An Electrolyte-Gated Graphene Field-Effect Transistor for Detection of Gadolinium(III) in Aqueous Media

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
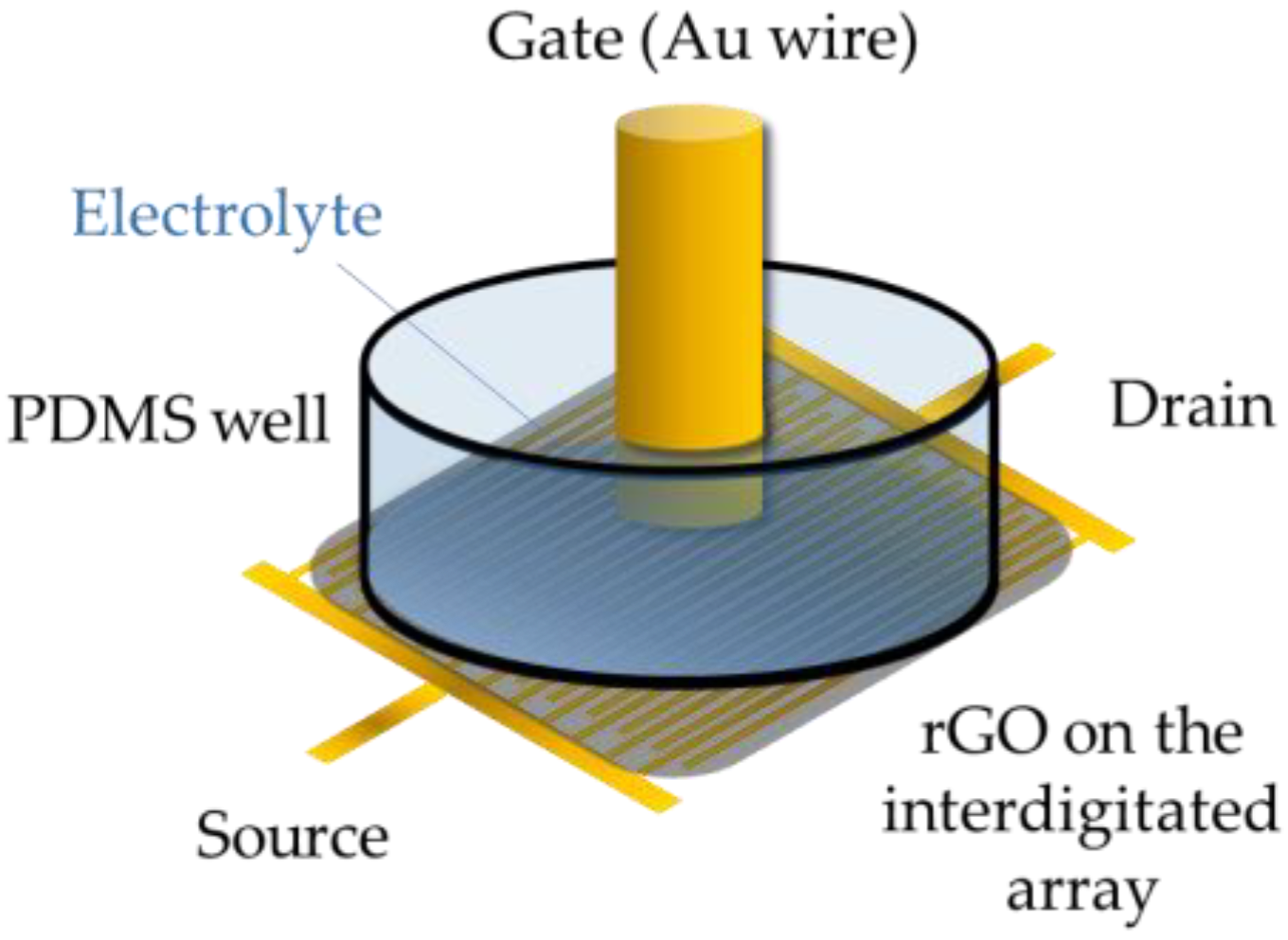
2.1. Instruments and Procedures
2.2. Protocol for DO3A-Alkyne Immobilization
3. Results and Discussion
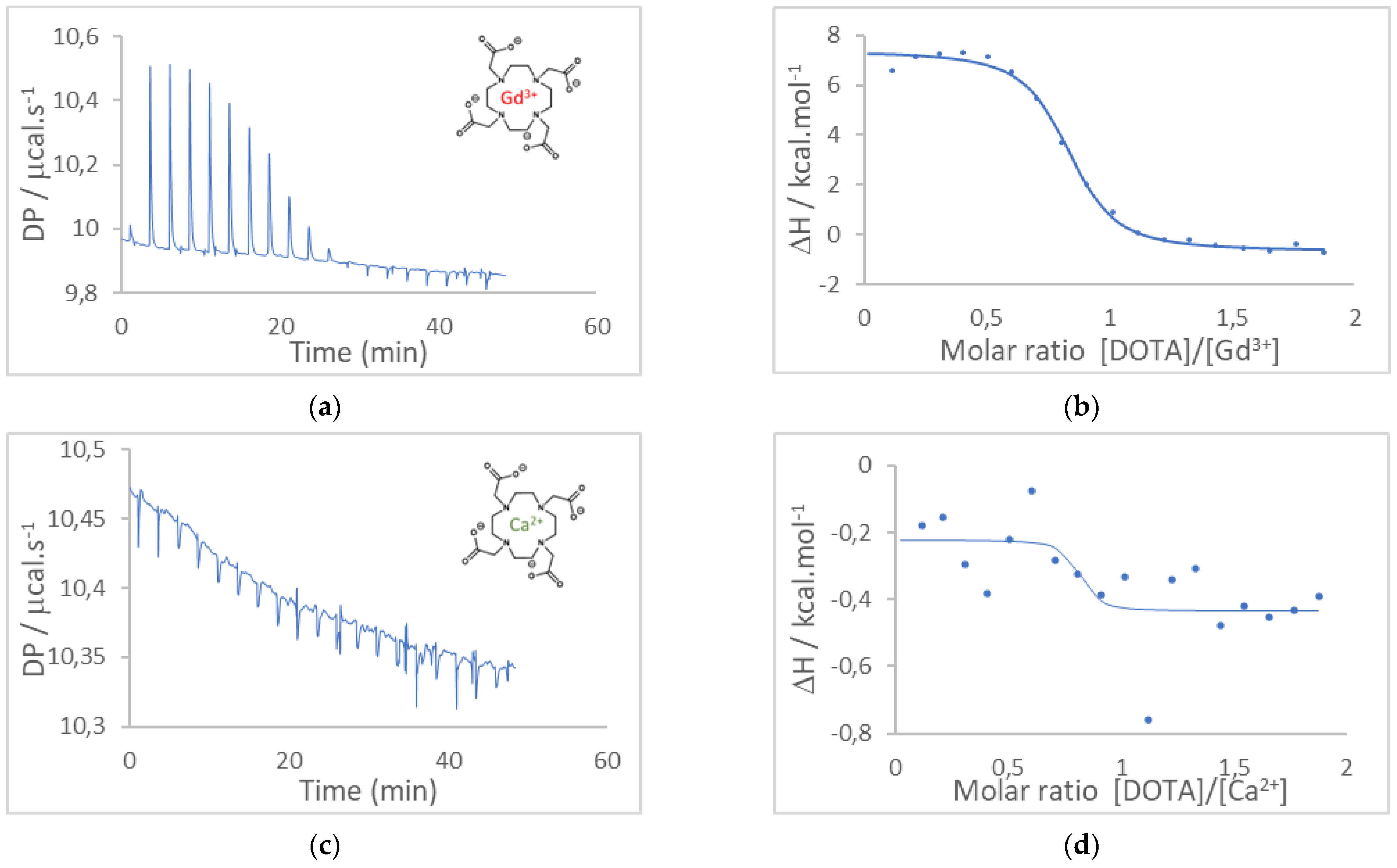
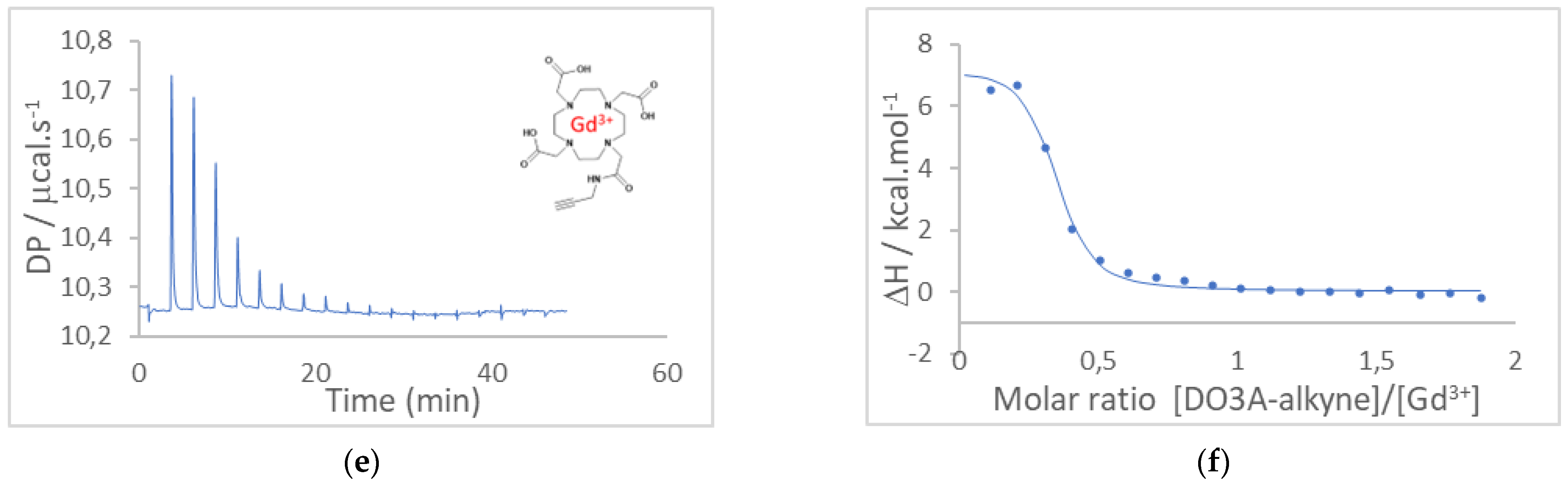
3.1. Determination of the Affinity Constants between Gd3+ and DOTA or DO3A-Alkyne
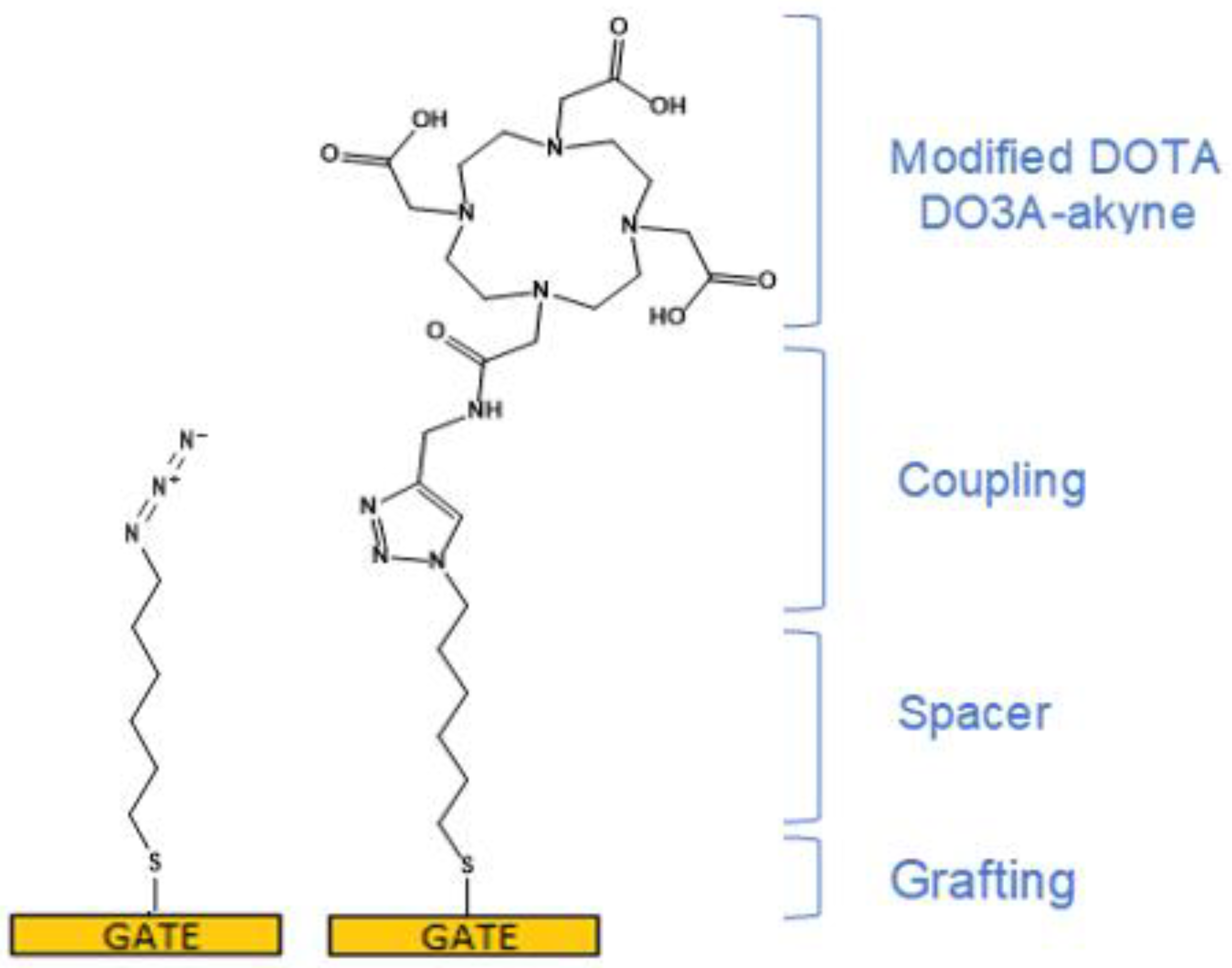
3.2. Functionalization of the Gate Electrode
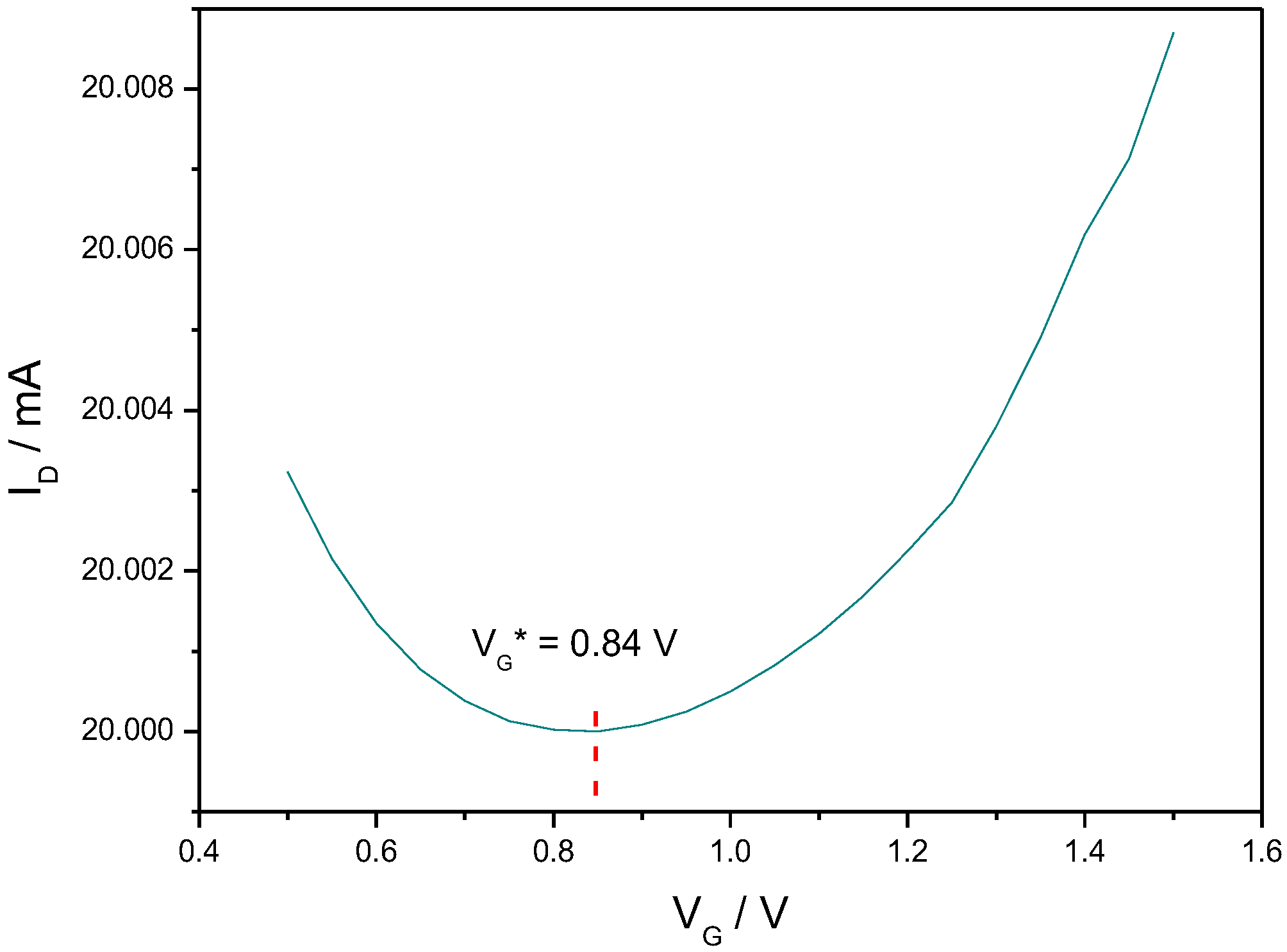
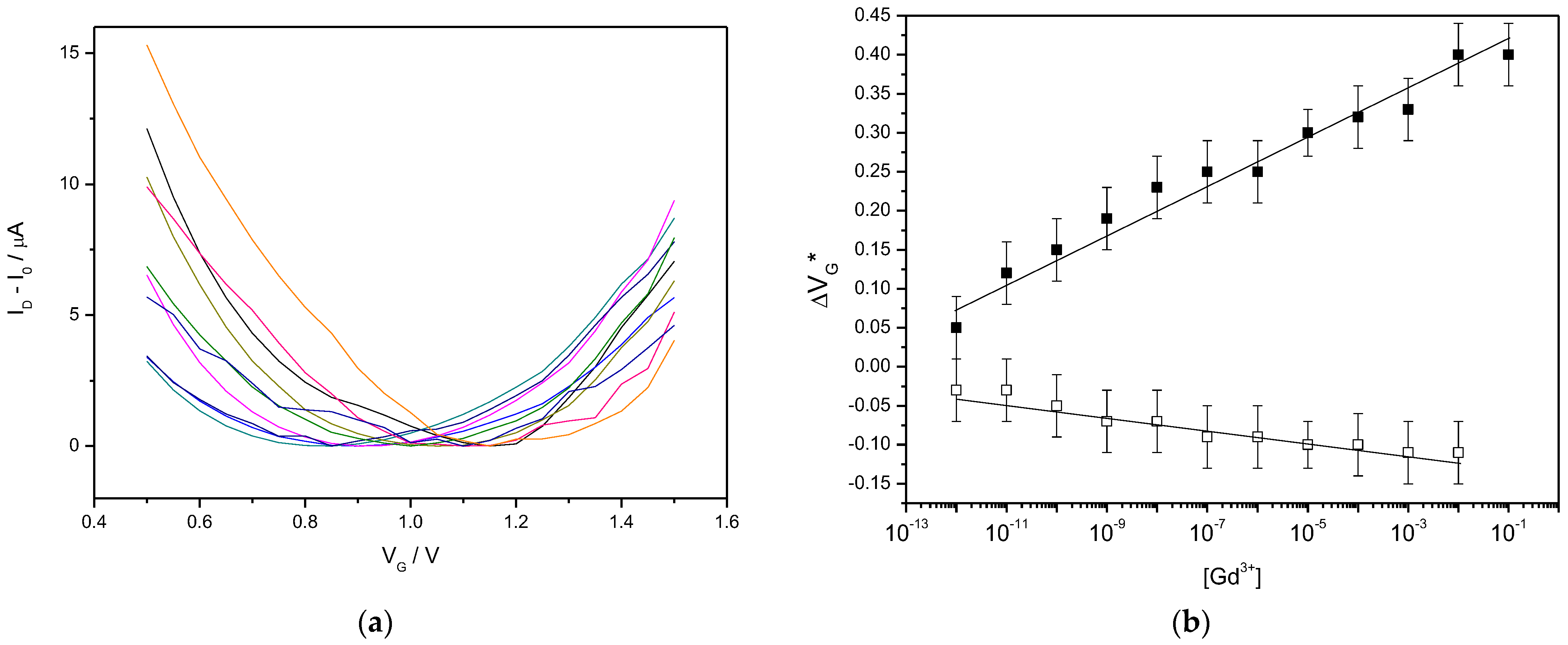
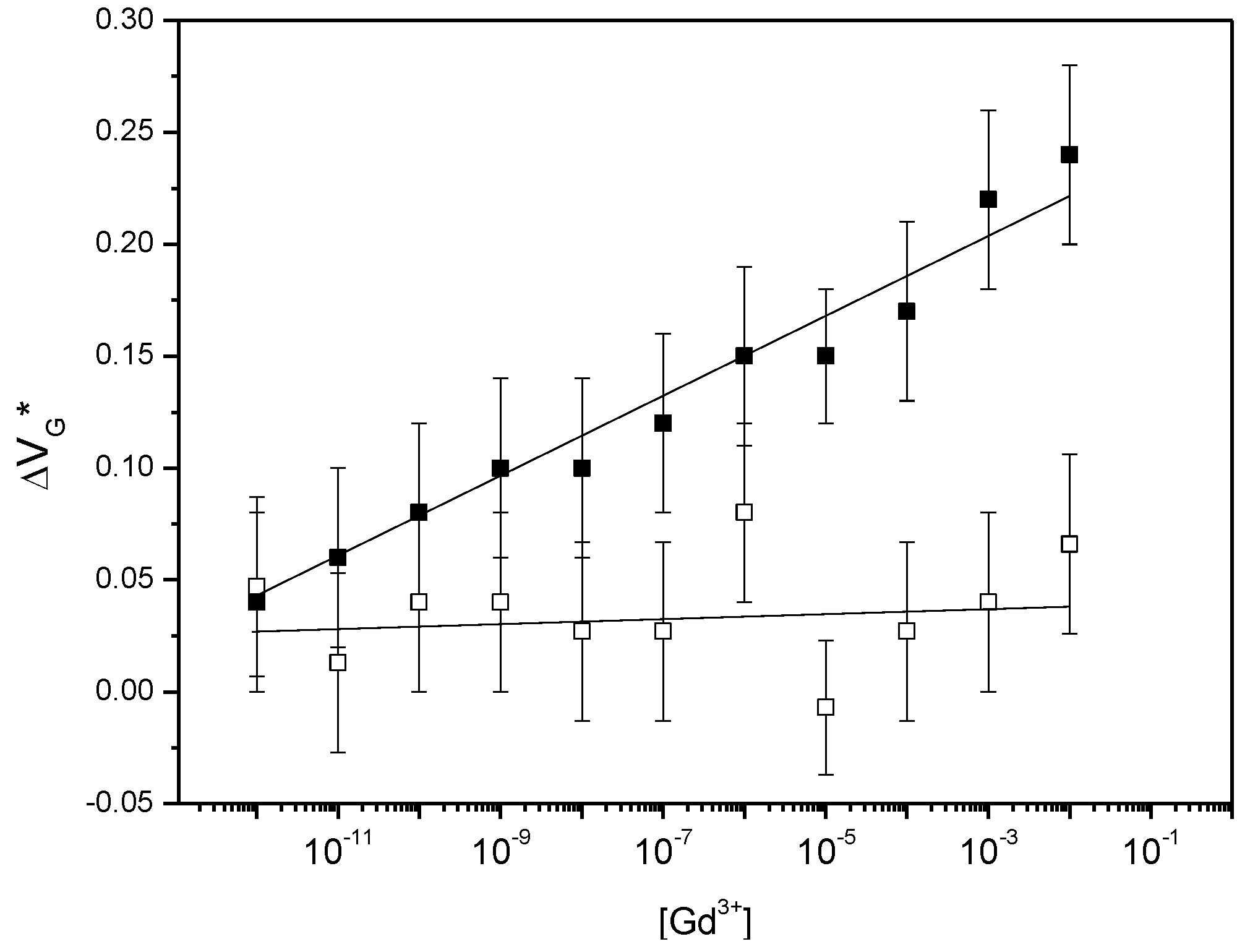
3.3. Gd3+ Sensing Using the EGGFET
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Chemicals, Reagents and Routine Procedures
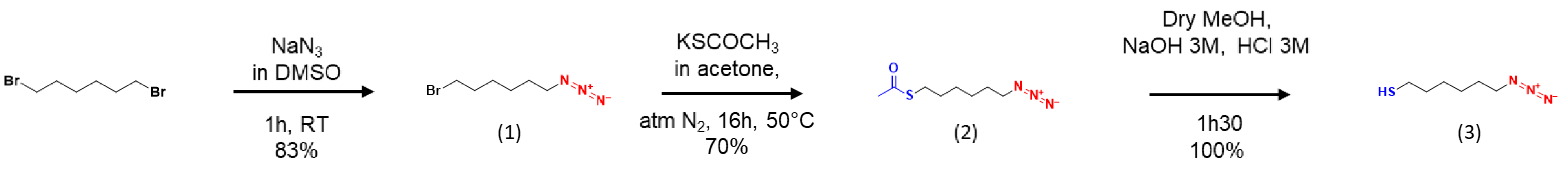
Appendix B. Synthesis Protocols


Appendix C. Characterizations
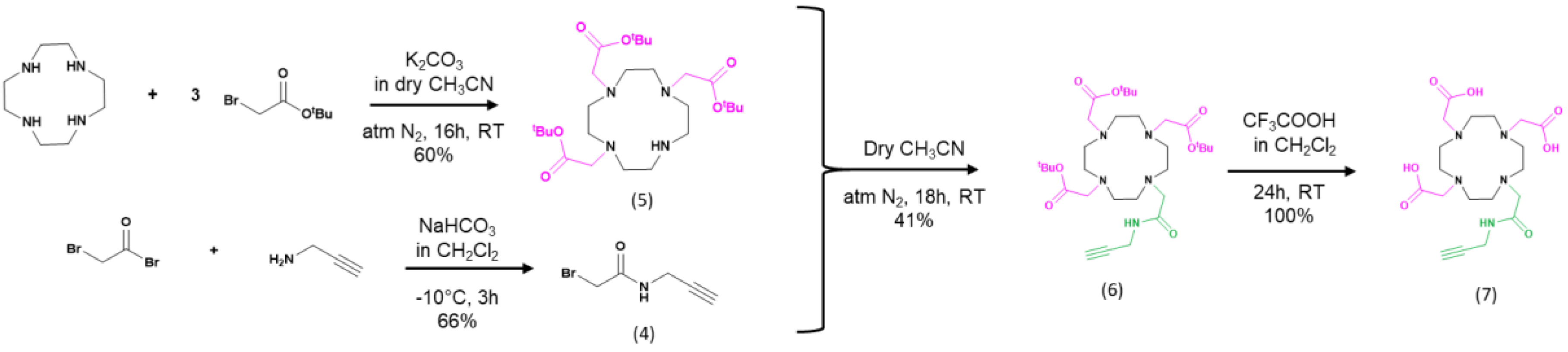
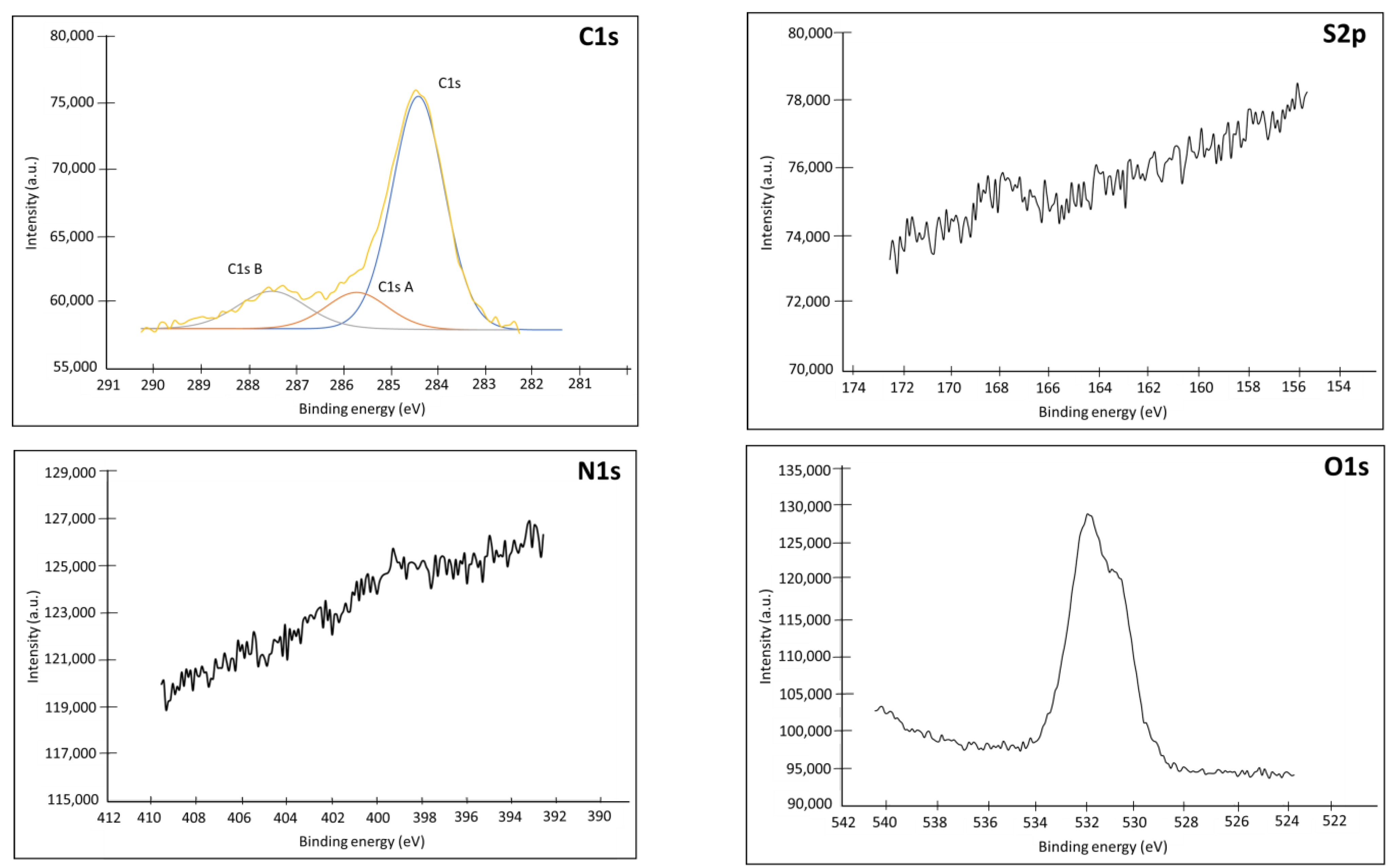
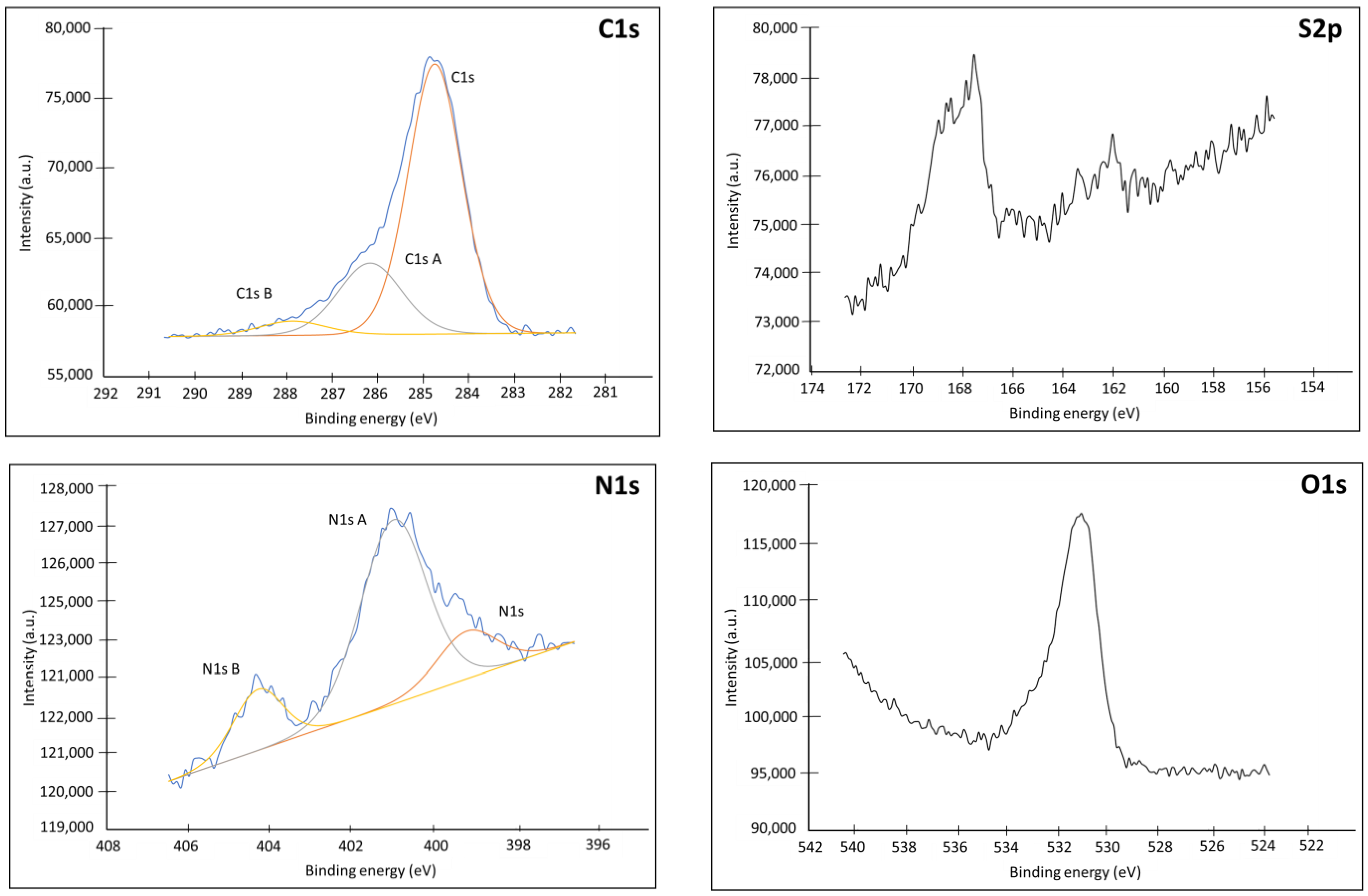
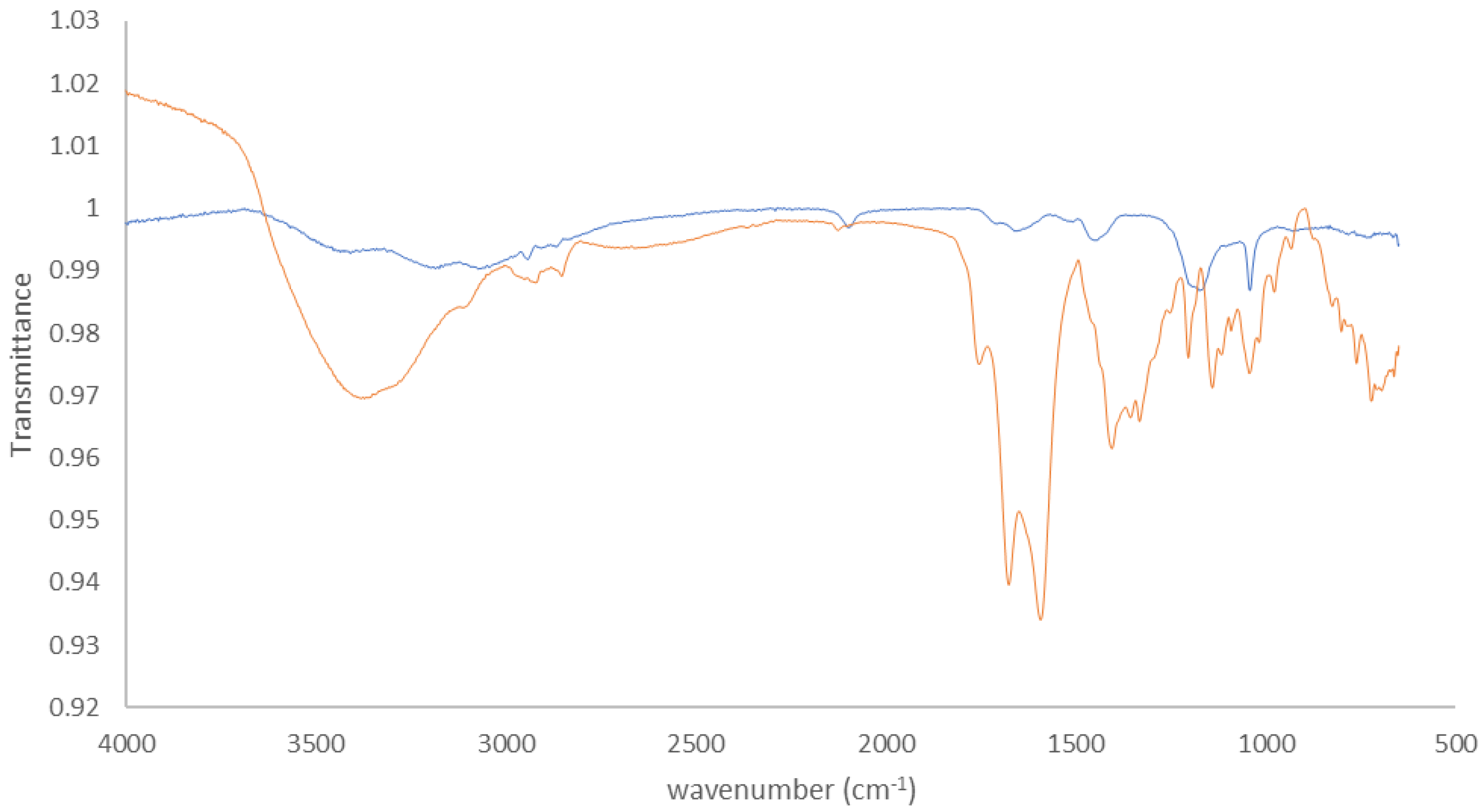
Appendix C.1. XPS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Peak BE | FWHM eV | Area (P) CPS.eV | Atomic % |
|---|---|---|---|---|
| Au4f | 84.24 | 0.77 | 1,622,127.82 | 46.89 |
| C1s | 284.42 | 1.31 | 24,840.75 | 16.77 |
| C1s A | 285.72 | 1.54 | 4611.59 | 3.12 |
| C1s B | 287.52 | 1.72 | 5281.57 | 3.57 |
| N1s | 399.25 | 0.4 | 2447.89 | 1.06 |
| O1s | 531.7 | 2.67 | 85,257.16 | 23.82 |
| S2p Ox | 167.76 | 0.28 | 1899.15 | 0.63 |

| Name | Peak BE | FWHM eV | Area (P) CPS.eV | Atomic % |
|---|---|---|---|---|
| Au4f | 84.18 | 0.77 | 1,753,345.31 | 50.44 |
| C1s | 284.76 | 1.39 | 29,150.61 | 19.59 |
| C1s A | 286.18 | 1.65 | 9128.36 | 6.14 |
| C1s B | 287.88 | 1.73 | 1898.79 | 1.28 |
| N1s | 399.19 | 1.72 | 2320.82 | 1 |
| N1s A | 400.99 | 1.9 | 9963.94 | 4.32 |
| N1s B | 404.3 | 1.43 | 2497.32 | 1.09 |
| O1s | 531.13 | 1.76 | 43,714.73 | 12.15 |
| S2p | 161.99 | 0.83 | 1882.86 | 0.62 |
| S2p Ox | 167.79 | 1.37 | 7713.57 | 2.57 |

| Name | Peak BE | FWHM eV | Area (P) CPS.eV | Atomic % |
|---|---|---|---|---|
| Ag3d | 368.05 | 0.78 | 78,864.56 | 1.83 |
| Au4f | 84.2 | 0.75 | 616,969.53 | 12.84 |
| C1s | 285.65 | 1.69 | 81,148.36 | 39.47 |
| C1s A | 287.44 | 1.74 | 24,231.7 | 11.8 |
| C1s B | 289.32 | 1.65 | 7701.99 | 3.75 |
| N1s | 400.7 | 1.91 | 10,018.3 | 3.14 |
| O1s | 533.76 | 1.86 | 126,405.99 | 25.46 |
| S2p | 162.09 | 0.9 | 3894.05 | 0.93 |
| S2p Ox | 169.19 | 1.14 | 2720.03 | 0.65 |

Appendix C.2. FTIR

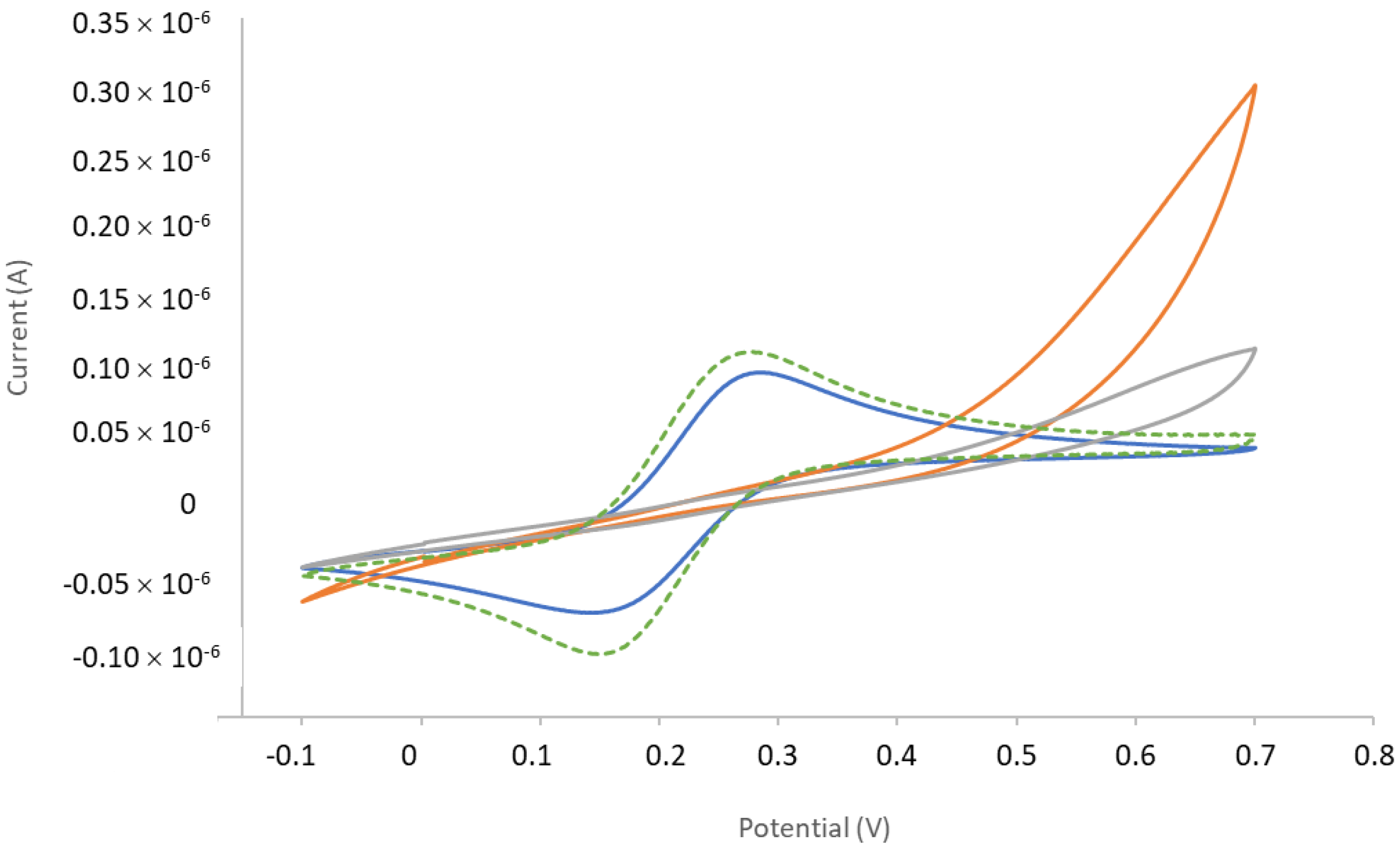
Appendix C.3. Cyclic Voltammetry

References
- Dekkers, I.A.; Roos, R.; van der Molen, A.J. Gadolinium retention after administration of contrast agents based on linear chelators and the recommendations of the European Medicines Agency. Eur. Radiol. 2018, 28, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Edogun, O.; Nguyen, N.H.; Halim, M. Fluorescent single-stranded DNA-based assay for detecting unchelated Gadolinium(III) ions in aqueous solution. Anal. Bioanal. Chem. 2016, 408, 4121–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallares, R.M.; An, D.D.; Tewari, P.; Wang, E.T.; Abergel, R.J. Rapid Detection of Gadolinium-Based Contrast Agents in Urine with a Chelated Europium Luminescent Probe. ACS Sens. 2020, 5, 1281–1286. [Google Scholar] [CrossRef]
- Ganjali, M.R.; Emami, M.; Rezapour, M.; Shamsipur, M.; Maddah, B.; Salavati-Niasari, M.; Hosseini, M.; Talebpoui, Z. Novel gadolinium poly(vinyl chloride) membrane sensor based on a new S–N Schiff’s base. Anal. Chim. Acta 2003, 495, 51–59. [Google Scholar] [CrossRef]
- Zamani, H.A.; Hosseini, M.; Haji-Mohammadrezazadeh, S.; Faridbod, F.; Ganjali, M.R.; Meghdadi, S.; Davoodnia, A. Gadolinium(III) ion selective sensor using a new synthesized Schiff’s base as a sensing material. Mater. Sci. Eng. C 2012, 32, 712–717. [Google Scholar] [CrossRef]
- Zamani, H.A.; Faridbod, F.; Ganjali, M.R. A new selectophore for gadolinium selective sensor. Mater. Sci. Eng. C 2014, 43, 488–493. [Google Scholar] [CrossRef]
- Gadhari, N.S.; Patil, S.S.; Gholave, J.V.; Patil, V.R.; Upadhyay, S.S. Highly efficient potentiometric sensing device for gadolinium based on Tetraazacyclododecane-1, 4, 7, 10-tetraaceticacid crown ether and multiwalled carbon nanotube composite. Microchem. J. 2022, 175, 107130. [Google Scholar] [CrossRef]
- Panzer, M.J.; Frisbie, C.D. Polymer Electrolyte-Gated Organic Field-Effect Transistors: Low-Voltage, High-Current Switches for Organic Electronics and Testbeds for Probing Electrical Transport at High Charge Carrier Density. J. Am. Chem. Soc. 2007, 129, 6599–6607. [Google Scholar] [CrossRef]
- Kergoat, L.; Piro, B.; Berggren, M.; Horowitz, G.; Pham, M.C. Advances in organic transistor-based biosensors: From organic electrochemical transistors to electrolyte-gated organic field-effect transistors. Anal. Bioanal. Chem. 2012, 402, 1813–1826. [Google Scholar] [CrossRef]
- Kawarada, H.; Araki, Y.; Sakai, T.; Ogawa, T.; Umezawa, H. Electrolyte-Solution-Gate FETs Using Diamond Surface for Biocompatible Ion Sensors. Phys. Status Solidi 2001, 185, 79–83. [Google Scholar] [CrossRef]
- Casalini, S.; Leonardi, F.; Cramer, T.; Biscarini, F. Organic field-effect transistor for label-free dopamine sensing. Org. Electron. 2013, 14, 156–163. [Google Scholar] [CrossRef]
- Leonardi, F.; Tamayo, A.; Casalini, S.; Mas-Torrent, M. Modification of the gate electrode by self-assembled monolayers in flexible electrolyte-gated organic field effect transistors: Work function vs. capacitance effects. RSC Adv. 2018, 8, 27509–27515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmoltner, K.; Kofler, J.; Klug, A.; List-Kratochvil, E.J.W. Electrolyte-Gated Organic Field-Effect Transistor for Selective Reversible Ion Detection. Adv. Mater. 2013, 25, 6895–6899. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.K.; Tran, H.V.; Vu, T.T.; Reisberg, S.; Noël, V.; Mattana, G.; Pham, M.C.; Piro, B. Peptide-modified electrolyte-gated organic field effect transistor. Application to Cu2+ detection. Biosens. Bioelectron. 2019, 127, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Béraud, A.; Sauvage, M.; Bazán, C.M.; Tie, M.; Bencherif, A.; Bouilly, D. Graphene field-effect transistors as bioanalytical sensors: Design, operation and performance. Analyst 2021, 146, 403–428. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.J.; Patridge, C.J.; Lee, V.; Jaye, C.; Lysaght, P.S.; Smith, C.; Barnett, J.; Fischer, D.A.; Prendergast, D.; Banerjee, S. Imaging local electronic corrugations and doped regions in graphene. Nat. Commun. 2011, 2, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehashi, K.; Sofue, Y.; Okamoto, S.; Ohno, Y.; Inoue, K.; Matsumoto, K. Selective ion sensors based on ionophore-modified graphene field-effect transistors. Sens. Actuators B Chem. 2013, 187, 45–49. [Google Scholar] [CrossRef]
- Wang, R.; Cao, Y.; Qu, H.; Wang, Y.; Zheng, L. Label-free detection of Cu(II) in fish using a graphene field-effect transistor gated by structure-switching aptamer probes. Talanta 2002, 237, 122965. [Google Scholar] [CrossRef]
- Li, P.; Tao, C.A.; Wang, B.; Huang, J.; Li, T.; Wang, J. Preparation of Graphene Oxide-Based Ink for Inkjet Printing. J. Nanosci. Nanotech. 2018, 18, 713–718. [Google Scholar] [CrossRef]
- Vasilijević, S.; Mattana, G.; Anquetin, G.; Battaglini, N.; Piro, B. Electrochemical tuning of reduced graphene oxide in printed electrolyte-gated transistors. Impact on charge transport properties. Electrochim. Acta 2021, 371, 137819. [Google Scholar] [CrossRef]
- Kumar, K.; Chang, C.A.; Francesconi, L.C.; Dischino, D.D.; Malley, M.F.; Gougoutas, J.Z.; Tweedle, M.F. Synthesis, Stability, and Structure of Gadolinium(III) and Yttrium(III) Macrocyclic Poly(amino carboxylates). Inorg. Chem. 1994, 33, 3567–3575. [Google Scholar] [CrossRef]
- Wang, X.; Jin, T.; Comblin, V.; Lopez-Mut, A.; Merciny, E.; Desreux, J.F. Detection of Bacterial Spores with Lanthanide-Macrocycle Binary Complexes. Inorg. Chem. 1992, 31, 1095–1099. [Google Scholar] [CrossRef]
- Burai, L.; Hietapelto, V.; Király, R.; Tóth, E.; Brücher, E. Stability constants and 1H relaxation effects of ternary complexes formed between Gd-DTPA, Gd-DTPA-BMA, Gd-dOTA, and Gd-EDTA and citrate, phosphate, and carbonate ions. Magnet. Res. Med. 1997, 38, 146–150. [Google Scholar] [CrossRef]
- Tosato, M.; Lazzari, L.; Di Marco, V. Revisiting Lead(II)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic Acid Coordination Chemistry in Aqueous Solutions: Evidence of an Underestimated Thermodynamic Stability. ACS Omega 2022, 7, 15596–15602. [Google Scholar] [CrossRef]
- van der Meer, S.B.; Loza, K.; Wey, K.; Heggen, M.; Beuck, C.; Bayer, P.; Epple, M. Click Chemistry on the Surface of Ultrasmall Gold Nanoparticles (2 nm) for Covalent Ligand Attachment Followed by NMR Spectroscopy. Langmuir 2019, 35, 7191–7204. [Google Scholar] [CrossRef] [PubMed]
- Worell, B.T.; Malik, J.A.; Fokin, V.V. Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, J.; Mouillard, F.; Le, T.N.; Vu, T.T.; Mattana, G.; Brayner, R.; Zrig, S.; Noël, V.; Piro, B. Monitoring photosynthetic microorganism activity with an electrolyte-gated organic field effect transistor. Biosens. Bioelectron. 2020, 157, 112166. [Google Scholar] [CrossRef]
- Lang, A.S.; Thelakkat, M. Modular synthesis of poly(perylene bisimides) using click chemistry: A comparative study. Polym. Chem. 2011, 2, 2213–2221. [Google Scholar] [CrossRef]
- Gobbo, P.; Novoa, S.; Biesinger, M.C.; Workentin, M.S. Interfacial strain-promoted alkyne–azidecycloaddition (I-SPAAC) for the synthesis of nanomaterial hybrids. Chem. Commun. 2013, 49, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Goswami, L.N.; Cai, Q.; Ma, L.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis, relaxation properties and in vivo assessment of a carborane-GdDOTA-monoamide conjugate as an MRI blood pool contrast agent. Org. Biomol. Chem. 2015, 13, 8912–8918. [Google Scholar] [CrossRef]
- Wan, F.; Liu, M.; Zhang, J.; Li, Y.; Jiang, L. Synthesis and characterization of DOTA-mono-adamantan-1-ylamide. Res. Chem. Intermed. 2015, 41, 5109–5119. [Google Scholar] [CrossRef]
- Prasuhn, D.E., Jr.; Yeh, R.M.; Obenaus, A.; Manchester, M.; Finn, M.G. Viral MRI contrast agents: Coordination of Gd by native virions and attachment of Gd complexes by azide–alkyne cycloaddition. Chem. Commun. 2007, 12, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Gouget-Laemmel, A.C.; Yang, J.; Lodhi, M.A.; Siriwardena, A.; Aureau, D.; Boukherroub, R.; Chazalviel, J.N.; Ozanam, F.; Szunerits, S. Functionalization of Azide-Terminated Silicon Surfaces with Glycans Using Click Chemistry: XPS and FTIR Study. J. Phys. Chem. C 2013, 117, 368–375. [Google Scholar] [CrossRef]
- Al-Hajj, N.; Mousli, Y.; Miche, A.; Humblot, V.; Hunel, J.; Heuzé, K.; Buffeteau, T.; Genin, E.; Vellutini, L. Influence of the grafting process on the orientation and the reactivity of azide-terminated monolayers onto silica surface. Appl. Surf. Sci. 2020, 527, 146778. [Google Scholar] [CrossRef]
- Spampinato, V.; Parracino, M.A.; La Spina, R.; Rossi, F.; Ceccone, G. Surface Analysis of Gold Nanoparticles Functionalized with Thiol-Modified Glucose SAMs for Biosensor Applications. Front. Chem. 2016, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Rieley, H.; Kendall, G.K.; Zemicael, F.W.; Smith, T.L.; Yang, S. X-ray Studies of Self-Assembled Monolayers on Coinage Metals. 1. Alignment and Photooxidation in 1,8-Octanedithiol and 1-Octanethiol on Au. Langmuir 1998, 14, 5147–5153. [Google Scholar] [CrossRef]
- Berner, S.; Lidbaum, H.; Ledung, G.; Åhlund, J.; Nilson, K.; Schiessling, J.; Gelius, U.; Bäckvall, J.E.; Puglia, C.; Oscarsson, S. Electronic and structural studies of immobilized thiol-derivatized cobalt porphyrins on gold surfaces. Appl. Surf. Sci. 2007, 253, 7540–7548. [Google Scholar] [CrossRef]
- Beshkov, G.; Dimitrov, D.B.; Georgiev, S.; Juan-Cheng, D.; Petrov, P.; Velchev, N.; Krastev, V. XPS spectra of thin CNx films prepared by chemical vapor deposition. Diam. Relat. Mater. 1999, 8, 591–594. [Google Scholar] [CrossRef]
- Rouxhet, P.G.; Misselyn-Bauduin, A.M.; Ahimou, F.; Genet, M.J.; Adriaensen, Y.; Desille, T.; Bodson, P.; Deroanne, C. XPS analysis of food products: Toward chemical functions and molecular compounds. Surf. Interf. Anal. 2008, 40, 718–724. [Google Scholar] [CrossRef]
- Collman, J.P.; Devaraj, N.K.; Eberspacher, T.P.A.; Chidsey, C.E.D. Mixed Azide-Terminated Monolayers: A Platform for Modifying Electrode Surfaces. Langmuir 2006, 22, 2457–2464. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, J.; Cai, C.Z. Rapid Grafting of Azido-labeled Oligo(ethylene glycol)s onto an Alkynyl-terminated Monolayer on Non-oxidized Silicon via Microwave-assisted “Click” Reaction. Langmuir 2011, 27, 2437–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Steinhurst, D.A.; Carpenter, E.E.; Owrutsky, J.C. Fourier Transform Infrared Spectroscopy of Azide Ion in Reverse Micelles. Langmuir 2002, 18, 7401–7408. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadroy, C.; Boukraa, R.; Battaglini, N.; Le Derf, F.; Mofaddel, N.; Vieillard, J.; Piro, B. An Electrolyte-Gated Graphene Field-Effect Transistor for Detection of Gadolinium(III) in Aqueous Media. Biosensors 2023, 13, 363. https://doi.org/10.3390/bios13030363
Gadroy C, Boukraa R, Battaglini N, Le Derf F, Mofaddel N, Vieillard J, Piro B. An Electrolyte-Gated Graphene Field-Effect Transistor for Detection of Gadolinium(III) in Aqueous Media. Biosensors. 2023; 13(3):363. https://doi.org/10.3390/bios13030363
Chicago/Turabian StyleGadroy, Charlène, Rassen Boukraa, Nicolas Battaglini, Franck Le Derf, Nadine Mofaddel, Julien Vieillard, and Benoît Piro. 2023. "An Electrolyte-Gated Graphene Field-Effect Transistor for Detection of Gadolinium(III) in Aqueous Media" Biosensors 13, no. 3: 363. https://doi.org/10.3390/bios13030363
APA StyleGadroy, C., Boukraa, R., Battaglini, N., Le Derf, F., Mofaddel, N., Vieillard, J., & Piro, B. (2023). An Electrolyte-Gated Graphene Field-Effect Transistor for Detection of Gadolinium(III) in Aqueous Media. Biosensors, 13(3), 363. https://doi.org/10.3390/bios13030363