Enhanced Specificity in Loop-Mediated Isothermal Amplification with Poly(ethylene glycol)-Engrafted Graphene Oxide for Detection of Viral Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and DNA Polymerase
2.2. Primer DNAs
2.3. Total RNA Extraction and Reverse Transcription
2.4. Preparation of PEG-nGO
2.5. LAMP Assays
2.6. Gel Electrophoresis
2.7. ssDNA Adsorption on PEG-nGO
2.8. RT-qPCR
3. Results and Discussion
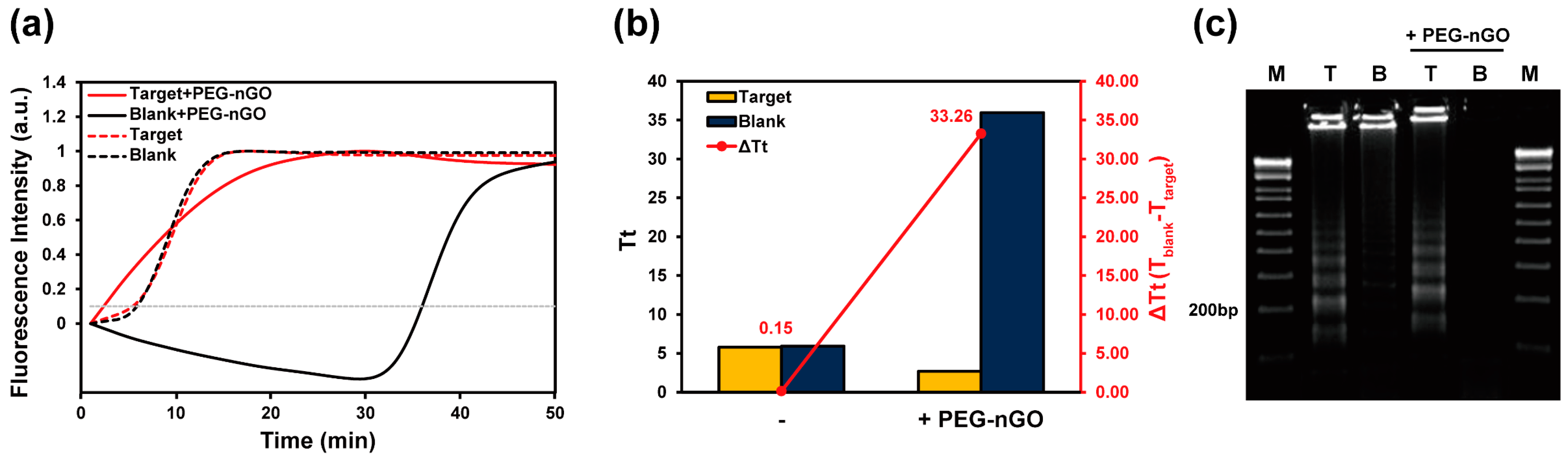
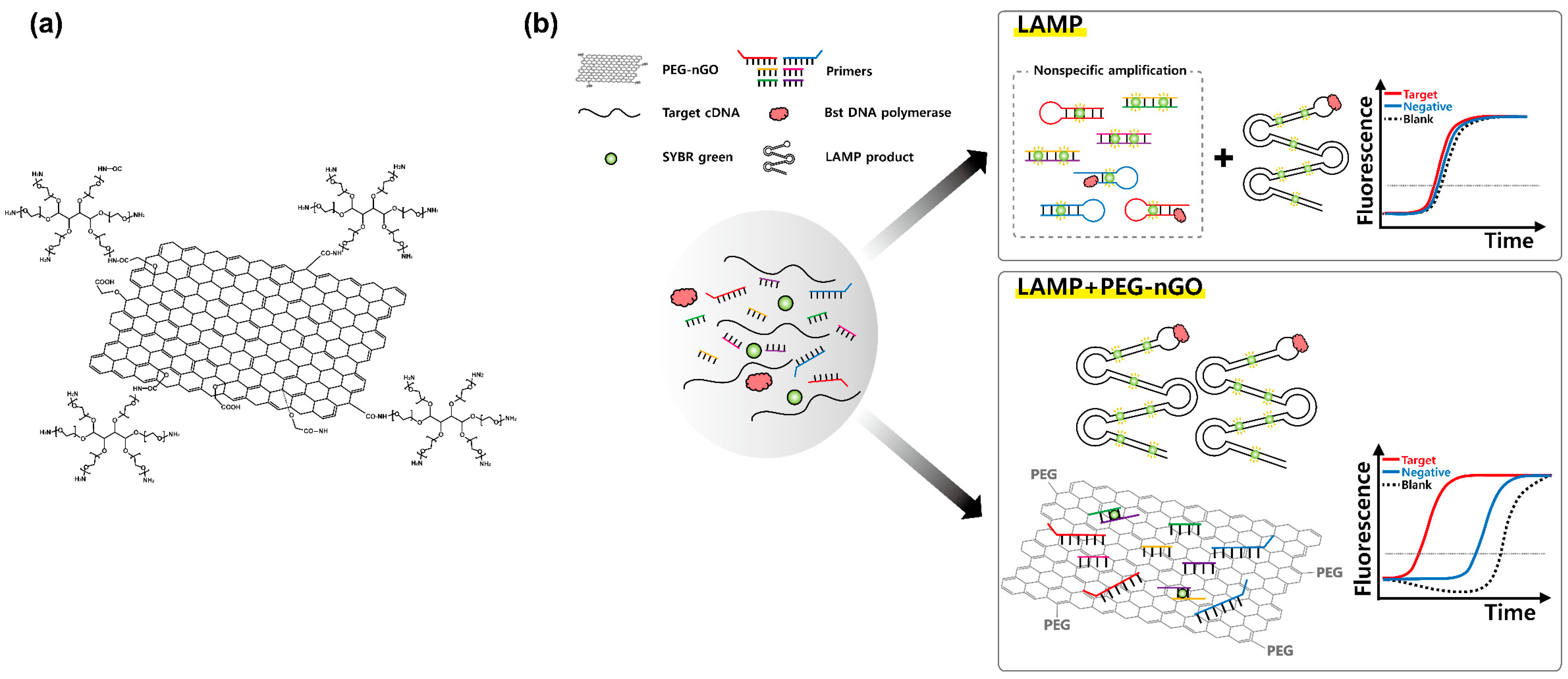
3.1. Working Mechanism of LAMP Facilitation by PEG-nGO
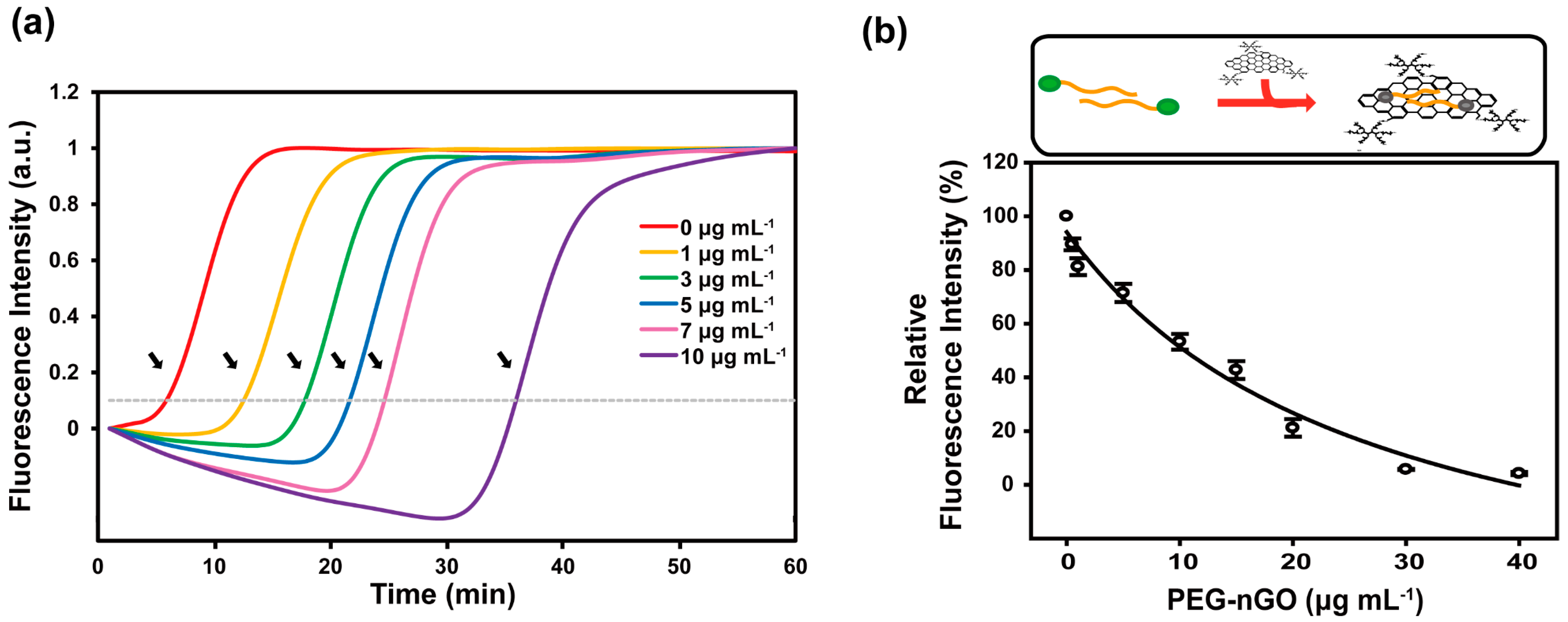
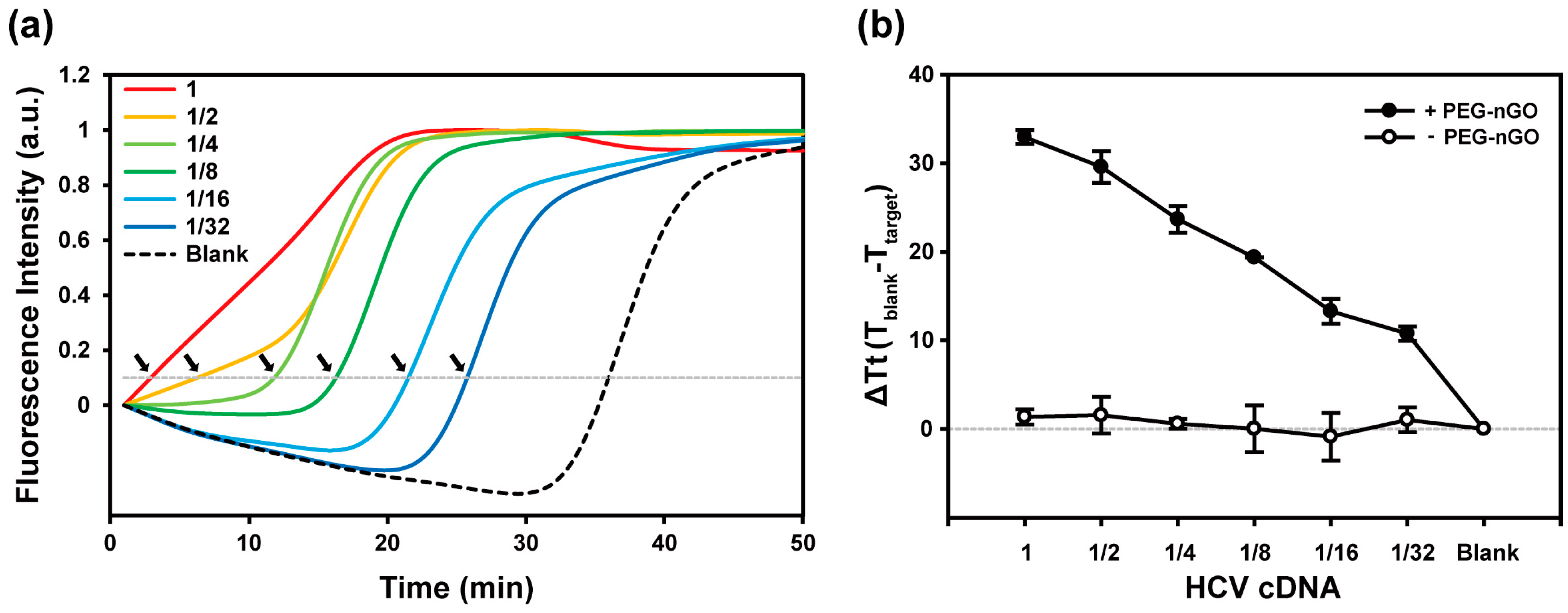
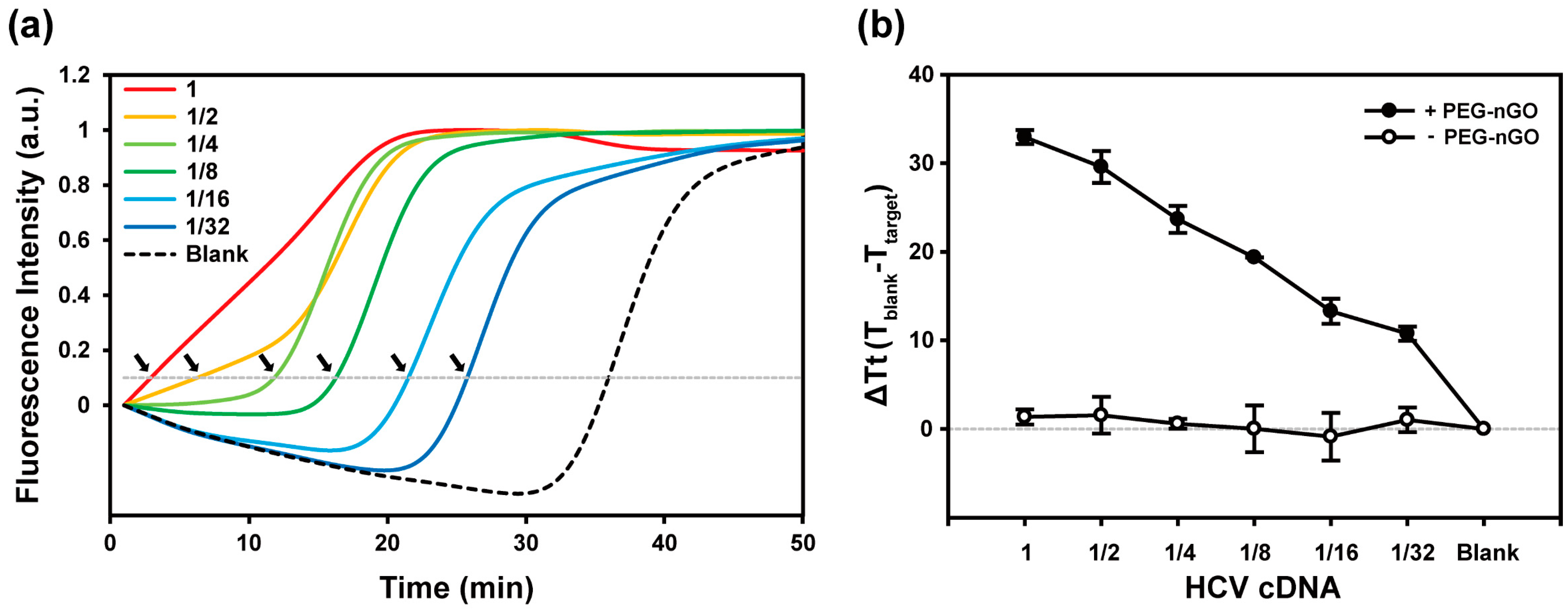
3.2. Specificity and Sensitivity of PEG-nGO-Based LAMP
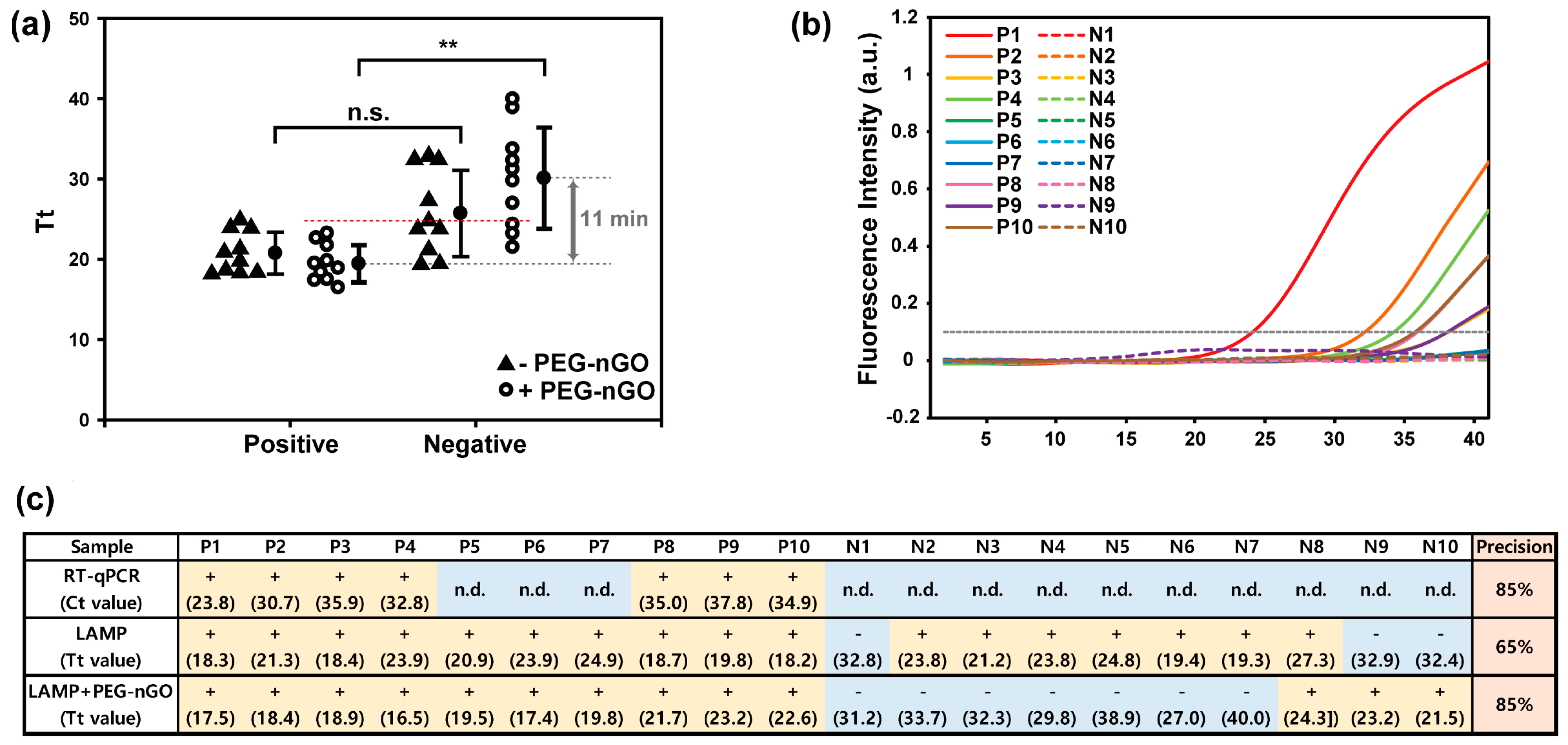
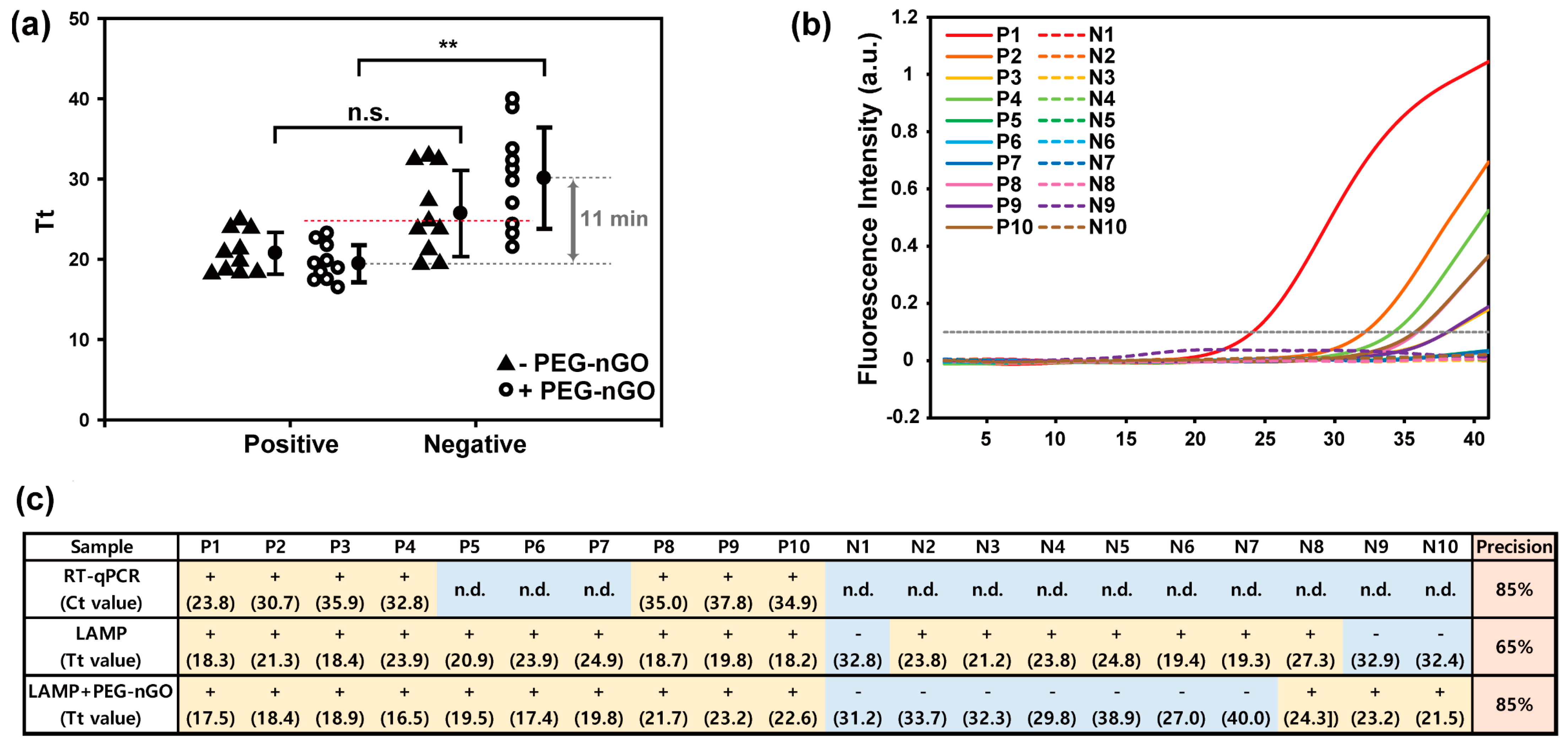
3.3. Detection of HCV-Positive and HCV-Negative Serum Samples by PEG-nGO-Based LAMP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, N.; Mori, Y.; Kanda, H.; Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 2008, 3, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, D.; Li, K.; Ye, C. Rapid and sensitive detection of Vibrio parahaemolyticus and Vibrio vulnificus by multiple endonuclease restriction real-time loop-mediated isothermal amplification technique. Molecules 2016, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell Probes 2002, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, K.; Kuzuhara, Y.; Notomi, T. Isolation of single-stranded DNA from loop-mediated isothermal amplification products. Biochem. Biophys. Res. Commun. 2002, 290, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.A.; Zhang, Y.; Evans, T.C., Jr. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. Biotechniques 2012, 53, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Jung, C.; Bhadra, S.; Ellington, A.D. Phosphorothioated primers lead to loop-mediated isothermal amplification at low temperatures. Anal. Chem. 2018, 90, 8290–8294. [Google Scholar] [CrossRef] [Green Version]
- Seevaratnam, D.; Ansah, F.; Aniweh, Y.; Awandare, G.A.; Hall, E.A.H. Analysis and validation of silica-immobilised BST polymerase in loop-mediated isothermal amplification (LAMP) for malaria diagnosis. Anal. Bioanal. Chem. 2022, 414, 6309–6326. [Google Scholar] [CrossRef]
- Jiang, Y.S.; Bhadra, S.; Li, B.; Wu, Y.R.; Milligan, J.N.; Ellington, A.D. Robust strand exchange reactions for the sequence-specific, real-time detection of nucleic acid amplicons. Anal. Chem. 2015, 87, 3314–3320. [Google Scholar] [CrossRef]
- Ball, C.S.; Light, Y.K.; Koh, C.Y.; Wheeler, S.S.; Coffey, L.L.; Meagher, R.J. Quenching of unincorporated amplification signal reporters in reverse-transcription loop-mediated isothermal amplification enabling bright, single-step, closed-tube, and multiplexed detection of RNA viruses. Anal. Chem. 2016, 88, 3562–3568. [Google Scholar] [CrossRef]
- Iwamoto, T.; Sonobe, T.; Hayashi, K. Loop-mediated isothermal amplification for direct detection of Mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. J. Clin. Microbiol. 2003, 41, 2616–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Yu, H.; Fang, J.; Sun, C.; Zhang, M. Employing DNA binding dye to improve detection of Enterocytozoon hepatopenaei in real-time LAMP. Sci. Rep. 2019, 9, 15860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, F.; Qin, J.; Wei, J.; Wu, W.; Zhao, Y. Engineered Janus probes modulate nucleic acid amplification to expand the dynamic range for direct detection of viral genomes in one microliter crude serum samples. Chem. Sci. 2018, 9, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ye, X.; Huang, Z.; Yang, B.; Fang, X.; Chen, H.; Kong, J. Graphene oxide-based suppression of nonspecificity in loop-mediated isothermal amplification enabling the sensitive detection of cyclooxygenase-2 mRNA in colorectal cancer. Anal. Chem. 2019, 91, 15694–15702. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.H.; Lin, Y.C.; Teng, P.H.; Chen, C.L.; Lee, P.Y. Real-time target-specific detection of loop-mediated isothermal amplification for white spot syndrome virus using fluorescence energy transfer-based probes. J. Virol. Methods 2011, 173, 67–74. [Google Scholar] [CrossRef]
- Hardinge, P.; Murray, J.A.H. Reduced false positives and improved reporting of loop-mediated isothermal amplification using quenched fluorescent primers. Sci. Rep. 2019, 9, 7400. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Mikosza, A.S.; Armstrong, T.; Enyaru, J.C.; Ndung’u, J.M.; Thompson, A.R. Loop-mediated isothermal amplification (LAMP) method for rapid detection of Trypanosoma brucei rhodesiense. PLoS Negl. Trop. Dis. 2008, 2, e147. [Google Scholar] [CrossRef]
- Varona, M.; Anderson, J.L. Visual etection of single-nucleotide polymorphisms using molecular beacon loop-mediated isothermal amplification with centrifuge-free DNA extraction. Anal. Chem. 2019, 91, 6991–6995. [Google Scholar] [CrossRef] [Green Version]
- Ghaith, D.M.; Abu Ghazaleh, R. Carboxamide and N-alkylcarboxamide additives can greatly reduce non specific amplification in loop-mediated isothermal amplification for foot-and-mouth disease virus (FMDV) using Bst 3.0 polymerase. J. Virol Methods 2021, 298, 114284. [Google Scholar] [CrossRef]
- Gao, X.; Sun, B.; Guan, Y. Pullulan reduces the non-specific amplification of loop-mediated isothermal amplification (LAMP). Anal. Bioanal. Chem. 2019, 411, 1211–1218. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, G.; Buss, J.; Barry, A.J.; Patton, G.C.; Tanner, N.A. Enhancing colorimetric loop-mediated isothermal amplification speed and sensitivity with guanidine chloride. Biotechniques 2020, 69, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.G.; Brewster, J.D.; Paul, M.; Tomasula, P.M. Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules 2015, 20, 6048–6059. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Fang, X.; Li, X.; Kong, J. Gold nanoparticle-mediated nucleic acid isothermal amplification with enhanced specificity. Anal. Chim. Acta 2018, 1043, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.S.; Paliwoda, R.E.; Zhang, H.; Le, X.C. Reduction of background generated from template-template hybridizations in the exponential amplification reaction. Anal. Chem. 2018, 90, 11033–11039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Cote, L.J.; Huang, J. Two dimensional soft material: New faces of graphene oxide. Acc. Chem. Res. 2012, 45, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Feng, H.; Li, J. Graphene oxide: Preparation, functionalization, and electrochemical applications. Chem. Rev. 2012, 112, 6027–6053. [Google Scholar] [CrossRef]
- Park, J.S.; Goo, N.I.; Kim, D.E. Mechanism of DNA adsorption and desorption on graphene oxide. Langmuir 2014, 30, 12587–12595. [Google Scholar] [CrossRef]
- Lei, H.; Mi, L.; Zhou, X.; Chen, J.; Hu, J.; Guo, S.; Zhang, Y. Adsorption of double-stranded DNA to graphene oxide preventing enzymatic digestion. Nanoscale 2011, 3, 3888–3892. [Google Scholar] [CrossRef]
- Kim, H.R.; Baek, A.; Lee, I.J.; Kim, D.E. Facilitation of polymerase hain reaction with poly(ethylene glycol)-engrafted graphene oxide analogous to a single-stranded-DNA binding protein. ACS Appl. Mater. Interfaces 2016, 8, 33521–33528. [Google Scholar] [CrossRef]
- Ye, N.; Wang, Z.; Wang, S.; Fang, H.; Wang, D. Aqueous aggregation and stability of graphene nanoplatelets, graphene oxide, and reduced graphene oxide in simulated natural environmental conditions: Complex roles of surface and solution chemistry. Env. Sci. Pollut. Res. Int. 2018, 25, 10956–10965. [Google Scholar] [CrossRef]
- Zhang, M.; Mao, X.; Wang, C.; Zeng, W.; Zhang, C.; Li, Z.; Fang, Y.; Yang, Y.; Liang, W.; Wang, C. The effect of graphene oxide on conformation change, aggregation and cytotoxicity of HIV-1 regulatory protein (Vpr). Biomaterials 2013, 34, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Katoch, J.; Kim, S.N.; Kuang, Z.; Farmer, B.L.; Naik, R.R.; Tatulian, S.A.; Ishigami, M. Structure of a peptide adsorbed on graphene and graphite. Nano Lett. 2012, 12, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Alava, T.; Mann, J.A.; Theodore, C.; Benitez, J.J.; Dichtel, W.R.; Parpia, J.M.; Craighead, H.G. Control of the graphene-protein interface is required to preserve adsorbed protein function. Anal. Chem. 2013, 85, 2754–2759. [Google Scholar] [CrossRef]
- Jia, J.; Sun, L.; Hu, N.; Huang, G.; Weng, J. Graphene enhances the specificity of the polymerase chain reaction. Small 2012, 8, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, J.; Ju, H.; Lu, H.; Wang, S.; Jin, S.; Hao, K.; Du, H.; Zhang, X. Highly sensitive multiple microRNA detection based on fluorescence quenching of graphene oxide and isothermal strand-displacement polymerase reaction. Anal. Chem. 2012, 84, 4587–4593. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ku, J.; Chauhan, K.; Hwang, S.-H.; Jeong, Y.-J.; Kim, D.-E. Enhanced Specificity in Loop-Mediated Isothermal Amplification with Poly(ethylene glycol)-Engrafted Graphene Oxide for Detection of Viral Genes. Biosensors 2022, 12, 661. https://doi.org/10.3390/bios12080661
Ku J, Chauhan K, Hwang S-H, Jeong Y-J, Kim D-E. Enhanced Specificity in Loop-Mediated Isothermal Amplification with Poly(ethylene glycol)-Engrafted Graphene Oxide for Detection of Viral Genes. Biosensors. 2022; 12(8):661. https://doi.org/10.3390/bios12080661
Chicago/Turabian StyleKu, Jamin, Khushbu Chauhan, Sang-Hyun Hwang, Yong-Joo Jeong, and Dong-Eun Kim. 2022. "Enhanced Specificity in Loop-Mediated Isothermal Amplification with Poly(ethylene glycol)-Engrafted Graphene Oxide for Detection of Viral Genes" Biosensors 12, no. 8: 661. https://doi.org/10.3390/bios12080661
APA StyleKu, J., Chauhan, K., Hwang, S.-H., Jeong, Y.-J., & Kim, D.-E. (2022). Enhanced Specificity in Loop-Mediated Isothermal Amplification with Poly(ethylene glycol)-Engrafted Graphene Oxide for Detection of Viral Genes. Biosensors, 12(8), 661. https://doi.org/10.3390/bios12080661