "Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons" Biosensors 12, no. 11: 929. https://doi.org/10.3390/bios12110929
APA StyleErb, A., Zissler, U. M., Oelsner, M., Chaker, A. M., Schmidt-Weber, C. B., & Jakwerth, C. A. (2022). Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons. Biosensors, 12(11), 929. https://doi.org/10.3390/bios12110929