Genetically Encoded Fluorescent Probe for Detection of Heme-Induced Conformational Changes in Cytochrome c


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
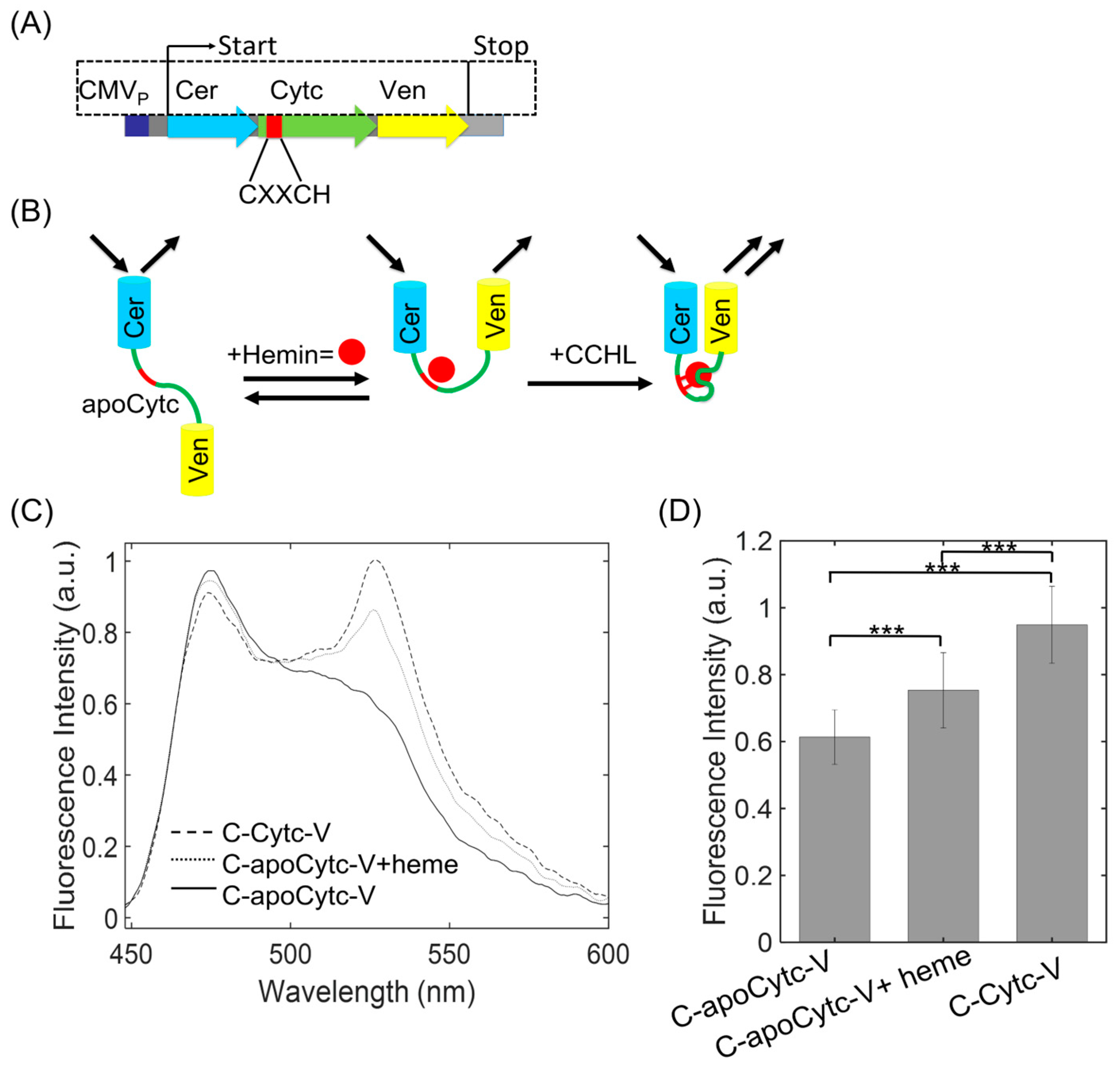
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. The Preparation of Cytc Expressing in the Cells
2.3. Spectroscopic Characterization of Cytc Constructs
2.4. Preparation of C-apoCytc-V Expression in Stable Cell Lines
2.5. Live Cell Imaging
2.6. Computation of Net and Normalized FRET Signals
2.7. Computation of FRET Signals
2.8. Statistical Tests
3. Results
3.1. Preparation and Characterization of C-apoCytc-V Construct
3.2. Characterization of Cytc Folding in the Cells by Using Timelapse Live-Cell Imaging
3.3. Time Course of Cytc Folding Change and Its Dependence on Heme Concentration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An Extreme Multifunctional Protein with a Key Role in Cell Fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Hanske, J.; Toffey, J.R.; Morenz, A.M.; Bonilla, A.J.; Schiavoni, K.H.; Pletneva, E.V. Conformational Properties of Cardiolipin-Bound Cytochrome, C. Proc. Natl. Acad. Sci. USA 2012, 109, 125–130. [Google Scholar] [CrossRef] [PubMed]
- González-Arzola, K.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Casado-Combreras, M.; Pérez-Mejías, G.; Díaz-Quintana, A.; Díaz-Moreno, I.; De la Rosa, M. New Moonlighting Functions of Mitochondrial Cytochrome c in the Cytoplasm and Nucleus. FEBS Lett. 2019, 593, 3101–3119. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Paggi, D.; Hannibal, L.; Castro, M.A.; Oviedo-Rouco, S.; Demicheli, V.; Tórtora, V.; Tomasina, F.; Radi, R.; Murgida, D.H. Multifunctional Cytochrome c: Learning New Tricks from an Old Dog. Chem. Rev. 2017, 117, 13382–13460. [Google Scholar] [CrossRef]
- Pérez-Mejías, G.; Díaz-Quintana, A.; Guerra-Castellano, A.; Díaz-Moreno, I.; De la Rosa, M.A. Novel Insights into the Mechanism of Electron Transfer in Mitochondrial Cytochrome c. Coord. Chem. Rev. 2022, 450, 214233. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c Phosphorylation: Control of Mitochondrial Electron Transport Chain Flux and Apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef]
- Moreno-Beltrán, B.; Guerra-Castellano, A.; Díaz-Quintana, A.; Del Conte, R.; García-Mauriño, S.M.; Díaz-Moreno, S.; González-Arzola, K.; Santos-Ocaña, C.; Velázquez-Campoy, A.; De la Rosa, M.A.; et al. Structural Basis of Mitochondrial Dysfunction in Response to Cytochrome c Phosphorylation at Tyrosine 48. Proc. Natl. Acad. Sci. USA 2017, 114, E3041–E3050. [Google Scholar] [CrossRef]
- Hamel, P.; Corvest, V.; Giegé, P.; Bonnard, G. Biochemical Requirements for the Maturation of Mitochondrial C-Type Cytochromes. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 125–138. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Márquez, I.; Pérez-Mejías, G.; Díaz-Quintana, A.; De la Rosa, M.A.; Díaz-Moreno, I. Post-Translational Modifications of Cytochrome c in Cell Life and Disease. Int. J. Mol. Sci. 2020, 21, 8483. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J.; Díaz-Moreno, I.; González-Arzola, K.; Janochas, S.; Navarro, J.A.; Hervás, M.; Bernhardt, R.; Velázquez-Campoy, A.; Díaz-Quintana, A.; De La Rosa, M.A. Structural and Functional Analysis of Novel Human Cytochrome c Targets in Apoptosis. Mol. Cell. Proteom. 2014, 13, 1439–1456. [Google Scholar] [CrossRef]
- Elena-Real, C.A.; Díaz-Quintana, A.; González-Arzola, K.; Velázquez-Campoy, A.; Orzáez, M.; López-Rivas, A.; Gil-Caballero, S.; De La Rosa, M.; Díaz-Moreno, I. Cytochrome c Speeds up Caspase Cascade Activation by Blocking 14-3-3ϵ-Dependent Apaf-1 Inhibition. Cell Death Dis. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Pierron, D.; Opazo, J.C.; Heiske, M.; Papper, Z.; Uddin, M.; Chand, G.; Wildman, D.E.; Romero, R.; Goodman, M.; Grossman, L.I. Silencing, Positive Selection and Parallel Evolution: Busy History of Primate Cytochromes c. PLoS ONE 2011, 6, e26269. [Google Scholar] [CrossRef]
- Diekert, K.; De Kroon, A.I.P.M.; Ahting, U.; Niggemeyer, B.; Neupert, W.; De Kruijff, B.; Lill, R. Apocytochrome c Requires the TOM Complex for Translocation across the Mitochondrial Outer Membrane. EMBO J. 2001, 20, 5626–5635. [Google Scholar] [CrossRef] [PubMed]
- Francisco, B.S.; Bretsnyder, E.C.; Kranz, R.G. Human Mitochondrial Holocytochrome c Synthase’s Heme Binding, Maturation Determinants, and Complex Formation with Cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Babbitt, S.E.; Sutherland, M.C.; Francisco, B.S.; Mendez, D.L.; Kranz, R.G. Mitochondrial Cytochrome c Biogenesis: No Longer an Enigma. Trends Biochem. Sci. 2015, 40, 446–455. [Google Scholar] [CrossRef]
- Kranz, R.G.; Richard-Fogal, C.; Taylor, J.-S.; Frawley, E.R. Cytochrome c Biogenesis: Mechanisms for Covalent Modifications and Trafficking of Heme and for Heme-Iron Redox Control. Microbiol. Mol. Biol. Rev. 2009, 73, 510–528. [Google Scholar] [CrossRef]
- Amacher, J.F.; Zhong, F.; Lisi, G.P.; Zhu, M.Q.; Alden, S.L.; Hoke, K.R.; Madden, D.R.; Pletneva, E.V. A Compact Structure of Cytochrome c Trapped in a Lysine-Ligated State: Loop Refolding and Functional Implications of a Conformational Switch. J. Am. Chem. Soc. 2015, 137, 8435–8449. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef]
- San Francisco, B.; Bretsnyder, E.C.; Rodgers, K.R.; Kranz, R.G. Heme Ligand Identification and Redox Properties of the Cytochrome c Synthetase, CcmF. Biochemistry 2011, 50, 10974–10985. [Google Scholar] [CrossRef]
- Mirkin, N.; Jaconcic, J.; Stojanoff, V.; Moreno, A. High Resolution X-ray Crystallographic Structure of Bovine Heart Cytochrome c and Its Application to the Design of an Electron Transfer Biosensor. Proteins Struct. Funct. Genet. 2008, 70, 83–92. [Google Scholar] [CrossRef]
- Babbitt, S.E.; Francisco, B.S.; Mendez, D.L.; Lukat-Rodgers, G.S.; Rodgers, K.R.; Bretsnyder, E.C.; Kranz, R.G. Mechanisms of Mitochondrial Holocytochrome c Synthase and the Key Roles Played by Cysteines and Histidine of the Heme Attachment Site, Cys-XX-Cys-His. J. Biol. Chem. 2014, 289, 28795–28807. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.E.; Cardillo, T.S.; Hayes, M.K.; Sherman, F. Role of Cytochrome c Heme Lyase in Mitochondrial Import and Accumulation of Cytochrome c in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 5487–5496. [Google Scholar]
- Kleingardner, J.G.; Bren, K.L. Comparing Substrate Specificity between Cytochrome c Maturation and Cytochrome c Heme Lyase Systems for Cytochrome c Biogenesis. Metallomics 2011, 3, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.M.G.; Maity, H.; Rumbley, J.N.; Lin, Y.; Englander, S.W. Order of Steps in the Cytochrome c Folding Pathway: Evidence for a Sequential Stabilization Mechanism. J. Mol. Biol. 2006, 359, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.R. Cytochrome c Folding Dynamics. Curr. Opin. Chem. Biol. 2004, 8, 169–174. [Google Scholar] [CrossRef]
- Paul, S.S.; Sil, P.; Haldar, S.; Mitra, S.; Chattopadhyay, K. Subtle Change in the Charge Distribution of Surface Residues May Affect the Secondary Functions of Cytochrome c. J. Biol. Chem. 2015, 290, 14476–14490. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, G.; Borsari, M.; De Rienzo, F.; Di Rocco, G.; Ranieri, A.; Sola, M. Free Energy of Transition for the Individual Alkaline Conformers of Yeast Iso-1-Cytochrome c. Biochemistry 2007, 46, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.M.G.; Lin, Y.; Rumbley, J.N.; Englander, S.W. Cooperative Omega Loops in Cytochrome c: Role in Folding and Function. J. Mol. Biol. 2003, 331, 29–36. [Google Scholar] [CrossRef]
- Zaidi, S.; Hassan, M.I.; Islam, A.; Ahmad, F. The Role of Key Residues in Structure, Function, and Stability of Cytochrome-C. Cell. Mol. Life Sci. 2014, 71, 229–255. [Google Scholar] [CrossRef]
- Parui, P.P.; Deshpande, M.S.; Nagao, S.; Kamikubo, H.; Komori, H.; Higuchi, Y.; Kataoka, M.; Hirota, S. Formation of Oligomeric Cytochrome c during Folding by Intermolecular Hydrophobic Interaction between N- and C-Terminal α-Helices. Biochemistry 2013, 52, 8732–8744. [Google Scholar] [CrossRef]
- Krishna, M.M.G.; Maity, H.; Rumbley, J.N.; Englander, S.W. Branching in the Sequential Folding Pathway of Cytochrome c. Protein Sci. 2007, 16, 1946–1956. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, G.; Corvest, V.; Meyer, E.H.; Hamel, P.P. Redox Processes Controlling the Biogenesis of C-Type Cytochromes. Antioxid. Redox Signal. 2010, 13, 1385–1401. [Google Scholar] [CrossRef] [PubMed]
- Babbitt, S.E.; San Francisco, B.; Bretsnyder, E.C.; Kranz, R.G. Conserved Residues of the Human Mitochondrial HoloCytochrome c Synthase Mediate Interactions with Heme. Biochemistry 2014, 53, 5261–5271. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Mitra, S.; Chattopadhyay, K. Role of Protein Stabilizers on the Conformation of the Unfolded State of Cytochrome c and Its Early Folding Kinetics: Investigation at Single Molecular Resolution. J. Biol. Chem. 2010, 285, 25314–25323. [Google Scholar] [CrossRef]
- Wimplinger, I.; Shaw, G.M.; Kutsche, K. HCCS Loss-of-Function Missense Mutation in a Female with Bilateral Microphthalmia and Sclerocornea: A Novel Gene for Severe Ocular Malformations? Mol. Vis. 2007, 13, 1475–1482. [Google Scholar]
- De Rocco, D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of Cytochrome c Identified in Patients with Thrombocytopenia THC4 Affect Both Apoptosis and Cellular Bioenergetics. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Goldes, M.E.; Jeakins-Cooley, M.E.; McClelland, L.J.; Mou, T.C.; Bowler, B.E. Disruption of a Hydrogen Bond Network in Human versus Spider Monkey Cytochrome c Affects Heme Crevice Stability. J. Inorg. Biochem. 2016, 158, 62–69. [Google Scholar] [CrossRef]
- Karsisiotis, A.I.; Deacon, O.M.; Wilson, M.T.; MacDonald, C.; Blumenschein, T.M.A.; Moore, G.R.; Worrall, J.A.R. Increased Dynamics in the 40-57 Ω-Loop of the G41S Variant of Human Cytochrome c Promote Its pro-Apoptotic Conformation. Sci. Rep. 2016, 6, 30447. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Grossman, L.I.; Doan, J.W.; Sanderson, T.H. Phosphorylation of Mammalian Cytochrome c and Cytochrome c Oxidase in the Regulation of Cell Destiny: Respiration, Apoptosis, and Human Disease. Adv. Exp. Med. Biol. 2012, 748, 237–264. [Google Scholar]
- Allen, J.W.A. Cytochrome c Biogenesis in Mitochondria—Systems III and V. FEBS J. 2011, 278, 4198–4216. [Google Scholar] [CrossRef]
- Hoang, L.; Maity, H.; Krishna, M.M.G.; Lin, Y.; Englander, S.W. Folding Units Govern the Cytochrome C Alkaline Transition. J. Mol. Biol. 2003, 331, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kan, Z.Y.; Mayne, L.; Englander, S.W. Cytochrome c Folds through Foldon-Dependent Native-like Intermediates in an Ordered Pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Tomasina, F.; Capdevila, D.A.; Demicheli, V.; Tórtora, V.; Alvarez-Paggi, D.; Jemmerson, R.; Murgida, D.H.; Radi, R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 2016, 55, 407–428. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.V.; Firmino, M.O.; Costa, N.L.; Louro, R.O.; Paquete, C.M. Investigation of the Molecular Mechanisms of the Eukaryotic Cytochrome-c Maturation System. Biomolecules 2022, 12, 549. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.L.; Lowder, E.P.; Tillman, D.E.; Sutherland, M.C.; Collier, A.L.; Rau, M.J.; Fitzpatrick, J.A.J.; Kranz, R.G. Cryo-EM of CcsBA Reveals the Basis for Cytochrome c Biogenesis and Heme Transport. Nat. Chem. Biol. 2022, 18, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, H.; Khan, M.K.A.; Hassan, M.I.; Islam, A.; Moosavi-Movahedi, A.A.; Ahmad, F. Heterogeneity of Equilibrium Molten Globule State of Cytochrome c Induced by Weak Salt Denaturants under Physiological Condition. PLoS ONE 2015, 10, e0120465. [Google Scholar] [CrossRef]
- Chen, E.; Goldbeck, R.; Kliger, D. Probing Early Events in Ferrous Cytochrome c Folding with Time-Resolved Natural and Magnetic Circular Dichroism Spectroscopies. Curr. Protein Pept. Sci. 2009, 10, 464–475. [Google Scholar] [CrossRef]
- Mara, M.W.; Hadt, R.G.; Reinhard, M.E.; Kroll, T.; Lim, H.; Hartsock, R.W.; Alonso-Mori, R.; Chollet, M.; Glownia, J.M.; Nelson, S.; et al. Metalloprotein Entatic Control of Ligand-Metal Bonds Quantified by Ultrafast X-ray Spectroscopy. Science 2017, 356, 1276–1280. [Google Scholar] [CrossRef]
- Weinkam, P.; Zimmermann, J.; Sagle, L.B.; Matsuda, S.; Dawson, P.E.; Wolynes, P.G.; Romesberg, F.E. Characterization of Alkaline Transitions in Ferricytochrome c Using Carbon-Deuterium Infrared Probes. Biochemistry 2008, 47, 13470–13480. [Google Scholar] [CrossRef]
- Sagle, L.B.; Zimmermann, J.; Dawson, P.E.; Romesberg, F.E. Direct and High Resolution Characterization of Cytochrome c Equilibrium Folding. J. Am. Chem. Soc. 2006, 128, 14232–14233. [Google Scholar] [CrossRef] [PubMed]
- Weinkam, P.; Zong, C.; Wolynes, P.G. A Funneled Energy Landscape for Cytochrome c Directly Predicts the Sequential Folding Route Inferred from Hydrogen Exchange Experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 12401–12406. [Google Scholar] [CrossRef] [PubMed]
- Scrosati, P.M.; Yin, V.; Konermann, L. Hydrogen/Deuterium Exchange Measurements May Provide an Incomplete View of Protein Dynamics: A Case Study on Cytochromec. Anal. Chem. 2021, 93, 14121–14129. [Google Scholar] [CrossRef] [PubMed]
- Kleingardner, J.G.; Bowman, S.E.J.; Bren, K.L. The Influence of Heme Ruffling on Spin Densities in Ferricytochromes C Probed by Heme Core 13C NMR. Inorg. Chem. 2013, 52, 12933–12946. [Google Scholar] [CrossRef]
- Bren, K.L.; Kellogg, J.A.; Kaur, R.; Wen, X. Folding, Conformational Changes, and Dynamics of Cytochromes C Probed by NMR Spectroscopy. Inorg. Chem. 2004, 43, 7934–7944. [Google Scholar] [CrossRef] [PubMed]
- Lalli, D.; Rosa, C.; Allegrozzi, M.; Turano, P. Distal Unfolding of Ferricytochrome c Induced by the F82K Mutation. Int. J. Mol. Sci. 2020, 21, 2134. [Google Scholar] [CrossRef]
- Yin, V.; Shaw, G.S.; Konermann, L. Cytochrome c as a Peroxidase: Activation of the Precatalytic Native State by H2O2-Induced Covalent Modifications. J. Am. Chem. Soc. 2017, 139, 15701–15709. [Google Scholar] [CrossRef]
- Galinato, M.G.I.; Kleingardner, J.G.; Bowman, S.E.J.; Alp, E.E.; Zhao, J.; Bren, K.L.; Lehnert, N. Heme-Protein Vibrational Couplings in Cytochrome c Provide a Dynamic Link That Connects the Heme-Iron and the Protein Surface. Proc. Natl. Acad. Sci. USA 2012, 109, 8896–8900. [Google Scholar] [CrossRef]
- Kidokoro, S.I.; Nakamura, S. IATC, DSC, and PPC Analysis of Reversible and Multistate Structural Transition of Cytochrome c. Methods Enzymol. 2016, 567, 391–412. [Google Scholar]
- Goldbeck, R.A.; Chen, E.; Kliger, D.S. Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c. Int. J. Mol. Sci. 2009, 10, 1476–1499. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Cox Cammer, N.; Huisman, T.M.; Verkhusha, V.V.; Hodgson, L. Direct Multiplex Imaging and Optogenetics of Rho GTPases Enabled by Near-Infrared FRET Article. Nat. Chem. Biol. 2018, 14, 591–600. [Google Scholar] [CrossRef]
- Hanna, D.A.; Harvey, R.M.; Martinez-Guzman, O.; Yuan, X.; Chandrasekharan, B.; Raju, G.; Outten, F.W.; Hamz, I.; Reddi, A.R. Heme Dynamics and Trafficking Factors Revealed by Genetically Encoded Fluorescent Heme Sensors. Proc. Natl. Acad. Sci. USA 2016, 113, 7539–7544. [Google Scholar] [CrossRef]
- Skruzny, M.; Pohl, E.; Abella, M. FRET Microscopy in Yeast. Biosensors 2019, 9, 122. [Google Scholar] [CrossRef]
- Ahmad, M.; Ameen, S.; Siddiqi, T.O.; Khan, P.; Ahmad, A. Live Cell Monitoring of Glycine Betaine by FRET-Based Genetically Encoded Nanosensor. Biosens. Bioelectron. 2016, 86, 169–175. [Google Scholar] [CrossRef]
- Yano, T.; Oku, M.; Akeyama, N.; Itoyama, A.; Yurimoto, H.; Kuge, S.; Fujiki, Y.; Sakai, Y. A Novel Fluorescent Sensor Protein for Visualization of Redox States in the Cytoplasm and in Peroxisomes. Mol. Cell. Biol. 2010, 30, 3458–3766. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, H.; Fields, A.P.; Kralj, J.M.; Spudich, J.L.; Rothschild, K.J.; Cohen, A.E. Ultrasensitive Measurements of Microbial Rhodopsin Photocycles Using Photochromic FRET. Photochem. Photobiol. 2012, 88, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szöllősi, J. Understanding FRET as a Research Tool for Cellular Studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef]
- Kumari, R.; Dkhar, D.S.; Mahapatra, S.; Divya; Kumar, R.; Chandra, P. Nano-Bioengineered Sensing Technologies for Real-Time Monitoring of Reactive Oxygen Species in in Vitro and in Vivo Models. Microchem. J. 2022, 180, 107615. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving Brightness and Photostability of Green and Red Fluorescent Proteins for Live Cell Imaging and FRET Reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef] [PubMed]
- Arsenovic, P.T.; Mayer, C.R.; Conway, D.E. SensorFRET: A Standardless Approach to Measuring Pixel-Based Spectral Bleed-through and FRET Efficiency Using Spectral Imaging. Sci. Rep. 2017, 7, 15609. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, O.; Soleja, N.; Khan, P.; Imtaiyaz Hassan, M.; Mohsin, M. Visualization of Thiamine in Living Cells Using Genetically Encoded Fluorescent Nanosensor. Biochem. Eng. J. 2019, 146, 170–178. [Google Scholar] [CrossRef]
- Mastop, M.; Bindels, D.S.; Shaner, N.C.; Postma, M.; Gadella, T.W.J.; Goedhart, J. Characterization of a Spectrally Diverse Set of Fluorescent Proteins as FRET Acceptors for MTurquoise2. Sci. Rep. 2017, 7, 11999. [Google Scholar]
- Manioglu, S.; Atis, M.; Aas, M.; Kiraz, A.; Bayraktar, H. Direct Conversion of Cytochrome c Spectral Shifts to Fluorescence Using Photochromic FRET. Chem. Commun. 2014, 50, 12333–12336. [Google Scholar]
- Qureshi, H.; Ozlu, N.; Bayraktar, H. Adaptive Tracking Algorithm for Trajectory Analysis of Cells and Layer-by-Layer Assessment of Motility Dynamics. Comp. Biol. Med. 2022, 150, 106193. [Google Scholar] [CrossRef]
- Xia, Z.; Liu, Y. Reliable and Global Measurement of Fluorescence Resonance Energy Transfer Using Fluorescence Microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Berney, C.; Danuser, G. FRET or No FRET: A Quantitative Comparison. Biophys. J. 2003, 84, 3992–4010. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Rombola, C.; Jyothikumar, V.; Periasamy, A. Förster Resonance Energy Transfer Microscopy and Spectroscopy for Localizing Protein-Protein Interactions in Living Cells. Cytom. Part A 2013, 83, 780–793. [Google Scholar] [CrossRef]
- Colombo, S.; Longoni, E.; Gnugnoli, M.; Busti, S.; Martegani, E. Fast Detection of PKA Activity in Saccharomyces cerevisiae Cell Population Using AKAR Fluorescence Resonance Energy Transfer Probes. Cell. Signal. 2022, 92, 110262. [Google Scholar] [CrossRef] [PubMed]
- San Martín, A.; Arce-Molina, R.; Aburto, C.; Baeza-Lehnert, F.; Barros, L.F.; Contreras-Baeza, Y.; Pinilla, A.; Ruminot, I.; Rauseo, D.; Sandoval, P.Y. Visualizing Physiological Parameters in Cells and Tissues Using Genetically Encoded Indicators for Metabolites. Free Radic. Biol. Med. 2022, 182, 34–58. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genceroglu, M.Y.; Cavdar, C.; Manioglu, S.; Bayraktar, H. Genetically Encoded Fluorescent Probe for Detection of Heme-Induced Conformational Changes in Cytochrome c. Biosensors 2023, 13, 890. https://doi.org/10.3390/bios13090890
Genceroglu MY, Cavdar C, Manioglu S, Bayraktar H. Genetically Encoded Fluorescent Probe for Detection of Heme-Induced Conformational Changes in Cytochrome c. Biosensors. 2023; 13(9):890. https://doi.org/10.3390/bios13090890
Chicago/Turabian StyleGenceroglu, Mehmet Yunus, Cansu Cavdar, Selen Manioglu, and Halil Bayraktar. 2023. "Genetically Encoded Fluorescent Probe for Detection of Heme-Induced Conformational Changes in Cytochrome c" Biosensors 13, no. 9: 890. https://doi.org/10.3390/bios13090890