1. Introduction
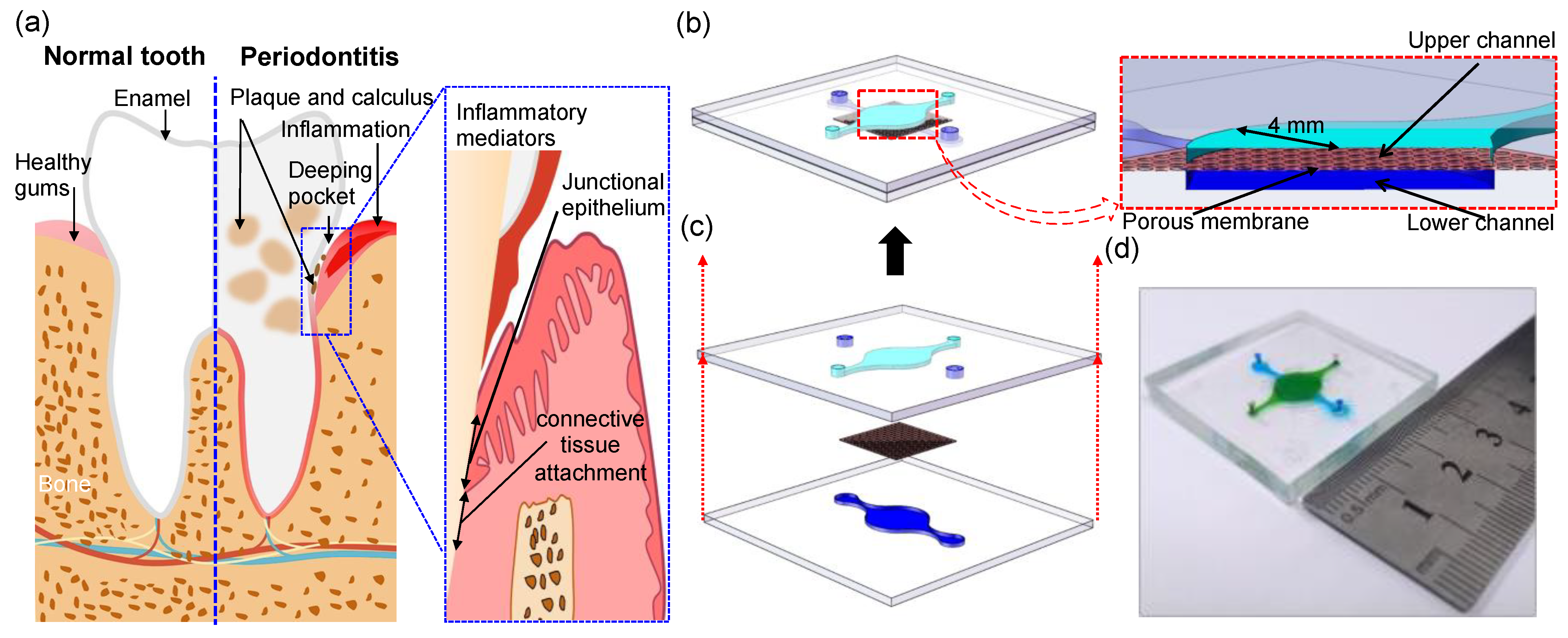
Periodontitis is a bacterially induced chronic inflammatory disease that compromises the integrity of the tooth-supporting tissues known as the periodontium [
1]. It includes the gingiva, periodontal ligament, and alveolar bone. The key presentations of periodontitis in the early stages are gingival bleeding and recession of the gingival margin. The chronic inflammation can develop gradually into the deep periodontal tissues [
2]. In severe cases, the total ulcerated area of the periodontal pocket wall can be as high as 72 cm
2 and become a focus of infection, which provides a favorable site for material exchange between periodontal pathogenic microorganisms and their metabolites with the blood vessels. Bacteria and their metabolites invade the gingival epithelium and the underlying connective tissue, which can activate periodontal cells, promote the release of cytokines and polymorphic granulocytes, and secrete a variety of pro-inflammatory mediators. Acting as a structural barrier between the underlying tissue and the outside environment [
3], the gingival epithelium provides an important contribution to the maintenance of periodontal tissue homeostasis [
4,
5]. It is not only a physical barrier against infection, but also actively participates in the response to infection. By interactions of epithelial cells with bacteria and metabolites, the gingival epithelium further stimulates host responses and integrates innate and acquired immunity in the innate host immune defense response [
6]. Once the junction epithelial and periodontal pocket wall integrity is destroyed, the disease is complicated by tooth migration, drifting, hypermobility, and even loss, further impacting the life quality of the affected individuals [
7,
8]. Furthermore, considerable evidence also points to the fact that relative bacteria and their metabolites, and even the inflammatory mediators originating in the inflamed periodontium, could go beyond the oral cavity via hematogenous dissemination and thus become a risk for various systemic diseases, including atherosclerosis, adverse pregnancy outcomes, rheumatoid arthritis, aspiration pneumonia, and cancer [
1,
2]. Therefore, research on the gingival epithelial barrier is beneficial for establishing the ecological network in terms of periodontal tissue structure and function as well as the further exploration of host-pathogenic factors and therapeutic drugs.
In recent decades, various in vitro models have been reported to study and reproduce the gingival epithelial barrier and characterize the role of gingival epithelium cells in an immuno-inflammatory state. For example, animal models have long been the preferred method for simulating and predicting the response of periodontal tissues to drugs, pathogens, and environmental toxins. However, animal studies are costly, time-consuming, and controversial, and animal models cannot fully recapitulate the correct human physiology, as the subjects are not human [
9]. Despite continuous improvements in two-dimensional cell culture technology, with their simple cell types, the cell culture assays still lack the complexity compared to the living systems. They are incapable of organ–organ or tissue–tissue communication and unable to predict complex host immune response and the effect of metabolite activity on non-target tissues [
10]. Therefore, it is urgently necessary to establish more representative model systems, especially for typical human organs and diseases.
Microfluidic cell culture technology, often referred to as organ-on-a-chip, is rapidly progressing and promising in preparing in vitro models, as it could better recapitulate the key structural and functional aspects of human tissues and organs. The organ-on-a-chip systems could also enable multiple cell interactions and hence are more physiologically relevant [
11]. Although most of the developed organ-on-a-chip systems cannot be considered as organs, they could partially mimic the microarchitectures of the functional units in specific organs or tissue–tissue interfaces. In addition, they could integrate various mechanical and biochemical stimuli to build valid artificial engineering organs. Usually, the organ-on-a-chip systems can be combined with biological microelectromechanical systems (bioMEMS), microfluidics, and biomimetics [
10]. With the advances in microengineering technologies with microfluidic controls, these novel platforms could connect multi-organs and study their interactions on a single chip [
12]. To date, the study of epithelial barrier on-a-chip that could mimic the in vivo function and microenvironment is still lacking, which hinders the understanding of the key structure of gingival human periodontal soft tissue.
From the perspective of organ-on-a-chip construction, the oral cavity is a very special organ that is an open system that is constantly challenged by microorganisms and their toxic products and antigenic components. At the same time, the oral cavity is a complex micro-ecological environment, and a balanced relationship between host and microbes is a prerequisite for periodontal health. The effectiveness of a model depends on its ability to reproduce the key physiological and biological characteristics of its in vivo prototype. The key features of the periodontal soft tissue barrier include: (1) the first barrier, composed of tight junctions between epithelial cells, protecting deep tissues against invasion by foreign microorganisms; (2) co-culture of epithelial and endothelial cells, including material exchange and indirect cell contact, which plays an important role in regulating barrier function via cell–cell signaling; (3) the selective permeation of macromolecules, the regulatory function on the proteins expression in inflammatory states, and the maintenance of high resistive resistance values representing integrity and effectiveness of the structure.
This study discusses and provides procedures for developing epithelium–capillary interfaces on a chip, including the fabrication of bilayer polymers, cell culturing procedure, validation of the developed models through optical imaging, transient permeability tests, and the proteins of intercellular cell adhesion molecule (ICAM-1) and human beta defensin-2 (HBD2) measurement under inflammation with or without inhibitor. Overall, we demonstrate a realistic microfluidic platform, comprising the smallest epithelium–capillary interface on-a-chip, which is capable of mechanically and biochemically modulating the barrier function.
2. Materials and Methods
2.1. Isolation and Culture of Cells
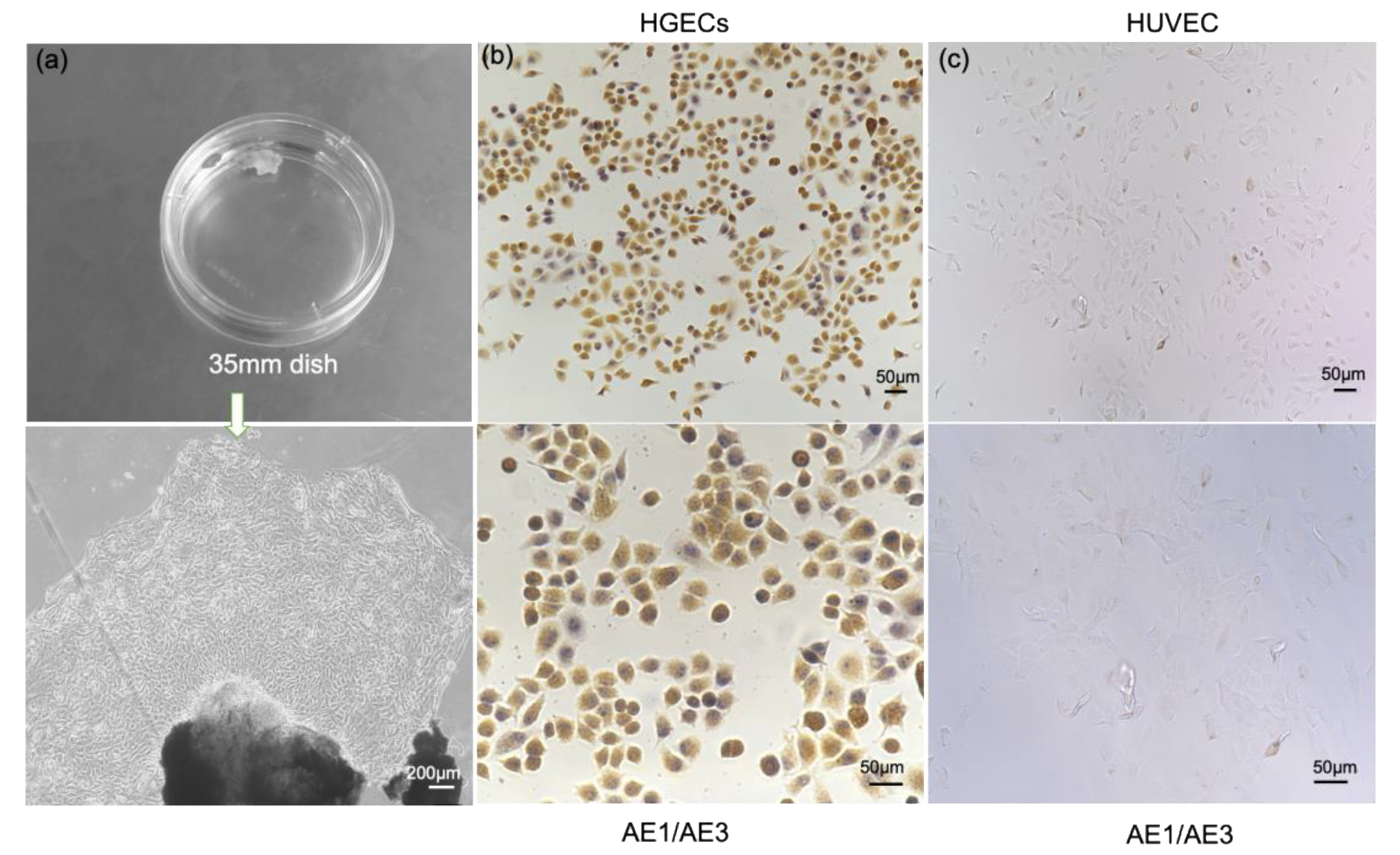
For human studies, approval from the Ethics Committee of Dalian Medical University as well as written informed consent from all participants was obtained. The study was performed in accordance with the principles of the Declaration of Helsinki. As previously reported [
13], with the informed consent of the patients, 1 to 2 mm of healthy gingival tissues were surgically dissected under aseptic conditions from patients aged 18 to 30 years who needed to have their wisdom teeth extracted due to wisdom tooth obstruction. After repeated rinsing in phosphate-buffered saline (PBS) without calcium and magnesium containing streptomycin-penicillin and fungicides, gingival tissues were incubated with 0.4% dispase at 4 °C for 16–18 h. The epithelium was separated from the lamina propria and trimmed into 0.3 cm
2 pieces, trypsinized for 10–15 min at 37 °C in 0.05%/0.53 mM trypsin EDTA, pipetted for 5–8 min, and the supernatant was removed by centrifugation at 700 rpm for 5 min. Next, the cell pellet and tissue fragments were collected and resuspended in keratinocyte growth medium (HuMedia-KG2, Kurabo, Osaka, Japan, KK-2150S). The cells were cultured at 37 °C with 5% CO
2 with the first 3-day fluid change and then every other day to observe the growth status of the cells and used for assays between 4 and 6 passages. Human vascular endothelial cells (Lonza, C2517A) were cultured in EBM-2 medium (Lonza, Japan). All cell culture-related reagents were purchased from Life Technologies Corporation, unless otherwise noted.
2.2. Primary Cell Identification
AE1/AE3 antibody was used to specifically characterize gingival epithelial cells. The digested gingival epithelial cells were inoculated on glass slides, washed with phosphate buffered saline (PBS) three times after culturing for 24 h, fixed with 4% (volume fraction) formaldehyde and incubated for 15 min at 25 °C, and 0.1% (volume fraction) polyethylene glycol octyl phenyl ether (Triton X-100) was also added for an incubation time of 15 min at 25 °C in order to penetrate cellular membranes. For treatment, 3% (volume fraction) hydrogen peroxide (H2O2) was also added. After the serum was blocked, mouse anti-human keratin monoclonal antibody AE1/AE3 (Beijing Zhongshan Jinqiao, China) was added overnight at 4 °C, rinsed with PBS, and then biotin-labeled secondary antibody (diluted by volume ratio 1:50) and horseradish enzyme were added in sequence. The labeled streptavidin was incubated at 25 °C for 30 min each, DAB developed, observed under a microscope, counterstained with hematoxylin, and mounted.
2.3. Measurement of the Transepithelial/Transendothelial Electrical Resistance
TEER was measured using 12 mm transwell inserts with 8 µm pore size polycarbonate membrane (Dow Corning, Midland, MI, USA). The values of TEER were obtained by transferring the transwell inserts into an Endohm chamber. Concentric counter electrodes above and below the membrane caused overlapping current densities to flow through the membrane, and EVOM2 (WPI, Sarasota, FL, USA) calculated the transmembrane resistance based on the currents. All TEER values were obtained after subtracting the background and time of insertion into the membrane [
14].
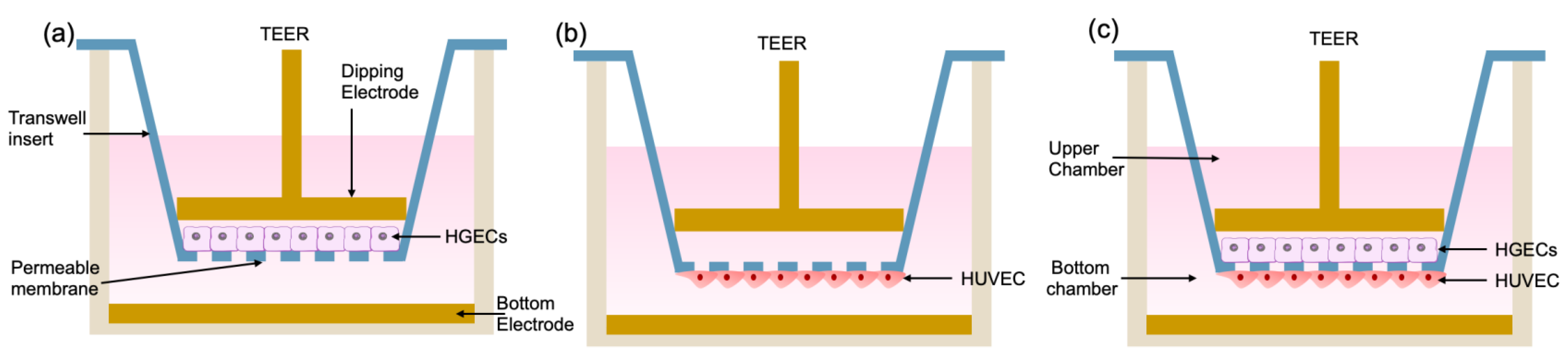
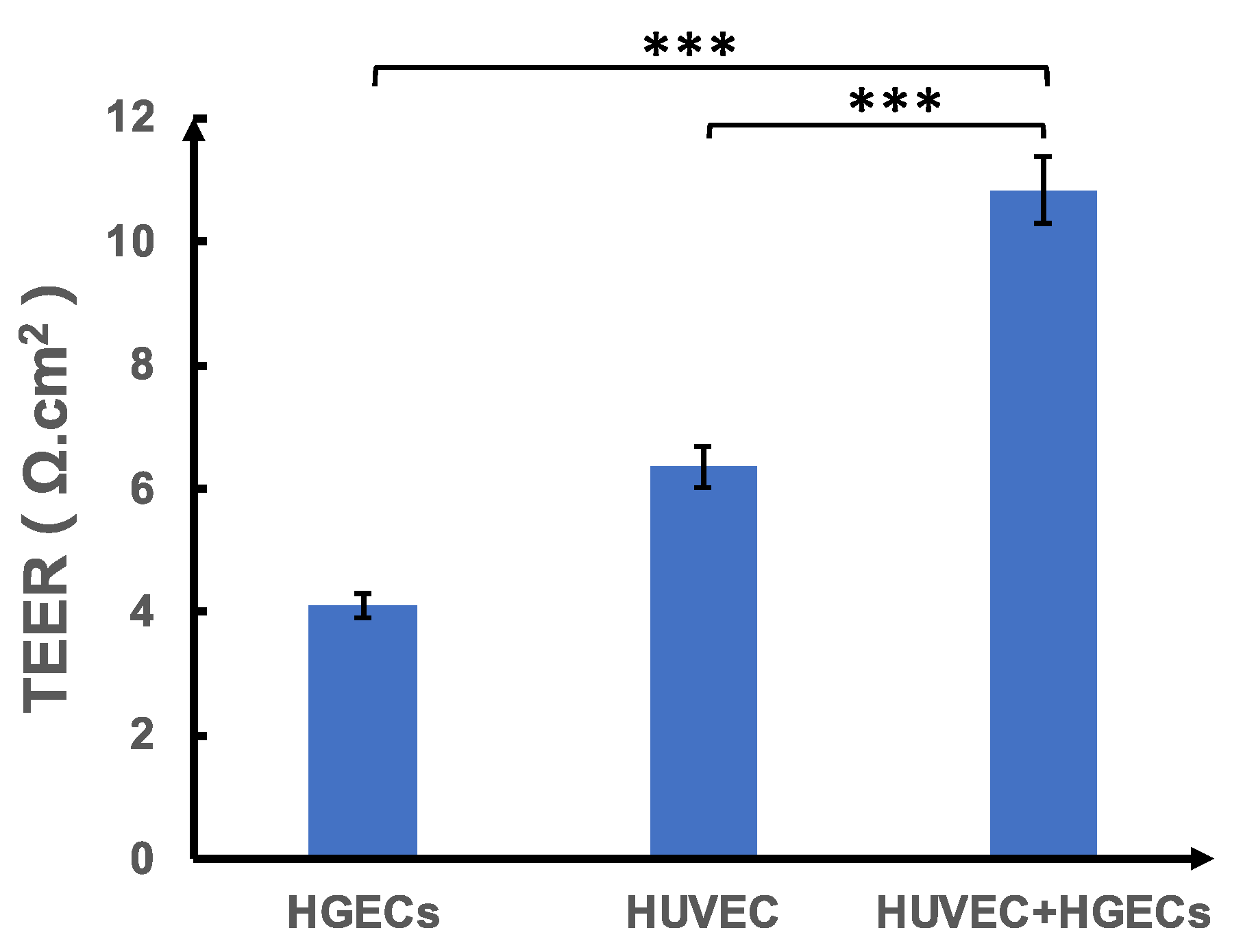
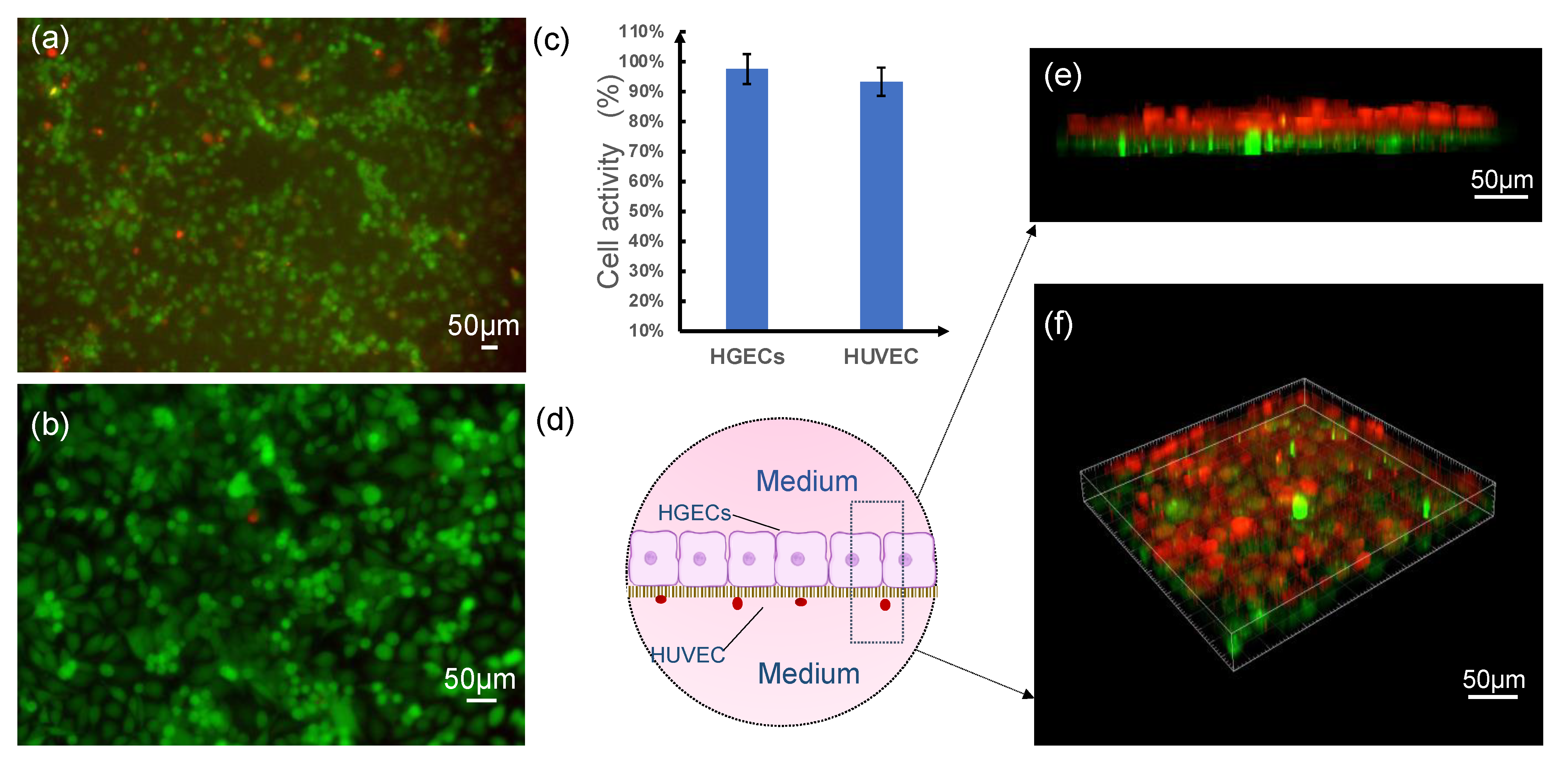
As shown in
Figure 1, we divided the test into three groups. The first group was inoculated with HGEC cells in the upper layer of the chamber, and the lower layer was blank (
Figure 1a); the second group was inoculated with HUVEC cells in the lower layer of the chamber, and the upper layer was blank (
Figure 1b). The third group was a co-culture chamber in which HGEC cells were inoculated in the upper layer, and HUVEC cells were inoculated in the lower layer (
Figure 1c).
2.4. Device Assembly and Operation
The upper and lower layers of the microfluidic device were produced using SU8–3050 negative photoresist (Dow Corning, Midland, MI, USA) and polydimethylsiloxane (PDMS, Dow Corning, Midland, MI, USA) according to the standard soft lithography and microfabrication methods [
15]. The channels of the two layers of the microfluidic device were separated by a thin (10 μm) semi porous polyester membrane (1 μm pores) that was purchased from Sterlitech Corporation (Auburn, WA, USA). The bonding of the device was carried out as previously described [
16]. PDMS and porous polyester (PETE) membranes were immersed for 20 min in a 1% (volume fraction) aqueous solution of 3-Glycidoxypropyltrimethoxysilane (GLMYO, Sigma-Aldrich, St. Louis, MO, USA) and a 5% (volume fraction) aqueous solution of 3-Aminopropyltriethoxysilane (APTS, Sigma-Aldrich, St. Louis, MO, USA), respectively, rinsed in water, and dried in a stream of compressed air. The porous PETE membrane was finally aligned and brought into contact with the PDMS layers comprising the basal microfluidic compartment that formed the microfluidic compartment and then the surfaces were pressed together. A porous PETE membrane was sandwiched between the two microfluidic channels during bonding. The assembled microfluidic chips were finally baked at 60 °C overnight.
2.5. In Vitro Co-Culture Epithelium–Capillary Interface Models and Assembly
To form a bilayer epithelial-capillary on the chip, the epithelial and endothelial cell suspensions were seeded on the upper and lower sides of the porous membrane in the device, respectively. Prior to cell inoculation, the chambers of the sterilized chip were filled with liquid and immersed in culture medium overnight. First, the chip was turned over so that its lower chamber could face upwards, and HUVEC cells were introduced into the lower chamber with a pipette at a concentration of 1 × 105 cells/mL. After resting for 2 h for cells to attach, the chip was turned over again so that its upper chamber faced upward and HGEC cells were introduced into the upper chamber at a concentration of 8.9 × 104 cells/mL. After HGEC cells adherence, the cell culture medium was refreshed every 24 h with the HGEC cells side facing up. The cells in both microchambers were grown to confluence within three days. Once the cells reached confluence, they were treated with culture medium containing either LPS (10 μg mL−1) or TNF-α (10 ng mL−1) to promote the formation of inflammation model in vitro. Inflammation stimulation experience and all cell cultivation was carried out in the incubator, which maintained a constant interior environment at 5% CO2 and 37 °C.
2.6. Cell Staining
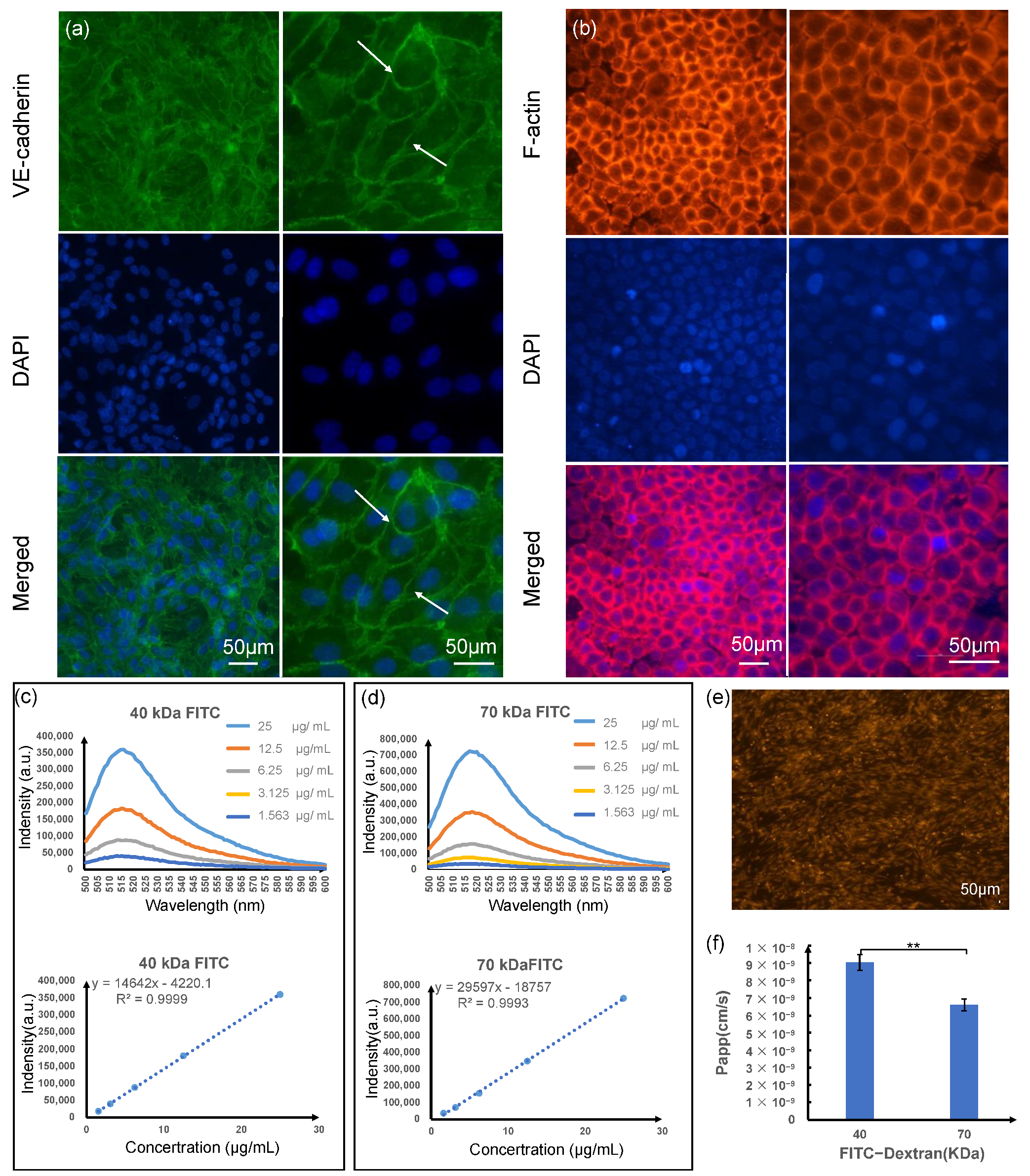
For morphological observations, light-phase and fluorescence imaging were used. Live/dead experiments were used to detect the cellular activity, and immunostaining was used to observe the expression of F-actin and tight junction marker proteins VE-cadherin. In order to analyze the viability of cultured cells, calcein AM and ethidium homodimer-1 of the Live/dead Kit (Invitrogen, Carlsbad, CA, USA) were mixed in a ratio of 1:4, incubated for 15 min and imaged using a fluorescence microscope. Immunostaining for both types of cells was fixed with 4% (volume fraction) formaldehyde and incubated for 10 min at 25 °C, and 0.1% Triton X-100 (volume fraction) was also added for an incubation time of 15 min at 25 °C in order to penetrate cellular membranes. For treatment, 3% (volume fraction) bovine serum (Invitrogen, Carlsbad, CA, USA) in a permeabilization buffer was also added. After the serum was blocked, the primary antibody was added in a blocking buffer overnight at 4 °C, and then a secondary antibody or a compatible counterstain for the cytoskeleton was added in sequence. The labeled streptavidin or a compatible counterstain for the cytoskeleton was incubated at 25 °C for 1 h, and then the nucleus was stained with DAPI (Sigma-Aldrich, St. Louis, MO, USA) for 5 min at 25 °C, protected from light. Rabbit anti-VE-cadherin (Invitrogen, Carlsbad, CA, USA) at 1/25 dilution was used in conjunction with Alexa Fluor 488 goat anti-rabbit secondary antibody (Invitrogen, Carlsbad, CA, USA) for HUVEC cells. Phalloidin- iFluor®594 (Abcam, Cambridge, UK) at a 1/1000 dilution in PBS was used for HGECs. Images was taken using a Nikon fluorescence microscope.
2.7. Characterization of Cell Layers
To assess barrier permeabilities to large compounds, fluxes of fluorescent tracers over a wide range of sizes were measured after steady-state HUVEC layers had been reached. The absorption and barrier capacities of the HUVEC layers were evaluated by measuring the apparent permeability (P
app) of labeled dextran (FITC-dextran, MW 40 kDa, and MW 70 kDa, Sigma-Aldrich, St. Louis, MO, USA) through the cell layers. A Horiba FluoroMax-4 spectrofluorometer and Orient KOJI Semi-Micro spectrofluorometer cuvettes were utilized to perform emission fluorescence measurements. Slit sizes were set at 1.5 nm for all monochromators. All processes were conducted at room temperature (25 ± 1 °C). According to the manufacturer’s requirements, we prepared a concentration gradient of FITC-dextran standard sample, measured the fluorescence intensity value at 518 nm, and made a standard curve to calculate the concentration of the sample to be tested; P
app was calculated using the equation:
where A = area of mass transfer, C
0 = donor concentration of reagent in upper medium, and ∆C/∆t = transmembrane transportation rate.
All permeability assays were conducted after day 3 of endothelial culture.
2.8. Analysis of the Protein Expression
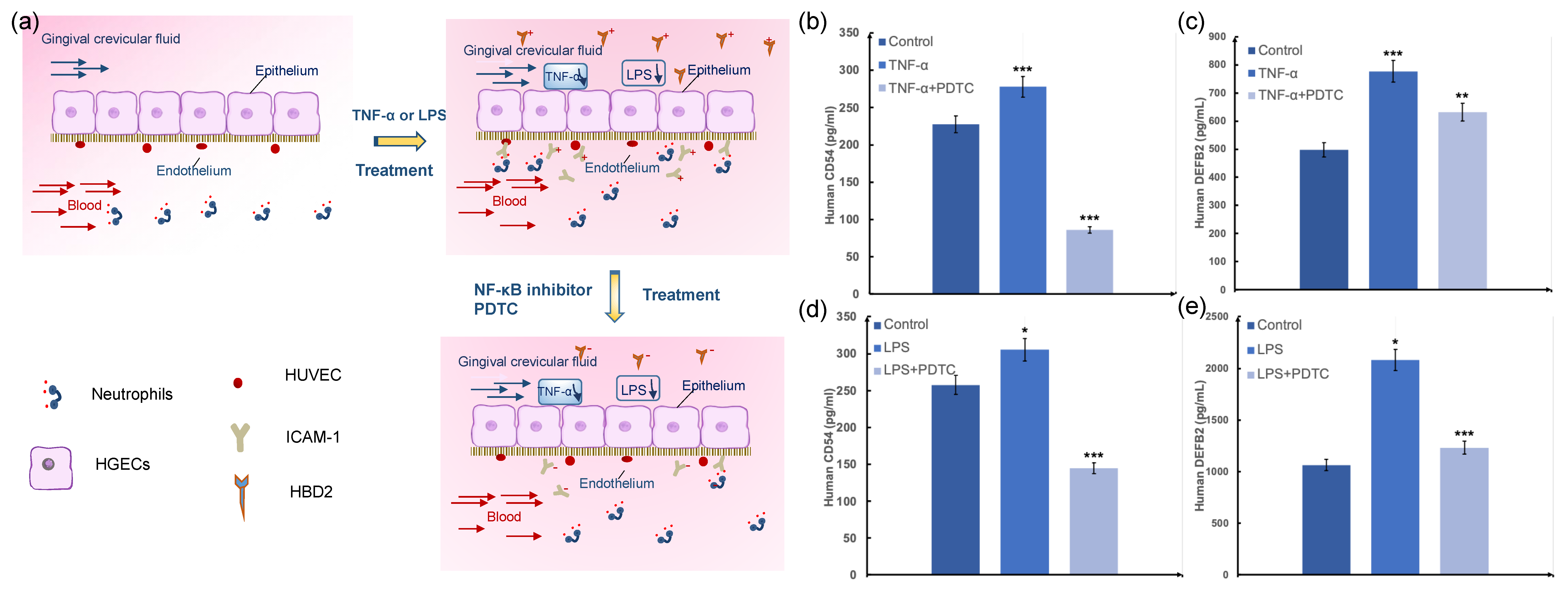
The effluent of the medium was analyzed for a panel of intercellular cell adhesion molecule (ICAM-1) and human beta defensin-2 (HBD2) using custom ELISA assay kits (Abbkine, Wuhan, Hubei, CN). Analyte concentrations were determined according to the manufacturer’s instructions using enzyme-labeled instrument coupled with Origin software (OriginLab, Northampton, MA, USA). For inflammation stimulation experiments, healthy epithelia were treated with LPS (10 μg mL−1) or TNF-α (10 ng mL−1) for 24 h, and amounts of secreted ICAM-1 and HBD2 were measured 24 h after treatment. For periodontitis drug studies in chips containing cocultures of human gingival epithelium and endothelium, the endothelial cells were treated with 25 mM NF-κB inhibitor PDTC (Ammonium pyrrolidinedithiocarbamate, Sigma-Aldrich, St. Louis, MO, USA) or blank medium under for 4 h before LPS (10 μg mL−1) or TNF-α (10 ng mL−1) was delivered into the epithelium channel for 24 h. The vascular effluents and epithelium effluents were then collected for ICAM-1 and HBD2 analysis.
2.9. Statistical Analysis
All results and error bars are presented as mean and SEM. Data were analyzed with an unpaired Student’s t-test using Origin (OriginLab, Northampton, MA, USA) or Excel software (Microsoft, Redmond, WA, USA). Differences between groups were considered statistically significant when * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
The host–microorganism homeostasis of the epithelial barrier within periodontal soft tissue determines how these key cells function and interact with their surrounding environment [
5]. In order to capture the abnormalities of the periodontitis and better replicate epithelium behavior than monolayer cultured cells in the host immune response, co-culturing of main cells such as epithelial cells and incorporation of a defined perfusable vasculature can help investigate the evolving epithelial barrier and its effect on drug bioavailability inside the vascularized periodontal soft tissue.
In this study, a highly integrated periodontal soft tissue chip model was constructed based on the anatomical structure and physiological functions of periodontal soft tissues. Cell spatial arrangement and distribution ratio are used to study periodontal soft tissue physiology and cell biological behavior under inflammatory conditions. The characteristic of this microfluid model is that it used many bionic designs, making the established in vitro model more compatible with in vivo physiology. Physiologically, epithelial tissue acts as the first barrier against foreign invasions. It plays an important role in controlling microbial infections, protecting subepithelial tissues, and maintaining periodontal tissue homeostasis [
23]. The cytoskeleton (F-actin) showed a good condition of HGEC cells as the cells were dispersed from one another and kept the stable cell numbers, ensuring the physical barrier function of the epithelial barrier. In addition, the gingival epithelium not only functions as a physical barrier, but it also produces antibacterial substances such as defensins. When periodontal tissue was invaded by microorganisms, the inflammatory mediators produced by epithelial cells also activated endothelial cells to enhance the expression of endothelial cell adhesion molecules, promoted the release of cytokines and polymorphonuclear granulocytes, and secreted a series of pro-inflammatory mediators that can cause periodontal tissue microvascular and tissue disease. This results in monocytes/macrophages infiltrating the blood vessel wall, causing damage to other organs caused by small vessel disease. Therefore, the barrier function of vascular endothelial cells cannot be ignored in the periodontal soft tissue barrier [
23,
24]. Here, the HUVEC cell monolayer can readily express tight junctions and higher permeability to tracers of lower stokes radius, indicating that smaller compounds pass through junctions more easily and maintain an effective endothelial barrier. In addition, endothelial and epithelial culture on the membranes indicated high cell viability (>90%) of HUVEC cells and HGEC cells cultured in the microdevice on day 3, the much thinner culture membrane, decreasing the distance between co-cultured cells from the epithelium-capillary interface model, and this made HGEs and HUVEC cells maintain a small population while mimicking the function of the gingival epithelial barrier. Overall, imaging indicated that the microdevice exhibited characteristics desirable for gingival epithelial barrier study, and cells are co-cultured in close contact with significantly lower costs and timescales than in vivo studies.
Compared with animal models and traditional flat-panel models, different types of functional cells and perfusion media can be easily and independently recovered from this device. Therefore, multiple biomarkers of each type of cell can be readily characterized by an off-line analysis, such as immunofluorescence staining. In the present study, we used this model to examine interactions between HGECs and HUVEC cells in periodontitis. We observed that both LPS and TNF-α could increase the expression with ICAM-1 and HBD2, the key factors in the activation of endothelial function and oral natural immunity. The adding of inhibitor PDTC can decrease in the chip and is consistent with the expression in animal experience, and this cell-parallel, controlled and repeated environment is not available with tradition models. Thus, the epithelium–capillary interface-on-a-chip may be a relevant model with making a dynamic microenvironment providing shear stress stimulation to the cells and allows the improved analysis of test compounds and controlled delivery compared to static models for periodontal soft tissue. However, it can also be adapted for studies of inflammation-based systemic disease such as meningitis, coronary heart disease, Alzheimer’s disease, and leukemia.
Limitations of this study include the use of HUVEC cells instead of periodontal endothelial cells derived from the periodontal microvasculature. We expect the effects on related medicine and cytokine secretion to be even more pronounced once periodontal associated microvasculature cells are used. Importantly, this work reproduced the key function interface of periodontal soft tissue and proposed the application of organ-on-a-chip in the oral cavity, which provides a novel in vitro platform for other oral diseases and other related organ diseases.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






