Dual-Domain Reporter Approach for Multiplex Identification of Major SARS-CoV-2 Variants of Concern in a Microarray-Based Assay

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Clinical Samples
2.3. RNA Extraction
2.4. Reverse Transcription and PCR Conditions
2.5. Silicon Chip Coating and Microarray Preparation
2.6. SARS-CoV-2 Variants Specific Hybridization in Solution
2.7. Microarray Hybridization and Image Scanning
3. Results and Discussion
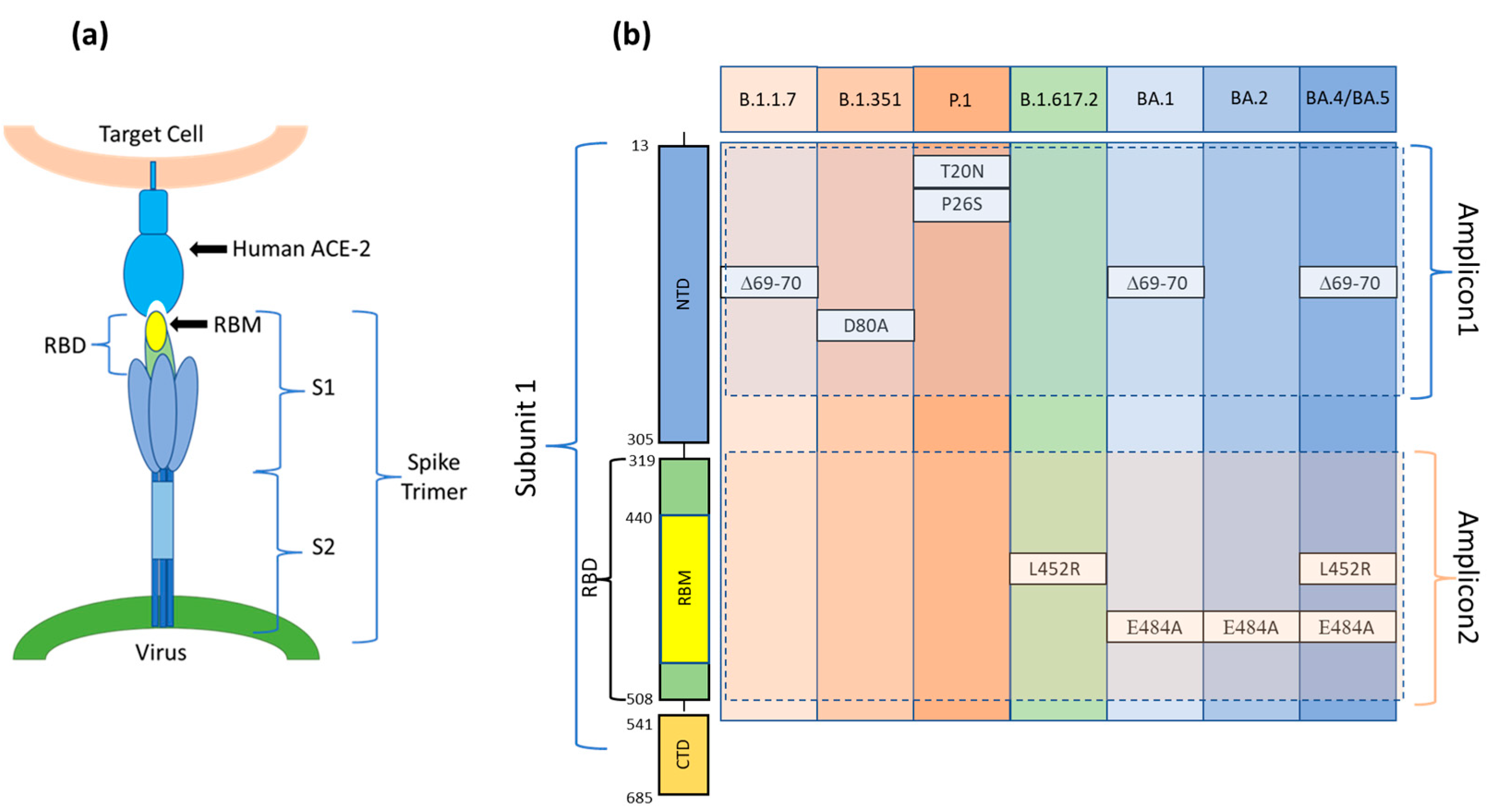
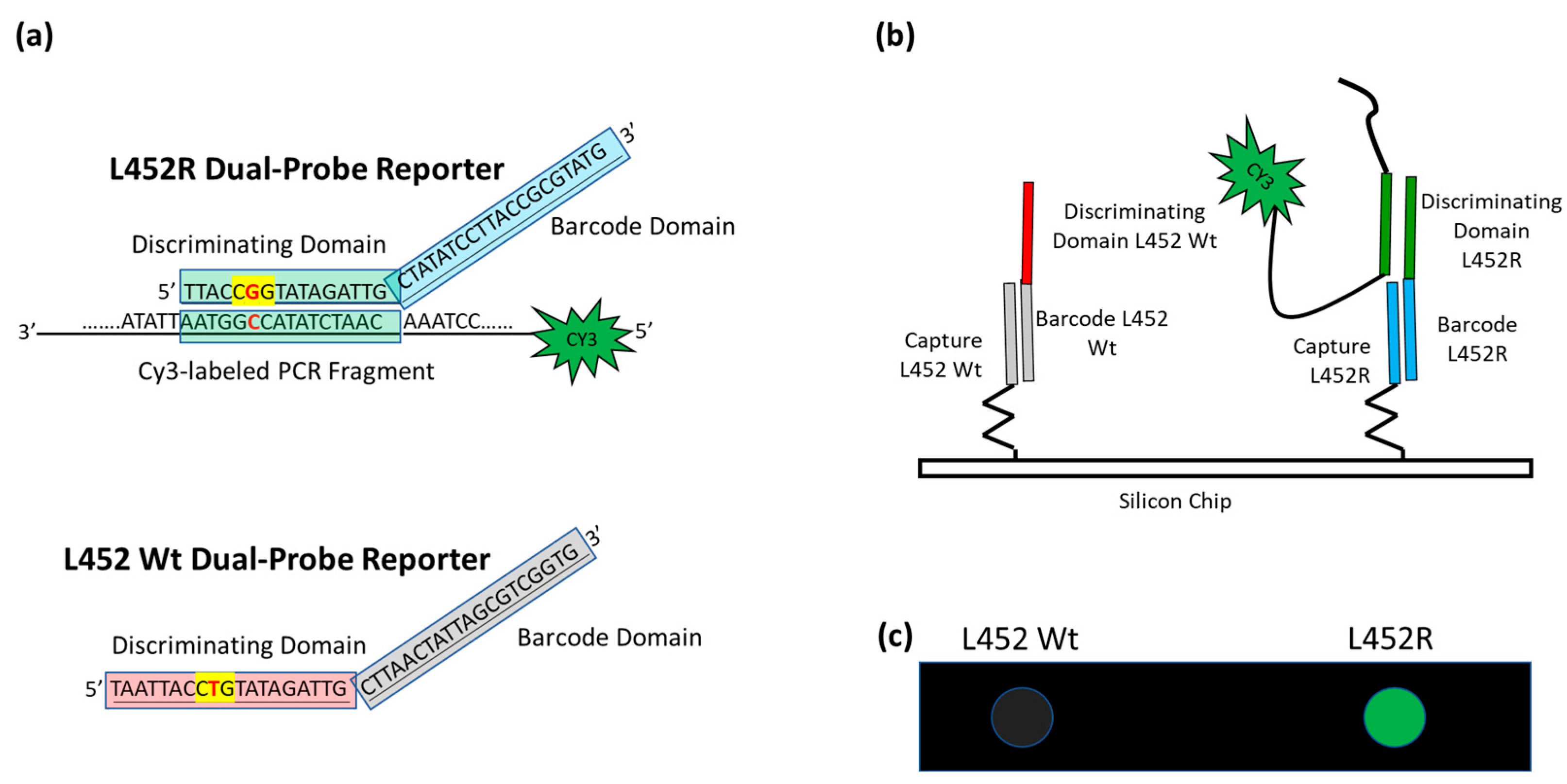
3.1. Design of the Assay for SARS-CoV-2 Variants
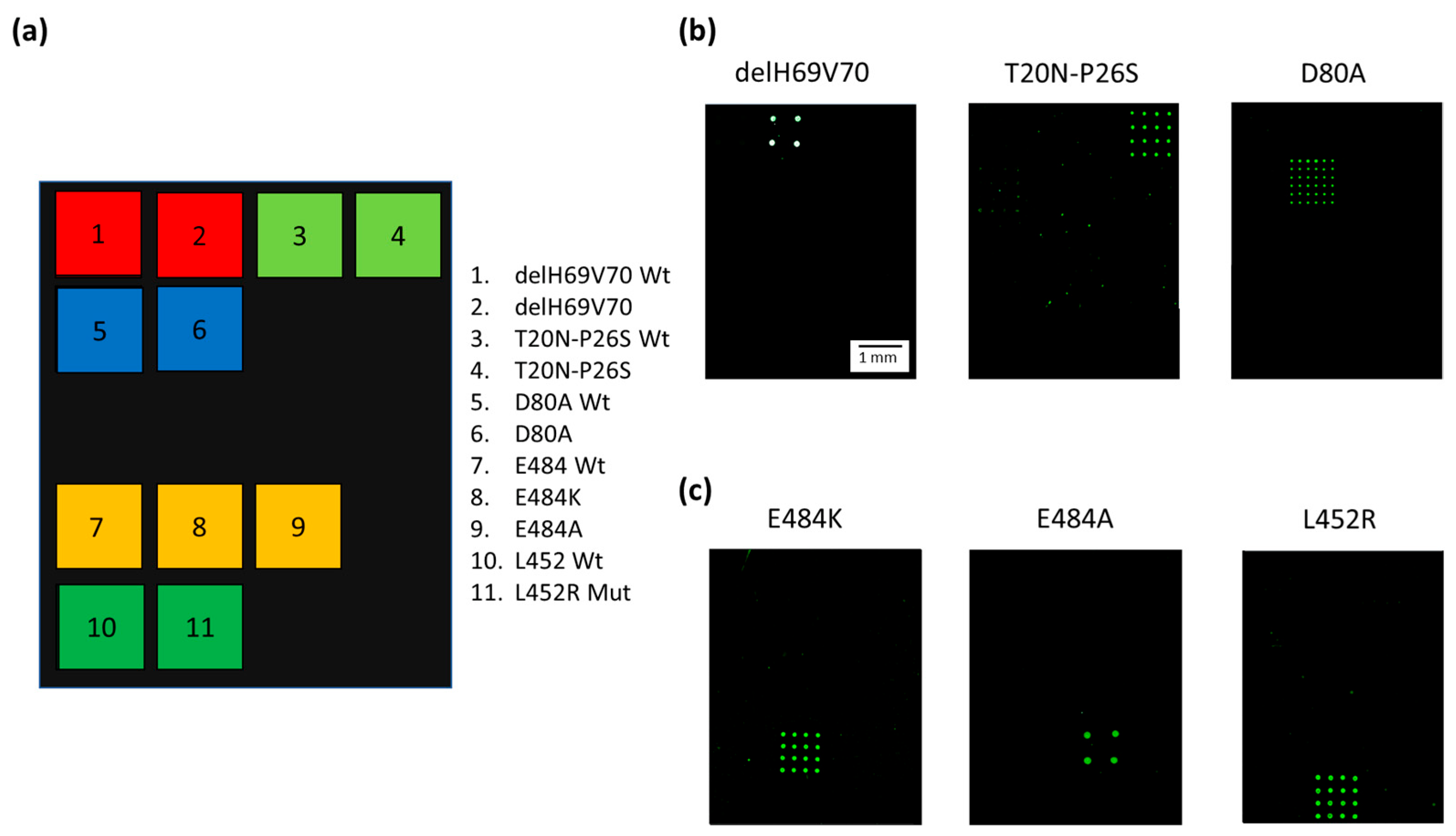
3.2. Specificity of the Variant-Microarray Technology
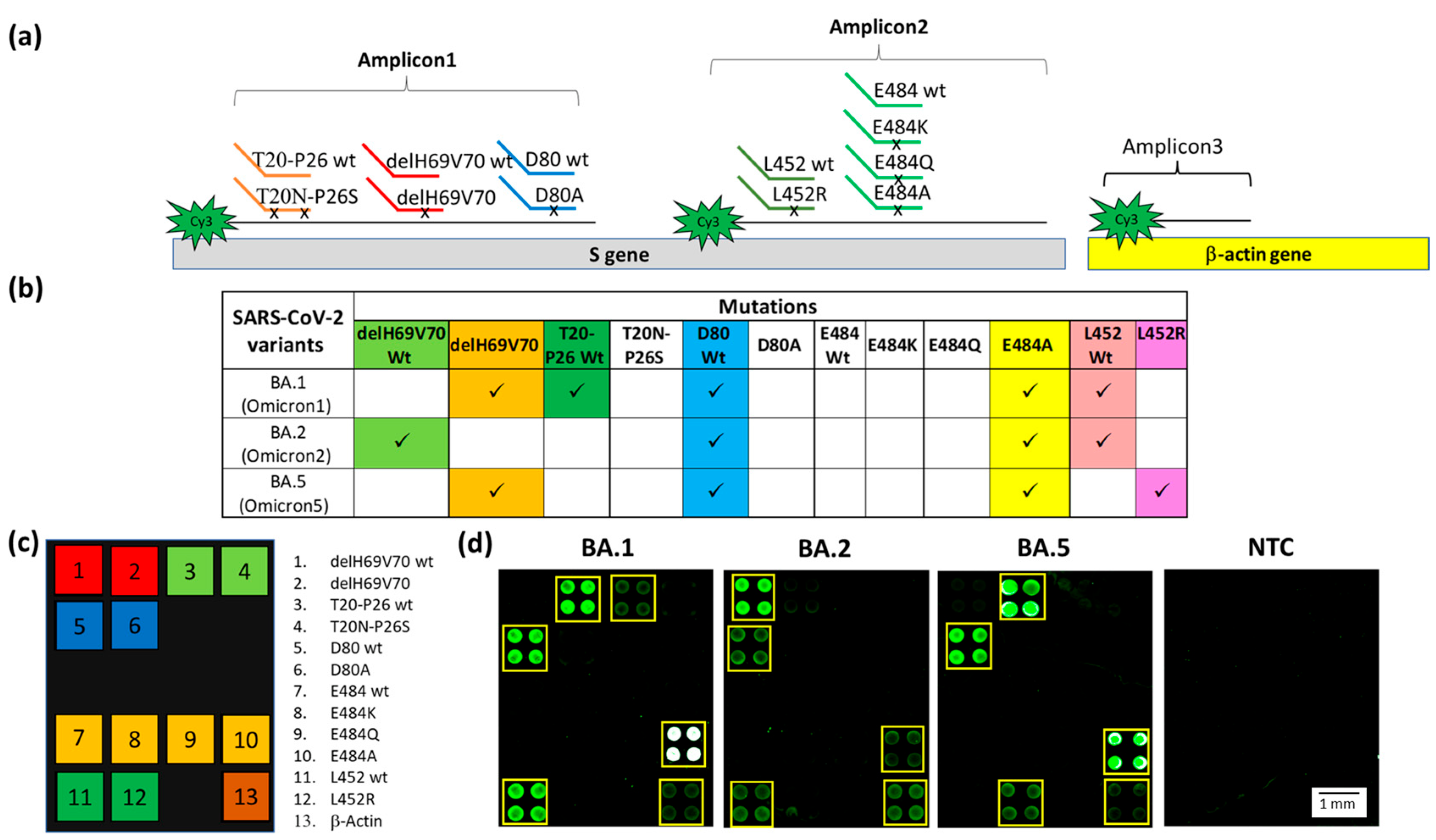
3.3. Discrimination of the Different Sub-Variants of the Omicron Lineage
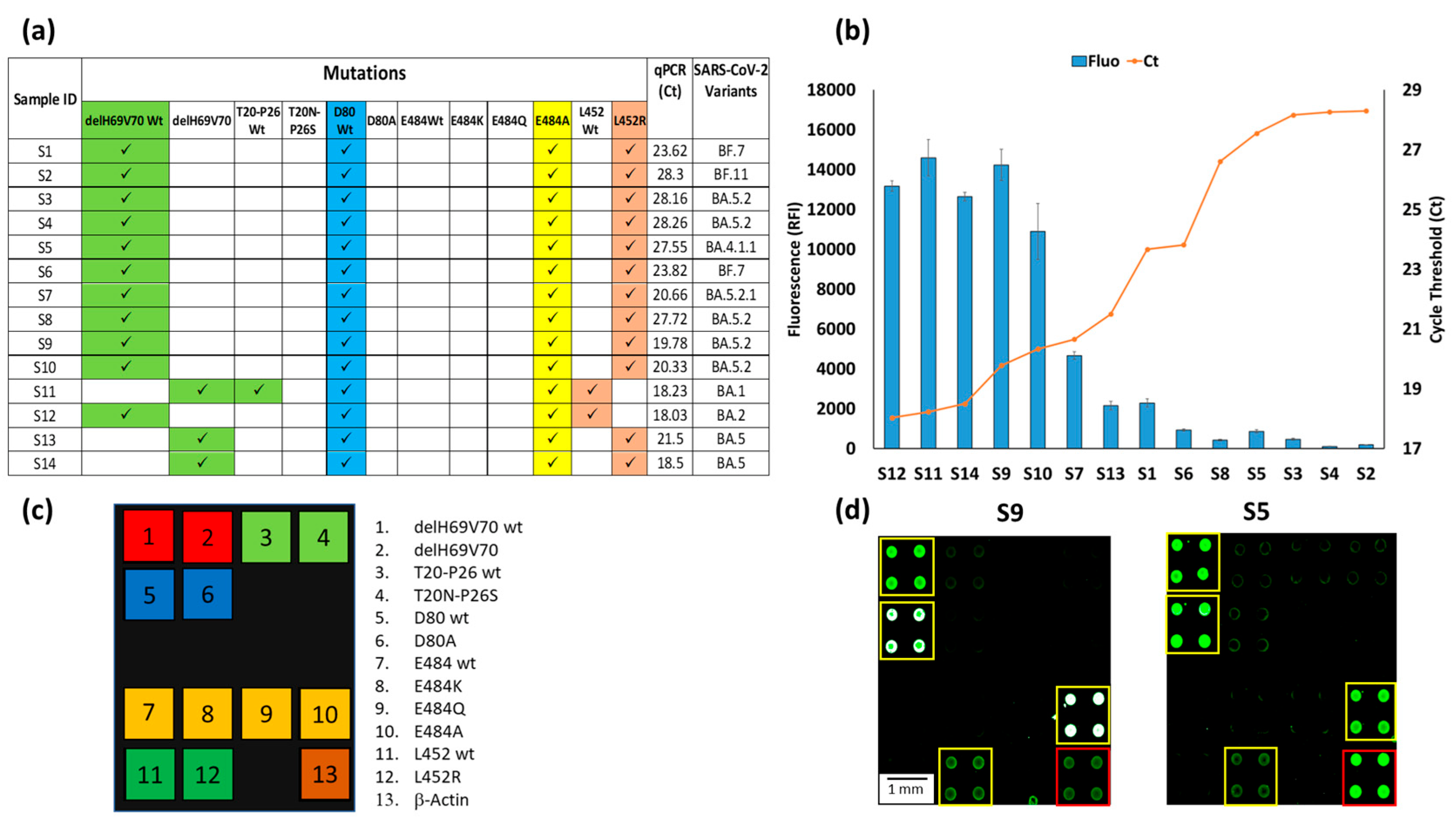
3.4. Clinical Verification
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 20 November 2022).
- Focosi, D.; Maggi, F. Neutralizing Antibody Escape of SARS-CoV-2 Spike Protein: Risk Assessment for Antibody-Based COVID-19 Therapeutics and Vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Boivin, S.; Cusack, S.; Ruigrok, R.W.; Hart, D.J. Influenza A virus polymerase: Structural insights into replication and host adaptation mechanisms. J. Biol. Chem. 2010, 285, 28411–28417. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Mutations Can Reveal How the Coronavirus Moves—But They’re Easy to Overinterpret. Available online: https://www.science.org/content/article/mutations-can-reveal-how-coronavirus-moves-they-re-easy-overinterpret (accessed on 10 September 2022).
- Ferron, F.; Subissi, L.; De Morais, A.T.S.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and Molecular Basis of Mismatch Correction and Ribavirin Excision from Coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021, 21, 340–341. [Google Scholar] [CrossRef]
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case study: Prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell 2020, 183, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus. Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; Rappuoli, R. SARS-CoV-2 Escaped Natural Immunity, Raising Questions About Vaccines and Therapies. Nat. Med. 2021, 27, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.M.; Boyton, R.J.; Beale, R. Immunity to Sars-Cov-2 Variants of Concern. Science 2021, 371, 1103–1104. [Google Scholar] [CrossRef]
- Wang, X.; Du, Z.; Johnson, K.E.; Pasco, R.F.; Fox, S.J.; Lachmann, M.; McLellan, J.S.; Meyers, L.A. Effects of COVID-19 Vaccination Timing and Risk Prioritization on Mortality Rates, United States. Emerg. Infect. Dis. 2021, 27, 1976–1979. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Here, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Rudi, E.; Martin, A.P.; Zurita, E.; Gonzalez Lopez, L.M.M.; Bottero, D.; Malito, J.; Gabrielli, M.; Gaillard, E.; Stuible, M.; Durocher, Y.; et al. Immunological study of COVID-19 vaccine candidate based on recombinant spike trimer protein from different SARS-CoV-2 variants of concern. Front. Immunol. 2022, 13, 1020159. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Ren, S.Y.; Wang, W.B.; Gao, R.D.; Zhou, A.M. Omicron variant (B.1.1.529) of SARS-CoV-2: Mutation, infectivity, transmission, and vaccine resistance. World J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Salman, M.; Khurshid, A.; Alam, M.; Badar, N.; Suleman, R.; Tahir, F.; Sharif, S.; Montgomery, J.; et al. Whole-genome sequencing of SARS-CoV-2 reveals the detection of G614 variant in Pakistan. PLoS ONE 2021, 16, e0248371. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Oude Munnink, B.B.; Schapendonk, C.M.E.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 circulation and diversity through community wastewater sequencing, The Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Durner, J.; Burggraf, S.; Czibere, L.; Tehrani, A.; Watts, D.C.; Becker, M. Fast and cost-effective screening for SARS-CoV-2 variants in a routine diagnostic setting. Dent. Mater. 2021, 37, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Banada, P.; Green, R.; Banik, S.; Chopoorian, A.; Streck, D.; Jones, R.; Chakravorty, S.; Alland, D. A simple RT-PCR melting temperature assay to rapidly screen for widely circulating SARS-CoV-2 variants. J. Clin. Microbiol. 2021, 59, e00845-e21. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Chen, J.; Wang, M.; Zhang, T.; Luo, W.; Li, Y.; Wu, Y.; Zeng, B.; Zhang, K.; et al. Detection of SARS-CoV-2 and its mutated variants via CRISPR-Cas13-based transcription amplification. Anal. Chem. 2021, 93, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- PerkinElmer. PKampTM VariantDetectTM SARS-CoV-2 RT-PCR Assay. Available online: https://perkinelemr-appliedgenomics.com/home/sars-cov-2-testing-solutions/pkamp-variantdetect-sars-cov-2-rt-pcr-assay/ (accessed on 16 September 2022).
- Damin, F.; Galbiati, S.; Gagliardi, S.; Cereda, C.; Dragoni, F.; Fenizia, C.; Savasi, V.; Sola, L.; Chiari, M. Covid Array: A Microarray-Based Assay with High Sensitivity for the Detection of Sars-Cov-2 in Nasopharyngeal Swabs. Sensors 2021, 21, 2490. [Google Scholar] [CrossRef] [PubMed]
- Damin, F.; Galbiati, S.; Soriani, N.; Burgio, V.; Ronzoni, M.; Ferrari, M.; Chiari, M. Analysis of KRAS, NRAS and BRAF mutational profile by combination of in-tube hybridization and universal tag-microarray in tumor tissue and plasma of colorectal cancer patients. PLoS ONE 2018, 13, e0207876. [Google Scholar] [CrossRef]
- Damin, F.; Galbiati, S.; Ferrari, M.; Chiari, M. DNA microarray-based solid-phase PCR on copoly (DMA–NAS–MAPS) silicon coated slides: An example of relevant clinical application. Biosens. Bioelectron. 2016, 78, 367–373. [Google Scholar] [CrossRef]
- Chiodi, E.; Damin, F.; Sola, L.; Ferraro, L.; Brambilla, D.; Unlu, M.S.; Chiari, M. A Reliable, Label Free Quality Control Method for the Production of DNA Microarrays with Clinical Applications. Polymers 2021, 13, 340. [Google Scholar] [CrossRef]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef]
- El Jaddaoui, I.; Allali, M.; Raoui, S.; Sehli, S.; Habib, N.; Chaouni, B.; Al Idrissi, N.; Benslima, N.; Maher, W.; Benrahma, H.; et al. A review on current diagnostic techniques for COVID-19H. Expert. Rev. Mol. Diagn. 2021, 21, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, X.; Wang, W.; Yang, Y.; Zhang, X.; Fang, W.; Zhang, L.; Li, S.; Gu, B. RT-LAMP assay for rapid detection of the R203M mutation in SARS-CoV-2 Delta variant. Emerg. Microbes Infect. 2022, 11, 978–987. [Google Scholar] [CrossRef]
- Safari, F.; Afarid, M.; Rastegari, B.; Borhani-Haghighi, A.; Barekati-Mowahed, M.; Behzad-Behbahani, A. CRISPR systems: Novel approaches for detection and combating COVID-19. Virus Res. 2021, 294, 198282. [Google Scholar] [CrossRef]
- Ravan, H.; Kashanian, S.; Sanadgol, N.; Badoei-Dalfard, A.; Karami, Z. Strategies for optimizing DNA hybridization on surfaces. Anal. Biochem. 2014, 444, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Pirri, G.; Damin, F.; Chiari, M.; Bontempi, E.; Depero, L.E. Characterization of a polymeric adsorbed coating for DNA microarray glass slides. Anal. Chem. 2004, 76, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Cretich, M.; di Carlo, G.; Longhi, R.; Gotti, C.; Spinella, N.; Coffa, S.; Galati, C.; Renna, L.; Chiari, M. High sensitivity protein assays on microarray silicon slides. Anal. Chem. 2009, 81, 5197–5203. [Google Scholar] [CrossRef] [PubMed]
- Sola, L.; Damin, F.; Gagni, P.; Consonni, R.; Chiari, M. Synthesis of Clickable Coating Polymers by Postpolymerization Modification: Applications in Microarray Technology. Langmuir 2016, 32, 10284–10295. [Google Scholar] [CrossRef]
- Hodcroft, E.B. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. Available online: https://covariants.org/ (accessed on 16 November 2022).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Liu, Y.; Lei, Z.; Dicker, J.; Cao, Y.; Zhang, X.F.; Im, W. Differential Interactions between Human ACE2 and Spike RBD of SARS-CoV-2 Variants of Concern. J. Chem. Theory Comput. 2021, 17, 7972–7979. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. What the latest Omicron subvariants mean for the pandemic. Nature 2022, 606, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Sola, L.; Damin, F.; Cretich, M.; Chiari, M. Novel polymeric coating with tailored hydrophobicity to control spot size and morphology in DNA microarray. Sens. Actuators B 2016, 231, 412–422. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Sequences | Fragment Lenght (bp) | |
|---|---|---|
| Amplicon1 (Spike gene) | Forward: 5′-TGCCACTAGTCTCTAGTCAGTGT-3′ Reverse: * 5′-CTCAGTGGAAGCAAAATAAACACCATC-3′ | 266 |
| Amplicon2 (Spike gene) | Forward: 5′-GATGAAGTCAGACAAATCGCTCCA-3′ Reverse: * 5′-AGAAAGTACTACTACTCTGTATGGTTGGTAA -3′ | 330 |
| Amplicon3 (human β-actin gene) | Forward: 5′-GCGAGAAGATGACCCAGATCATG-3′ Reverse: * 5′-AGAGGCGTACAGGGATAGCA-3′ | 89 |
| SARS-CoV-2 Variant Mutations | Capture Probes 1 (5′→3′) | Dual Domain Reporter Sequences |
|---|---|---|
| S: delH69V70 | ggctcacgtcttatttgggc | * CCATGCTATA TCTGGGACCAATGGTACTAAG-† gcccaaataagacgtgagcc |
| Wild-Type | cgagcacttaacattagagc | * CATGCTATACATGTCTCTGGGACCAATGGT-† gctctaatgttaagtgctcg |
| S: T20N-P26S | gcctcgggcaaacgactaa | * AACAGAACTCAATTACCCTCT-† tttagtcgtttgcccgaggc |
| Wild-Type | taatctaattctggtcgcgg | * ACCAGAACTCAATTACCCCCT-† ccgcgaccagaattagatta |
| S: D80A | attgaccaaactgcggtgcg | * TGCTAACCCTGTC-† cgcaccgcagtttggtcaat |
| Wild-Type | tcttctagttgtcgagcagg | * AGGTTTGATAACCCT-† cctgctcgacaactagaaga |
| S: L452R | catacgcggtaaggatata | * TTACCGGTATAGATTG-† ctatatccttaccgcgtatg |
| Wild-Type | caccgacgctaatagttaag | * TAATTACCTGTATAGATTG-† cttaactattagcgtcggtg |
| S: E484A | atcgtacttggcactggagt | * ATGGTGTTGCAGGTTT-† actccagtgccaagtacgat |
| S: E484K | aatgctcgggaaggctactc | * ATGGTGTTAAAGGTTT-† gagtagccttcccgagcatt |
| S: E484Q | tcttgacggaaaggtagac | * TGGTGTTCAAGGT-† tgtctacctttccgtcaaga |
| Wild-Type | atcccgtgagtcgatggttt | * TGGTGTTGAAGGT-† aaaccatcgactcacgggat |
| β-actin sequence | tgagaccttcaacaccccagccatgta | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damin, F.; Galbiati, S.; Clementi, N.; Ferrarese, R.; Mancini, N.; Sola, L.; Chiari, M. Dual-Domain Reporter Approach for Multiplex Identification of Major SARS-CoV-2 Variants of Concern in a Microarray-Based Assay. Biosensors 2023, 13, 269. https://doi.org/10.3390/bios13020269
Damin F, Galbiati S, Clementi N, Ferrarese R, Mancini N, Sola L, Chiari M. Dual-Domain Reporter Approach for Multiplex Identification of Major SARS-CoV-2 Variants of Concern in a Microarray-Based Assay. Biosensors. 2023; 13(2):269. https://doi.org/10.3390/bios13020269
Chicago/Turabian StyleDamin, Francesco, Silvia Galbiati, Nicola Clementi, Roberto Ferrarese, Nicasio Mancini, Laura Sola, and Marcella Chiari. 2023. "Dual-Domain Reporter Approach for Multiplex Identification of Major SARS-CoV-2 Variants of Concern in a Microarray-Based Assay" Biosensors 13, no. 2: 269. https://doi.org/10.3390/bios13020269
APA StyleDamin, F., Galbiati, S., Clementi, N., Ferrarese, R., Mancini, N., Sola, L., & Chiari, M. (2023). Dual-Domain Reporter Approach for Multiplex Identification of Major SARS-CoV-2 Variants of Concern in a Microarray-Based Assay. Biosensors, 13(2), 269. https://doi.org/10.3390/bios13020269