Comparative Study of Fluorescence Emission of Fisetin, Luteolin and Quercetin Powders and Solutions: Further Evidence of the ESIPT Process

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. Computational Details
3. Results and Discussion
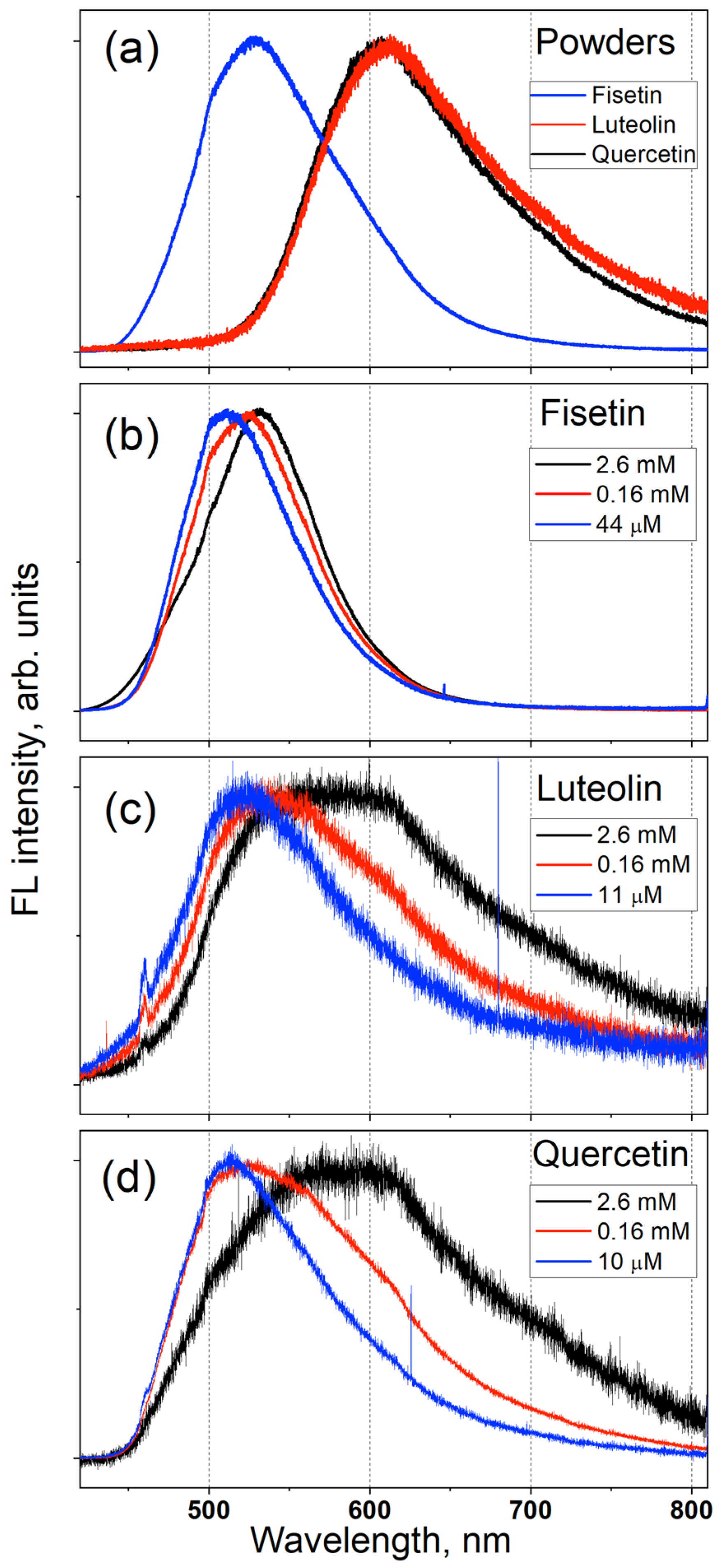
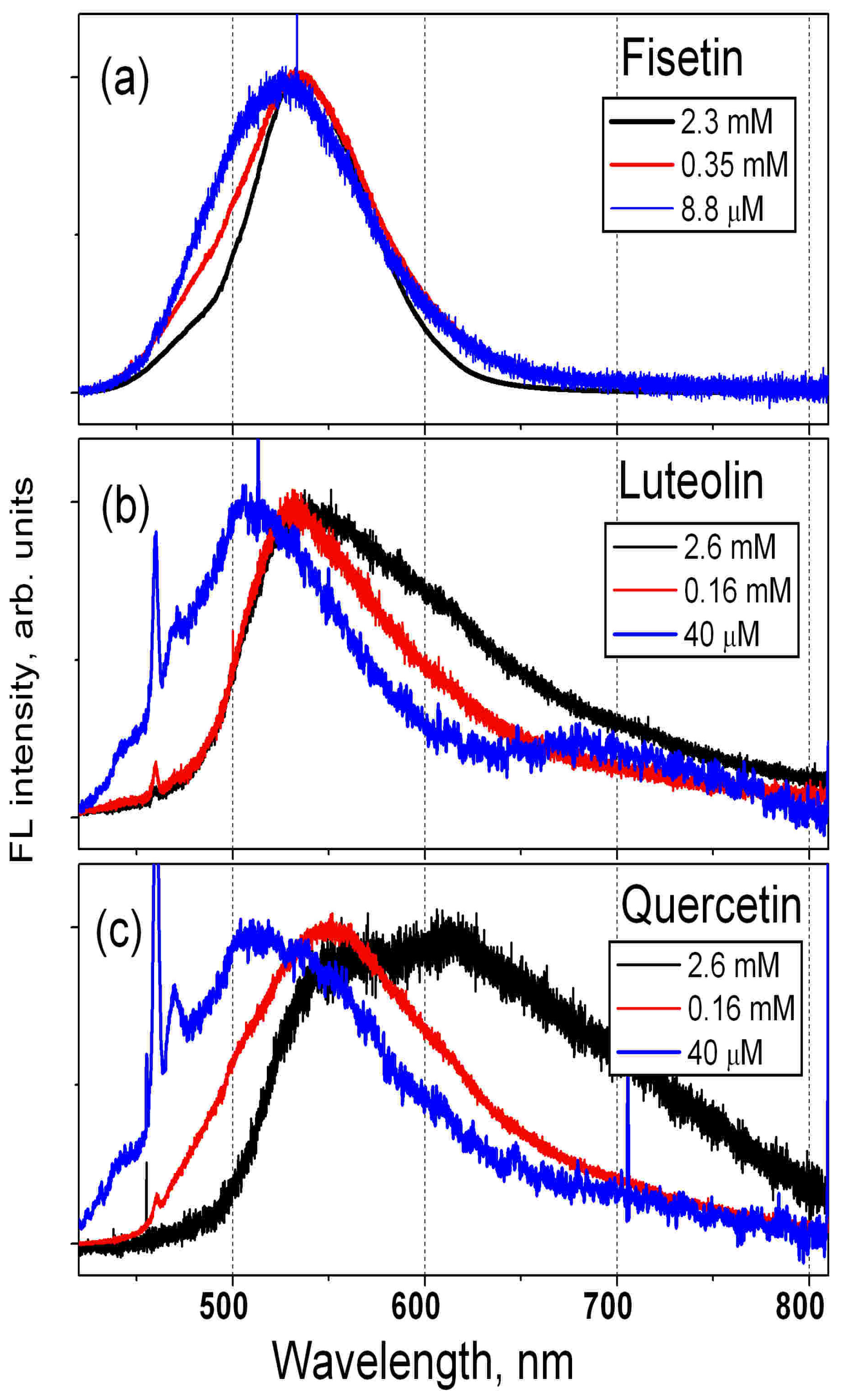
3.1. Measured FL Spectra of F, L and QDH Powders and Solutions
3.2. Calculation of Wavelengths of Absorption and Emission for F, L and Q Molecules
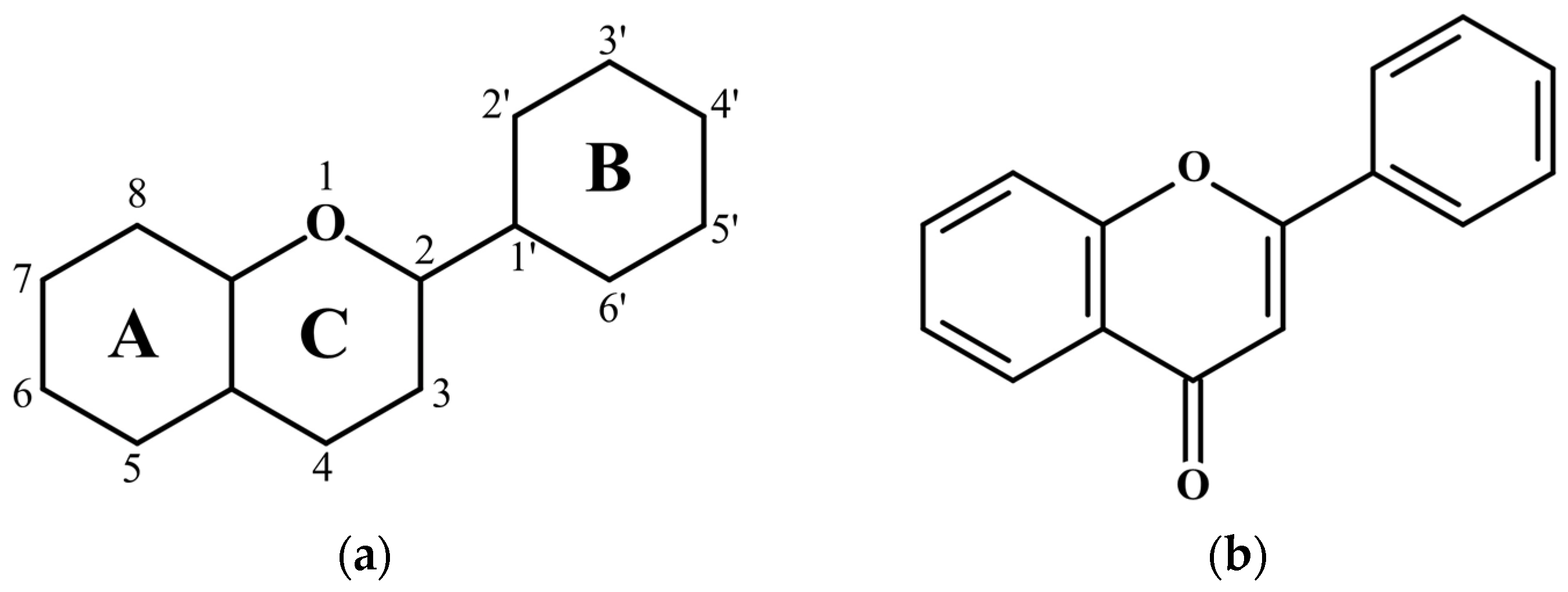
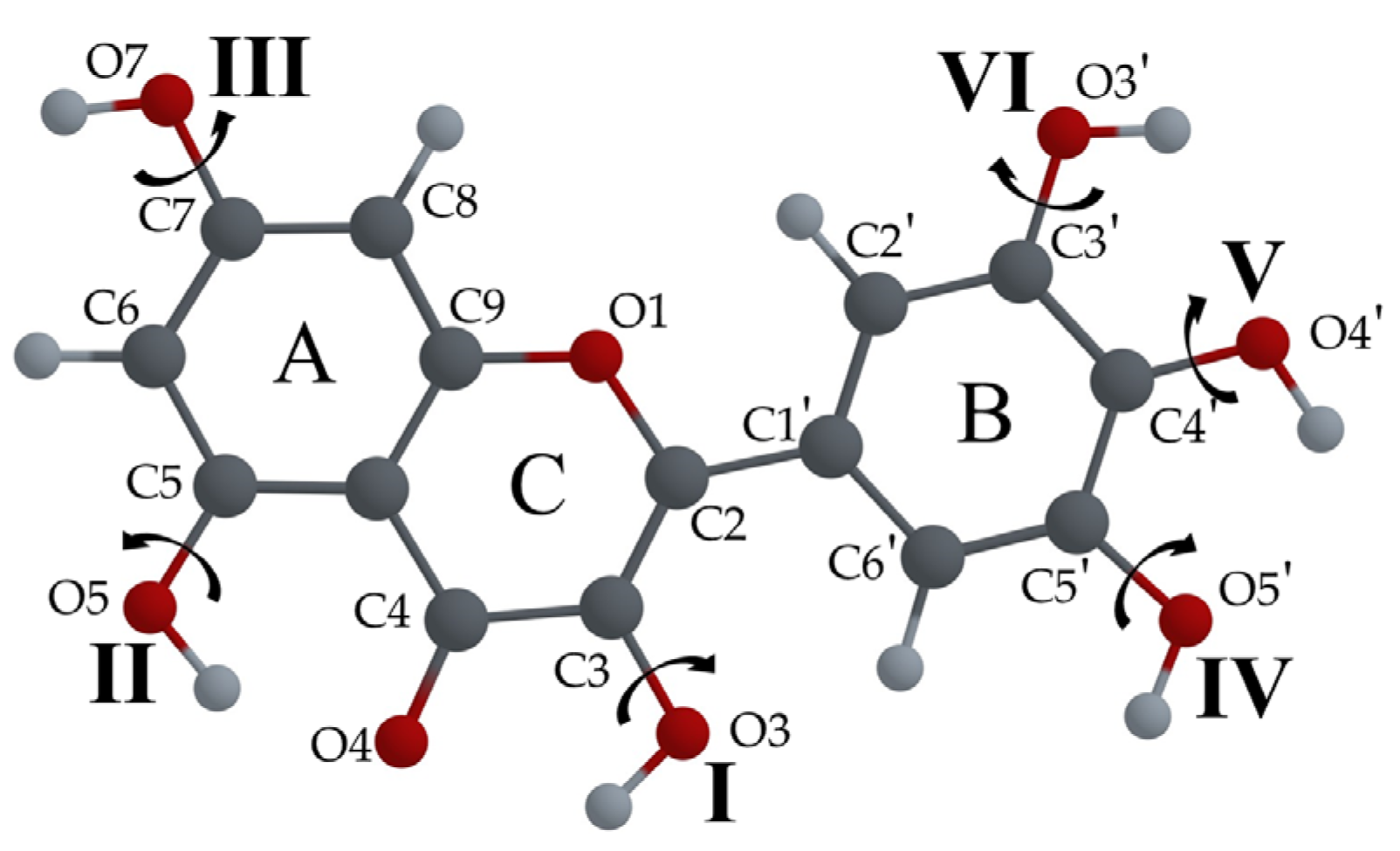
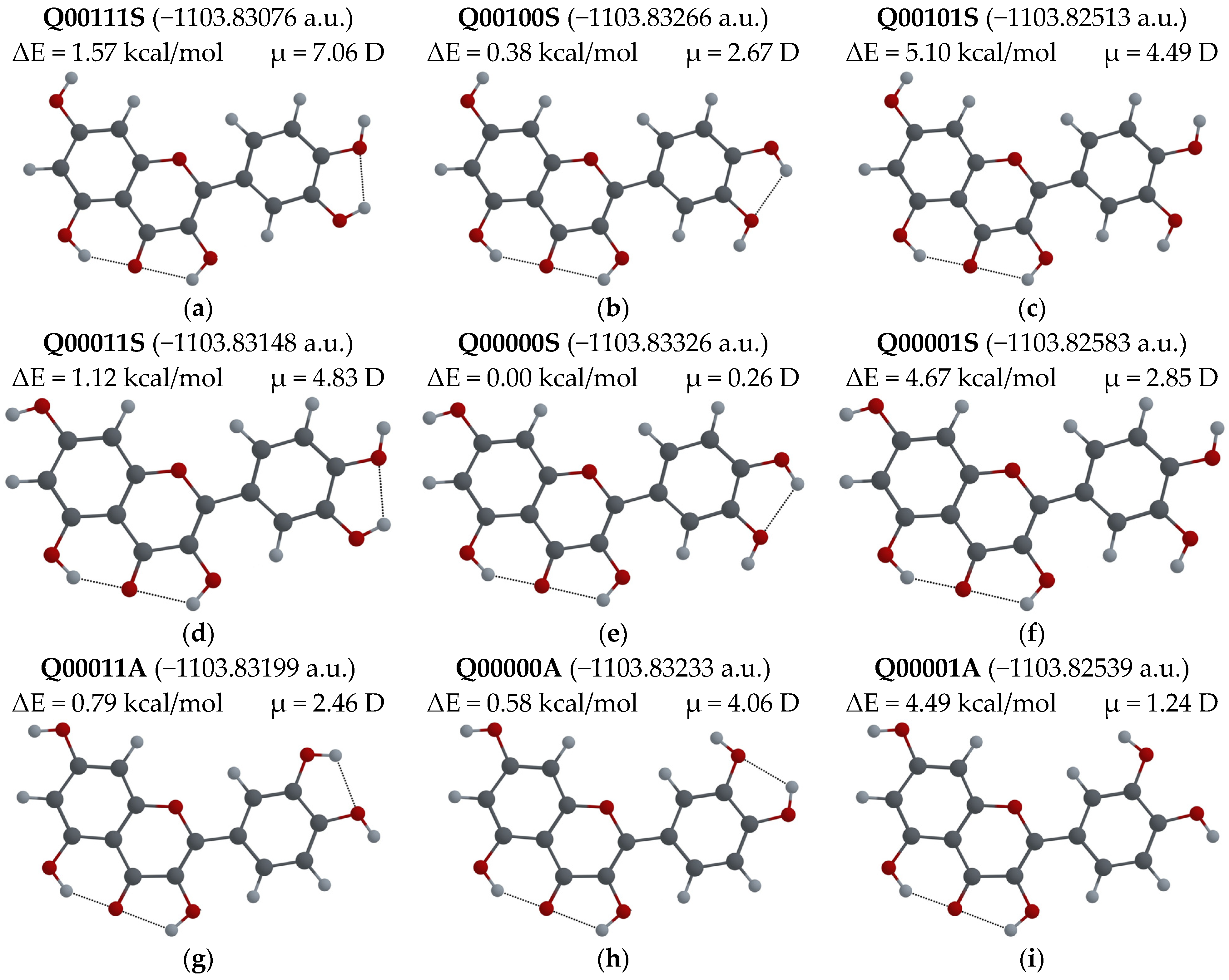
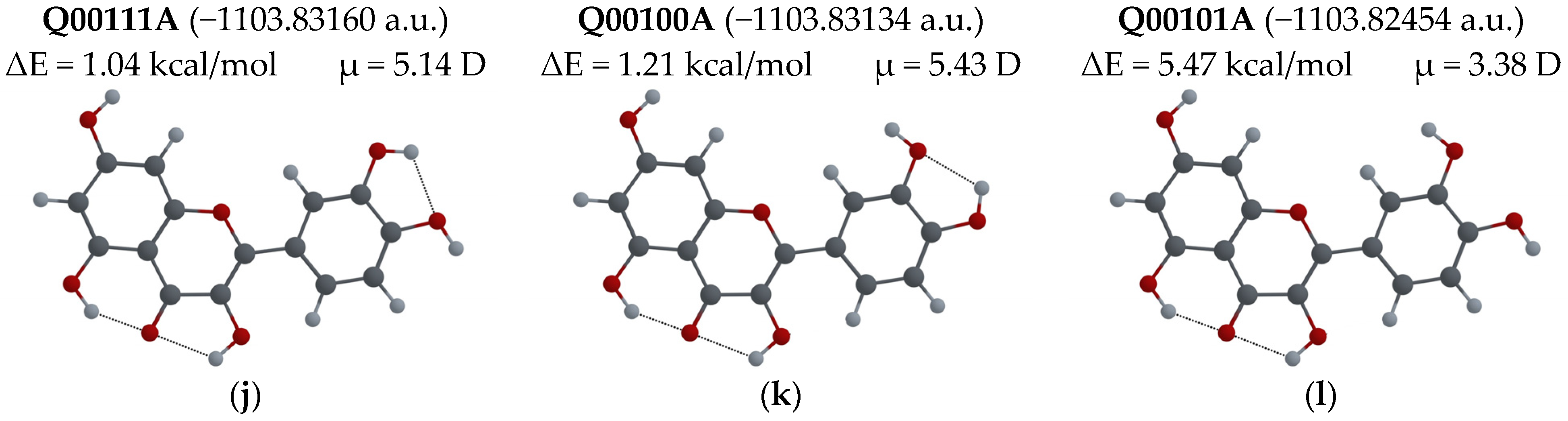
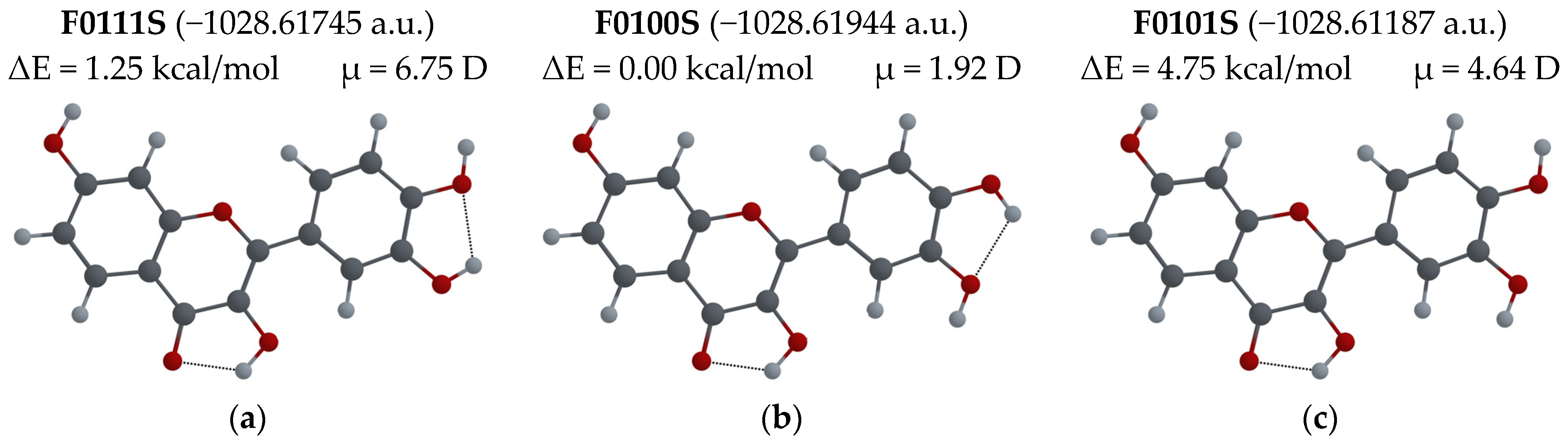
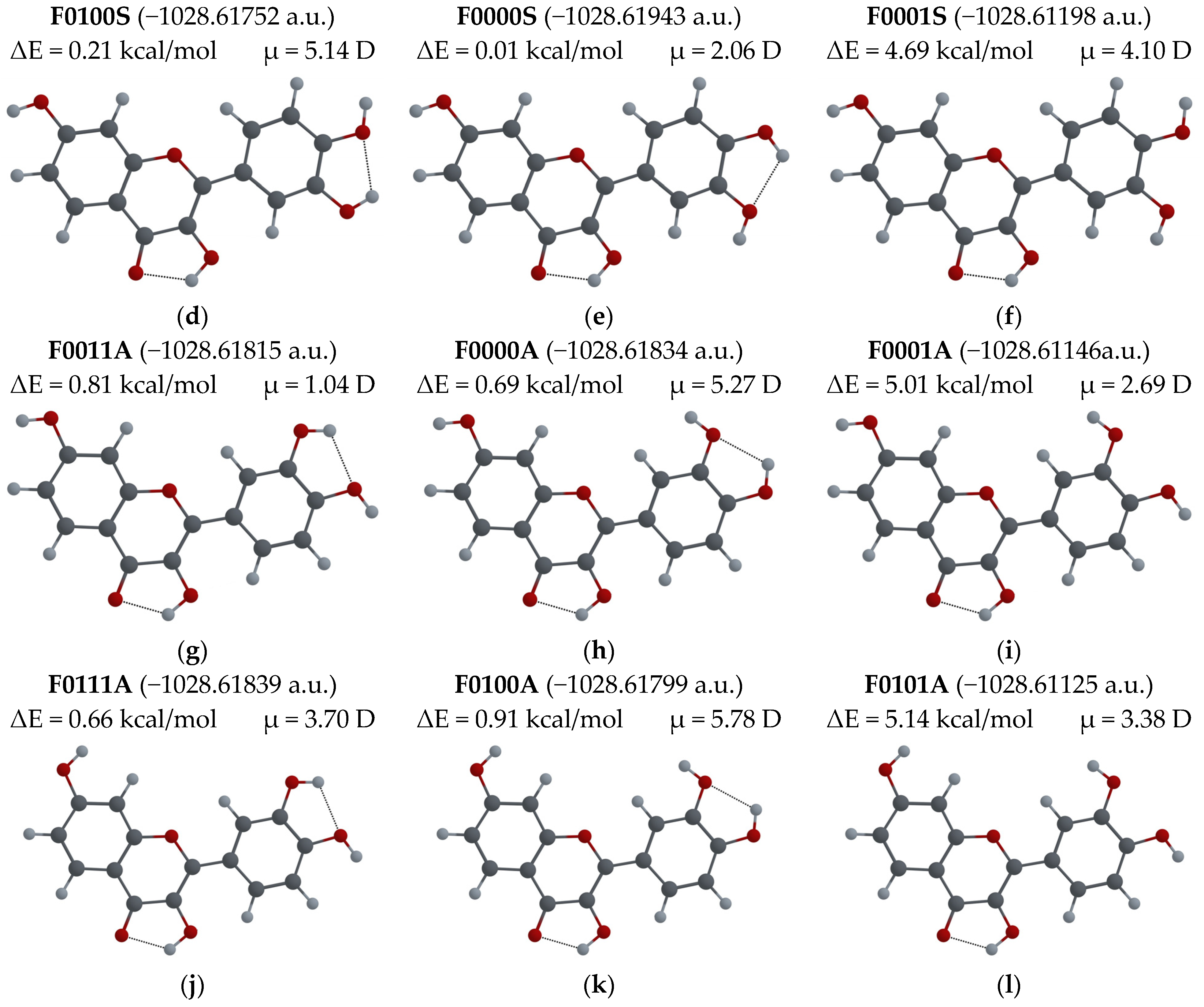
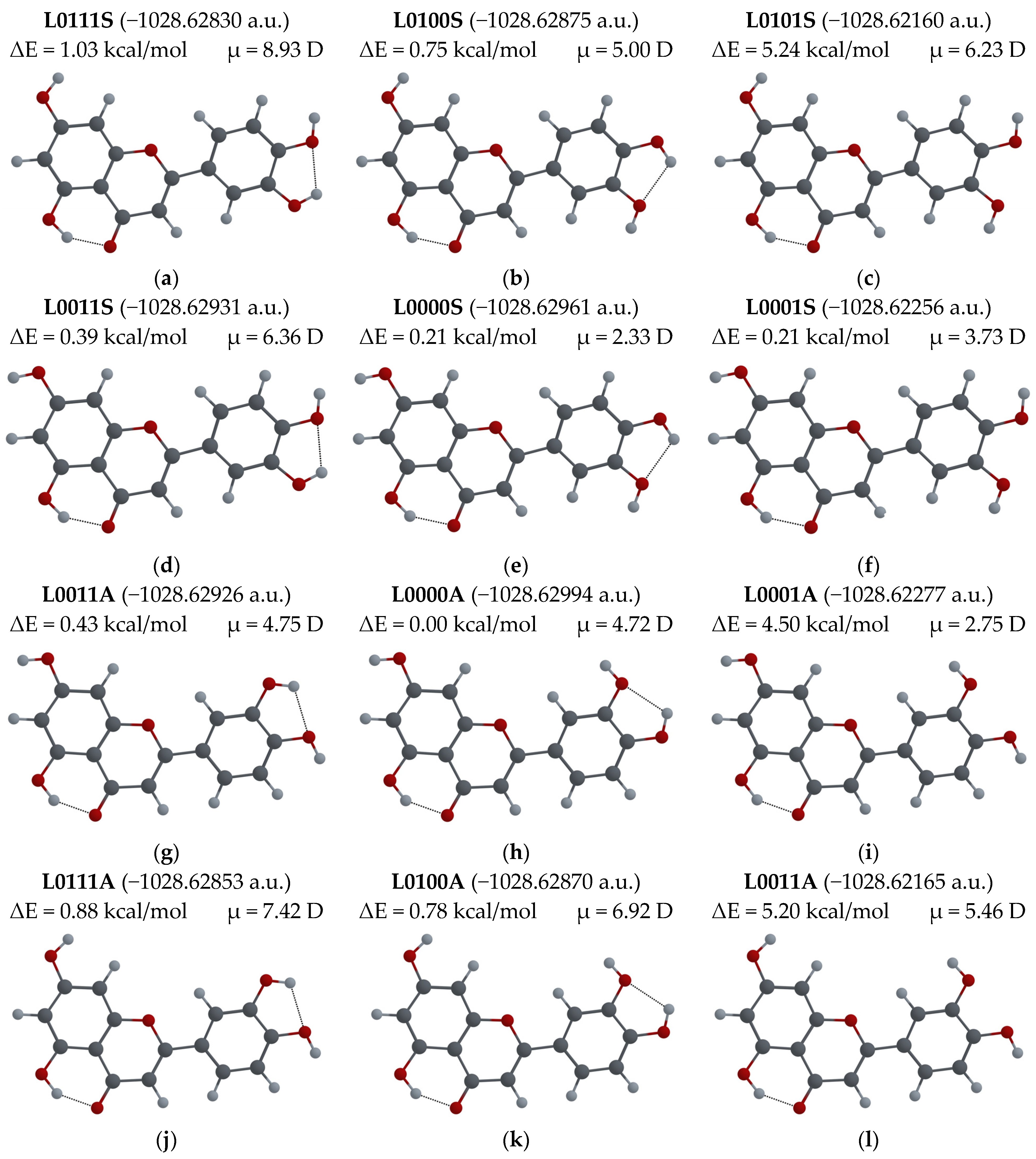
3.2.1. Different Configurations of Flavonoid Molecules: New System of Notation and Selection of the Configuration with the Lowest Potential Energy
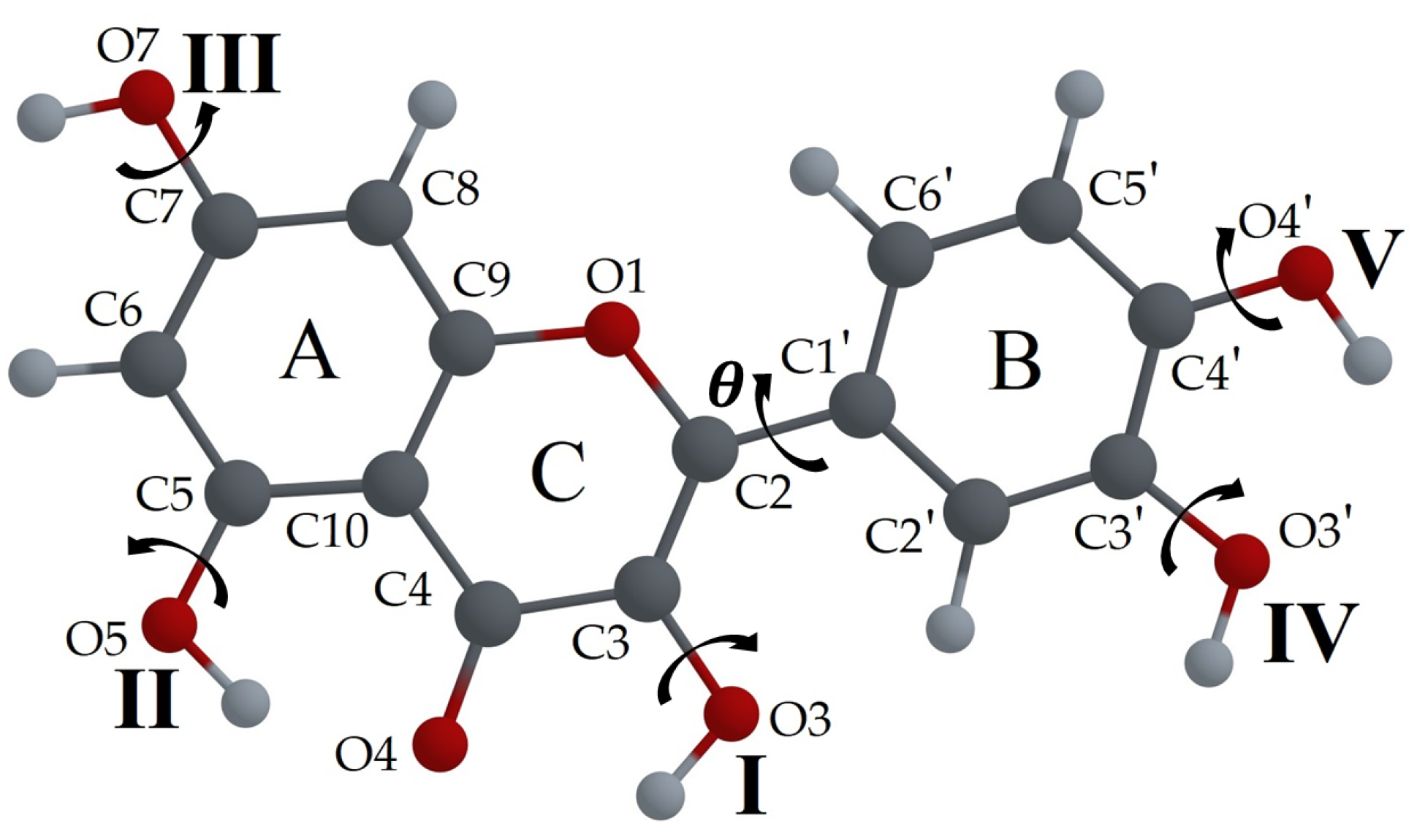
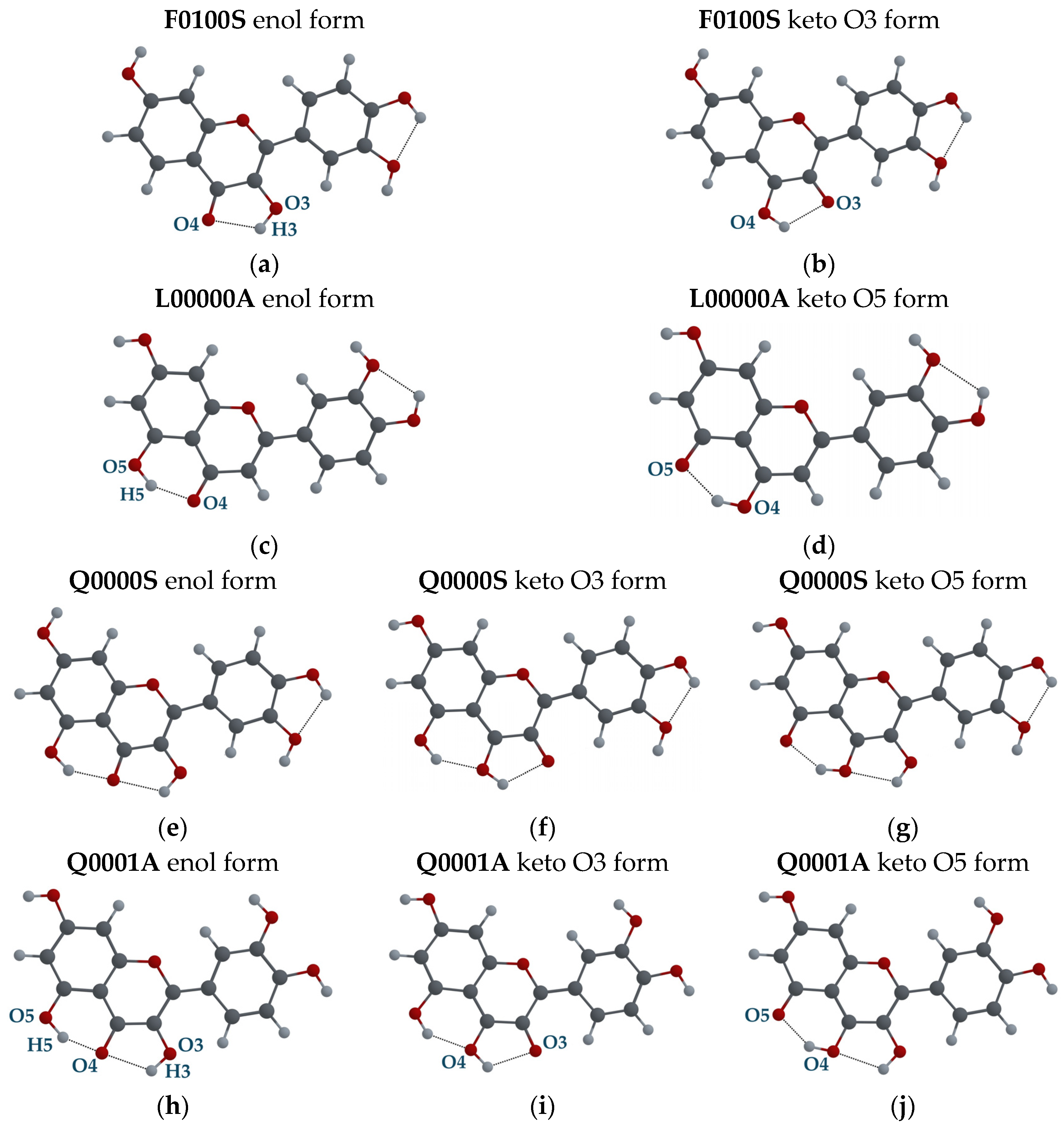
3.2.2. Tautomeric Forms (Enol and Keto) for F, L and Q Molecules
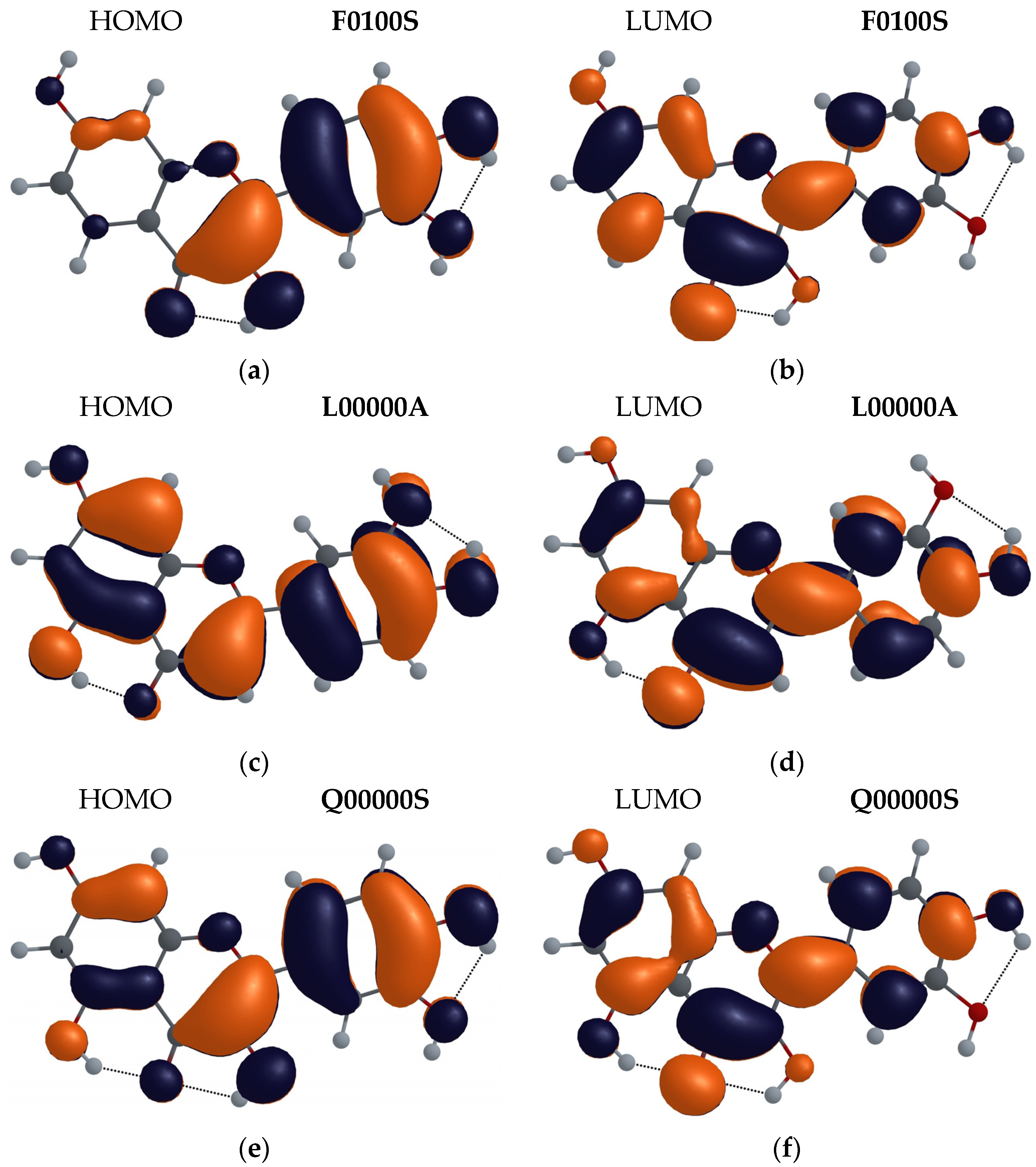
3.2.3. Molecular Orbital Analysis
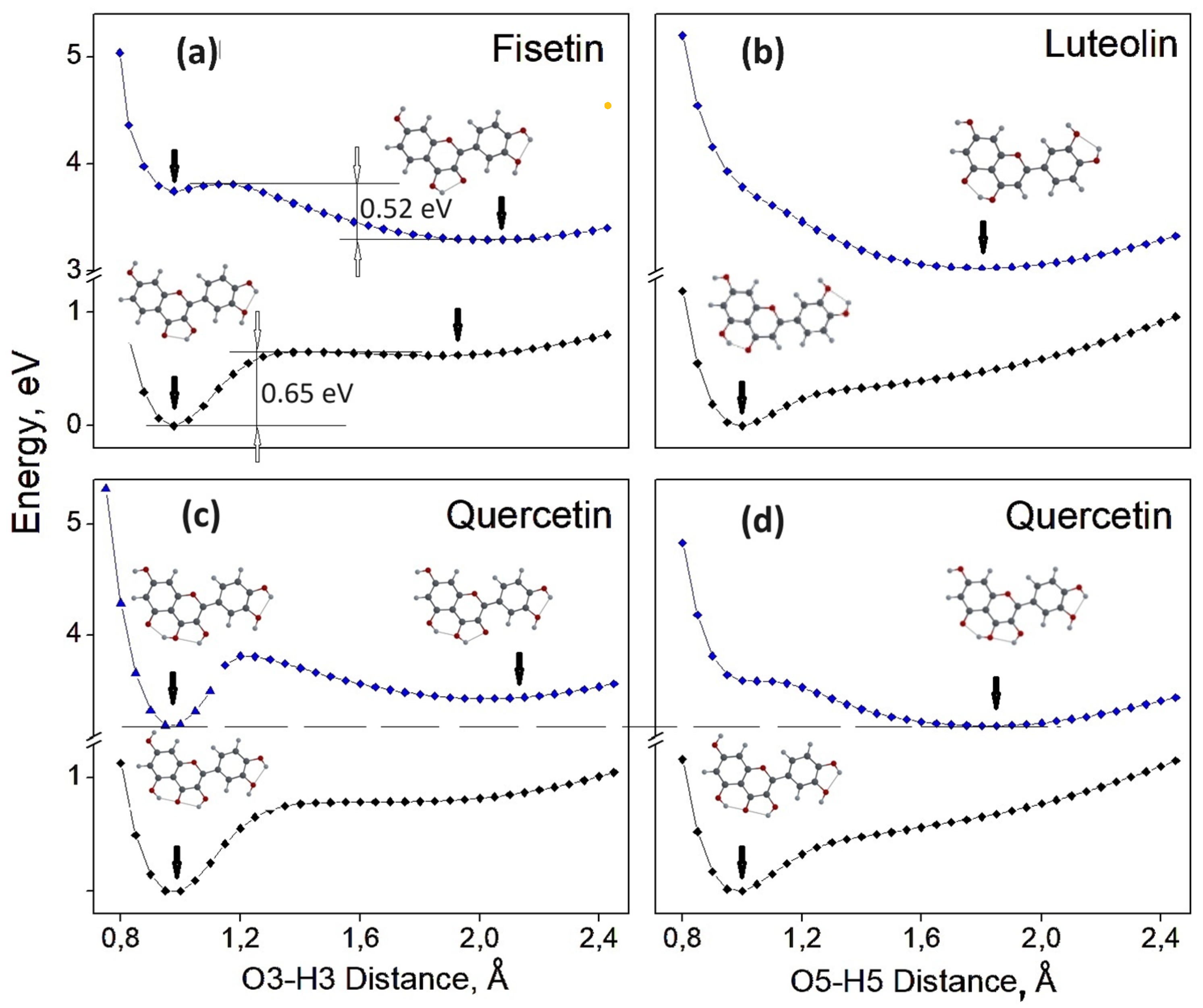
3.2.4. Relaxed Scan of the S0 and S1 State Energies as a Function of O3-H3 or O5-H5 Distance. Search for the Potential Energy Minima, Corresponding to Enol and Keto Forms
3.2.5. Absorption and Emission Characteristic Wavelengths of L, F and Q Molecules
4. Conclusions
- To better show the difference between the ESIPT processes involving different protons, F and L molecules were chosen, since only one proton can be transferred in each of them. We also analyzed the ESIPT in the Q molecule, where the transfer for two protons is possible.
- To easily distinguish between different configurations of flavonoid molecules, a new system of notation for all possible configurations of the molecules was developed.
- For several possible configurations of F, L and Q molecules, a search for the configuration with lowest potential energy was performed.
- Analysis of the electron populations of frontier molecular orbitals, HOMO and LUMO, performed for the lowest energy configurations of F, L and Q molecules, indicated that for all three molecules, the keto forms are favored in the first excited state.
- Relaxed scans of potential energy as a function of the distance between the corresponding O and H atoms were performed for the F, L, and Q molecules. It was found that in the Q molecule, the H5 proton transfer dominates, which is probably common for all flavonoid molecules that have both H3 and H5 protons.
- For the lowest energy configurations of the F, L and Q molecules, the characteristic wavelengths of absorption and emission were calculated using density functional theory (DFT), and the polarizable continuum model (PCM) was used to consider the influence of the solvents on the absorption and emission energy.
- The FL spectra of F, L and Q powders and their solutions in methanol and propylene glycol were measured. The calculation results for vacuum agreed well with the experimental FL spectra of powders and solutions with high concentration; and the calculation results for solutions, with those measured for solutions with low concentrations.
- The fact that the differences in the spectral positions of the maxima of the measured FL spectra of F and L powders correspond to the calculated emission wavelengths confirms the effectiveness of the ESIPT model used for the calculations and the accuracy of its predictions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ES0 (a.u) | ES1 (a.u) | ES1 − ES0 (eV) | λab (nm) | fab | μS0 (D) | ES1* (a.u) | ES0* (a.u) | ES1* − ES0* (eV) | λem (nm) | fem | μS1 (D) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S0 Opt | S0 Opt | S1 Opt | S1 Opt | |||||||||
| F0000A enol | −1028.61834 | −1028.47404 | 3.9267 | 315.75 | 0.6253 | 5.2699 | −1028.48288 | −1028.60976 | 3.4524 | 359.13 | 0.6719 | 5.6120 |
| F0000A keto | −1028.59333 | −1028.49146 | 2.7720 | 447.28 | 0.5319 | 7.5186 | −1028.49710 | −1028.58840 | 2.4843 | 499.08 | 0.5110 | 7.1521 |
| F0000S enol | −1028.61943 | −1028.47404 | 3.9563 | 313.38 | 0.6245 | 2.0566 | −1028.48303 | −1028.61063 | 3.4722 | 357.08 | 0.6709 | 1.9217 |
| F0000S keto | −1028.59616 | −1028.49317 | 2.8025 | 442.41 | 0.5292 | 4.8021 | −1028.49880 | −1028.59120 | 2.5145 | 493.08 | 0.5154 | 4.3103 |
| F0011A enol | −1028.61815 | −1028.47136 | 3.9943 | 310.41 | 0.5901 | 1.0451 | −1028.48044 | −1028.60909 | 3.5006 | 354.19 | 0.6381 | 1.4394 |
| F0011A keto | −1028.59368 | −1028.49008 | 2.8189 | 439.84 | 0.5104 | 2.6407 | −1028.49604 | −1028.58849 | 2.5159 | 492.81 | 0.4889 | 2.3030 |
| F0011S enol | −1028.61752 | −1028.47003 | 4.0133 | 308.94 | 0.5136 | 5.1418 | −1028.47969 | −1028.60824 | 3.4979 | 354.46 | 0.6264 | 5.6228 |
| F0011S keto | −1028.59204 | −1028.48909 | 2.8014 | 442.58 | 0.4885 | 6.7982 | −1028.49523 | −1028.58680 | 2.4918 | 497.58 | 0.4724 | 6.5741 |
| F0100S enol | −1028.61944 | −1028.47277 | 3.9911 | 310.65 | 0.6232 | 1.9192 | −1028.48200 | −1028.61049 | 3.4962 | 354.63 | 0.6733 | 2.3551 |
| F0100S keto | −1028.59672 | −1028.49262 | 2.8325 | 437.72 | 0.5277 | 3.6813 | −1028.49846 | −1028.59158 | 2.5341 | 489.28 | 0.5148 | 3.1464 |
| F0100A enol | −1028.61799 | −1028.47226 | 3.9656 | 312.65 | 0.6195 | 5.7780 | −1028.48149 | −1028.60922 | 3.4756 | 356.73 | 0.6750 | 6.2600 |
| F0100A keto | −1028.59346 | −1028.49050 | 2.8017 | 442.53 | 0.5306 | 7.4229 | −1028.49633 | −1028.58836 | 2.5041 | 495.12 | 0.5114 | 7.0050 |
| F0111A enol | −1028.61839 | −1028.47031 | 4.0296 | 307.69 | 0.5893 | 3.6960 | −1028.47962 | −1028.60918 | 3.5256 | 351.67 | 0.6422 | 4.0375 |
| F0111A keto | −1028.59443 | −1028.48972 | 2.8493 | 435.14 | 0.5096 | 3.0845 | −1028.49586 | −1028.58908 | 2.5368 | 488.75 | 0.4896 | 2.8868 |
| F0111S enol | −1028.61745 | −1028.46848 | 4.0535 | 305.87 | 0.5692 | 6.7527 | −1028.47848 | −1028.60794 | 3.5228 | 351.95 | 0.6293 | 7.2079 |
| F0111S keto | −1028.59245 | −1028.48836 | 2.8327 | 437.70 | 0.4871 | 7.5223 | −1028.49470 | −1028.58702 | 2.5122 | 493.54 | 0.4716 | 7.2479 |
| F0001A enol | −1028.61146 | −1028.46703 | 3.9301 | 315.47 | 0.6180 | 2.6925 | −1028.47576 | −1028.60298 | 3.4618 | 358.16 | 0.6692 | 2.8792 |
| F0001A keto | −1028.58676 | −1028.48446 | 2.7836 | 445.41 | 0.5344 | 5.2281 | −1028.49013 | −1028.58186 | 2.4960 | 496.73 | 0.5131 | 4.8196 |
| F0001S enol | −1028.61198 | −1028.46658 | 3.9564 | 313.38 | 0.6118 | 4.1028 | −1028.47554 | −1028.60322 | 3.4744 | 356.86 | 0.6616 | 4.3161 |
| F0001S keto | −1028.58819 | −1028.48541 | 2.7968 | 443.31 | 0.5191 | 6.5756 | −1028.49112 | −1028.58320 | 2.5056 | 494.82 | 0.5062 | 6.1663 |
| F0101A enol | −1028.61125 | −1028.46539 | 3.9689 | 312.39 | 0.6129 | 3.0937 | −1028.47451 | −1028.60259 | 3.4854 | 355.72 | 0.6725 | 3.5565 |
| F0101A keto | −1028.58703 | −1028.48364 | 2.8133 | 440.72 | 0.5333 | 4.6620 | −1028.48950 | −1028.58198 | 2.5164 | 492.70 | 0.5139 | 4.1789 |
| F0101S enol | −1028.61187 | −1028.46512 | 3.9933 | 310.48 | 0.6083 | 4.6401 | −1028.47438 | −1028.60295 | 3.4987 | 354.37 | 0.6635 | 5.0297 |
| F0101S keto | −1028.58864 | −1028.48474 | 2.8272 | 438.54 | 0.5167 | 6.3011 | −1028.49067 | −1028.58347 | 2.5253 | 490.98 | 0.5052 | 5.8538 |
| ES0 (a.u) | ES1 (a.u) | ES1 − ES0 (eV) | λab (nm) | fab | μS0 (D) | ES1* (a.u) | ES0* (a.u) | ES1* − ES0* (eV) | λem (nm) | fem | μS1 (D) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S0 Opt | S0 Opt | S1 Opt | S1 Opt | |||||||||
| L0000A enol | −1028.62994 | −1028.47479 | 4.2217 | 293.68 | 0.4830 | 4.7157 | - | - | - | - | - | - |
| L0000A keto | - | - | - | - | - | - | −1028.51678 | −1028.59237 | 2.0570 | 602.7 | 0.0541 | 7.0613 |
| L0000S enol | −1028.62961 | −1028.47377 | 4.2405 | 292.38 | 0.4699 | 2.3281 | - | - | - | - | - | - |
| L0000S keto | - | - | - | - | - | - | −1028.51661 | −1028.59140 | 2.0351 | 609.2 | 0.0484 | 7.3474 |
| L0011A enol | −1028.62926 | −1028.47299 | 4.2522 | 291.58 | 0.3734 | 4.7458 | - | - | - | - | - | - |
| L0011A keto | - | - | - | - | - | - | −1028.51660 | −1028.59104 | 2.0256 | 612.0 | 0.3734 | 9.6384 |
| L0011S enol | −1028.62931 | −1028.47206 | 4.2790 | 289.76 | 0.3408 | 6.3643 | - | - | - | - | - | - |
| L0011S keto | - | - | - | - | - | - | −1028.51635 | −1028.59135 | 2.0407 | 607.5 | 0.0479 | 9.4537 |
| L0100S enol | −1028.62875 | −1028.47180 | 4.2706 | 290.32 | 0.4091 | 5.0034 | - | - | - | - | - | - |
| L0100S keto | - | - | - | - | - | - | −1028.51536 | −1028.58785 | 1.9727 | 628.5 | 0.0360 | 9.8519 |
| L0100A enol | −1028.62870 | −1028.47239 | 4.2534 | 291.50 | 0.3954 | 6.9163 | - | - | - | - | - | - |
| L0100A keto | - | - | - | - | - | - | −1028.51520 | −1028.58851 | 1.9948 | 621.5 | 0.0406 | 9.9094 |
| L0111A enol | −1028.62853 | −1028.47150 | 4.2730 | 290.16 | 0.0011 | 7.4161 | - | - | - | - | - | - |
| L0111A keto | - | - | - | - | - | - | −1028.51556 | −1028.58772 | 1.9636 | 631.4 | 0.0362 | 12.1958 |
| L0111S enol | −1028.62830 | −1028.47079 | 4.2859 | 289.28 | 0.0527 | 8.9325 | - | - | - | - | - | - |
| L0111S keto | - | - | - | - | - | - | −1028.51500 | −1028.58776 | 1.9798 | 626.2 | 0.0360 | 12.3102 |
| L0001A enol | −1028.62277 | −1028.46780 | 4.2169 | 294.02 | 0.4742 | 2.7508 | - | - | - | - | - | - |
| L0001A keto | - | - | - | - | - | - | −1028.50962 | −1028.58504 | 2.0522 | 604.1 | 0.0532 | 7.0977 |
| L0001S enol | −1028.62256 | −1028.46659 | 4.2443 | 292.12 | 0.4753 | 3.7317 | - | - | - | - | - | - |
| L0001S keto | - | - | - | - | - | - | −1028.50938 | −1028.58440 | 2.0415 | 607.3 | 0.0364 | 7.0608 |
| L0101A enol | −1028.62165 | −1028.46553 | 4.2484 | 291.84 | 0.3954 | 5.4552 | - | - | - | - | - | - |
| L0101A keto | - | - | - | - | - | - | −1028.50814 | −1028.58125 | 1.9895 | 623.1 | 0.0399 | 9.7710 |
| L0101S enol | −1028.62160 | −1028.46454 | 4.2738 | 290.11 | 0.3947 | 6.2317 | - | - | - | - | - | - |
| L0101S keto | - | - | - | - | - | - | −1028.50806 | −1028.58073 | 1.9775 | 626.9 | 0.0364 | 9.8860 |
References
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A Dietary Antioxidant for Health Promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant Flavonoids: Classification, Distribution, Biosynthesis, and Antioxidant Activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Tong, C.; Shi, F.; Tong, X.; Shi, S.; Ali, I.; Guo, Y. Shining Natural Flavonols in Sensing and Bioimaging. TrAC Trends Anal. Chem. 2021, 137, 116222. [Google Scholar] [CrossRef]
- Forzato, C.; Vida, V.; Berti, F. Biosensors and Sensing Systems for Rapid Analysis of Phenolic Compounds from Plants: A Comprehensive Review. Biosensors 2020, 10, 105. [Google Scholar] [CrossRef]
- Trabelsi, H.; Koch, M.; Faulon, J. Building a Minimal and Generalizable Model of Transcription Factor–Based Biosensors: Showcasing Flavonoids. Biotech. Bioeng. 2018, 115, 2292–2304. [Google Scholar] [CrossRef]
- Kumar, V.; Arora, K. Trends in Nano-Inspired Biosensors for Plants. Mater. Sci. Energy Technol. 2020, 3, 255–273. [Google Scholar] [CrossRef]
- Vishnu, S.; Maity, S.; Maity, A.C.; Kumar, M.S.; Dolai, M.; Nag, A.; Bylappa, Y.; Dutta, G.; Mukherjee, B.; Kumar Das, A. Development of a Fluorescent Scaffold by Utilizing Quercetin Template for Selective Detection of Hg2+: Experimental and Theoretical Studies along with Live Cell Imaging. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2024, 315, 124249. [Google Scholar] [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-88460-3. [Google Scholar]
- Guharay, J.; Dennison, S.M.; Sengupta, P.K. Influence of Different Environments on the Excited-State Proton Transfer and Dual Fluorescence of Fisetin. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 1999, 55, 1091–1099. [Google Scholar] [CrossRef]
- Höfener, S.; Kooijman, P.C.; Groen, J.; Ariese, F.; Visscher, L. Fluorescence Behavior of (Selected) Flavonols: A Combined Experimental and Computational Study. Phys. Chem. Chem. Phys. 2013, 15, 12572. [Google Scholar] [CrossRef]
- Sengupta, B.; Pahari, B.; Blackmon, L.; Sengupta, P.K. Prospect of Bioflavonoid Fisetin as a Quadruplex DNA Ligand: A Biophysical Approach. PLoS ONE 2013, 8, e65383. [Google Scholar] [CrossRef]
- Ji, F.; Guo, Y.; Wang, M.; Wang, C.; Wu, Z.; Wang, S.; Wang, H.; Feng, X.; Zhao, G. New Insights into ESIPT Mechanism of Three Sunscreen Compounds in Solution: A Combined Experimental and Theoretical Study. Colloids Surf. B Biointerfaces 2021, 207, 112039. [Google Scholar] [CrossRef]
- Favaro, G.; Clementi, C.; Romani, A.; Vickackaite, V. Acidichromism and Ionochromism of Luteolin and Apigenin, the Main Components of the Naturally Occurring Yellow Weld: A Spectrophotometric and Fluorimetric Study. J. Fluoresc. 2007, 17, 707–714. [Google Scholar] [CrossRef]
- Han, Y.; Jia, Y.; Wang, X.; Chen, Z.; Jin, P.; Jia, M.; Pan, H.; Sun, Z.; Chen, J. Ultrafast Excited State Dynamics of Two Non-Emissive Flavonoids That Undergo Excited State Intramolecular Proton Transfer in Solution. Chem. Phys. Lett. 2023, 811, 140189. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited State Intramolecular Proton Transfer (ESIPT): From Principal Photophysics to the Development of New Chromophores and Applications in Fluorescent Molecular Probes and Luminescent Materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef] [PubMed]
- Weller, A. Über Die Fluoreszenz Der Salizylsäure Und Verwandter Verbindungen. Naturwissenschaften 1955, 42, 175–176. [Google Scholar] [CrossRef]
- Weller, A. Innermolekularer Protonenübergang im angeregten Zustand. Z. Für Elektrochem. Berichte Der Bunsenges. Für Phys. Chem. 1956, 60, 1144–1147. [Google Scholar] [CrossRef]
- Santos, G.C.; Rocha, I.O.; Stefanello, F.S.; Copetti, J.P.P.; Tisoco, I.; Martins, M.A.P.; Zanatta, N.; Frizzo, C.P.; Iglesias, B.A.; Bonacorso, H.G. Investigating ESIPT and Donor-Acceptor Substituent Effects on the Photophysical and Electrochemical Properties of Fluorescent 3,5-Diaryl-Substituted 1-Phenyl-2-Pyrazolines. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2022, 269, 120768. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, Y.; Shi, W.; Ma, F.; Li, Y. The Effects of Amino Group Meta- and Para-Substitution on ESIPT Mechanisms of Amino 2-(2′-Hydroxyphenyl) Benzazole Derivatives. J. Lumin. 2020, 218, 116836. [Google Scholar] [CrossRef]
- Zhao, G.; Shi, W.; Xin, X.; Yang, Y.; Ma, F.; Li, Y. Insights from Computational Analysis: Excited-State Hydrogen-Bonding Interactions and ESIPT Processes in Phenothiazine Derivatives. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2023, 286, 121935. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, M.; Zhao, G. Combined TDDFT and AIM Insights into Photoinduced Excited State Intramolecular Proton Transfer (ESIPT) Mechanism in Hydroxyl- and Amino-Anthraquinone Solution. Sci. Rep. 2017, 7, 13766. [Google Scholar] [CrossRef] [PubMed]
- Kenfack, C.A.; Klymchenko, A.S.; Duportail, G.; Burger, A.; Mély, Y. Ab Initio Study of the Solvent H-Bonding Effect on ESIPT Reaction and Electronic Transitions of 3-Hydroxychromone Derivatives. Phys. Chem. Chem. Phys. 2012, 14, 8910. [Google Scholar] [CrossRef]
- Kaya, S.; Aydın, H.G.; Keskin, S.; Ekmekci, Z.; Menges, N. Exploring of Indole Derivatives for ESIPT Emission: A New ESIPT-Based Fluorescence Skeleton and TD-DFT Calculations. J. Photochem. Photobiol. A Chem. 2021, 420, 113487. [Google Scholar] [CrossRef]
- Stoerkler, T.; Pariat, T.; Laurent, A.D.; Jacquemin, D.; Ulrich, G.; Massue, J. Excited-State Intramolecular Proton Transfer Dyes with Dual-State Emission Properties: Concept, Examples and Applications. Molecules 2022, 27, 2443. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.K.; Kasha, M. Excited State Proton-Transfer Spectroscopy of 3-Hydroxyflavone and Quercetin. Chem. Phys. Lett. 1979, 68, 382–385. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Li, Y. Theoretical Study of the ESIPT Process for a New Natural Product Quercetin. Sci. Rep. 2016, 6, 32152. [Google Scholar] [CrossRef]
- Prutskij, T.; Deriabina, A.; Melendez, F.J.; Castro, M.E.; Castillo Trejo, L.; Vazquez Leon, G.D.; Gonzalez, E.; Perova, T.S. Concentration-Dependent Fluorescence Emission of Quercetin. Chemosensors 2021, 9, 315. [Google Scholar] [CrossRef]
- Prutskij, T.; Deriabina, A.; Vázquez Leon, G.D.; Castillo Trejo, L. Excited-State Proton Transfer Based Fluorescence in Kaempferol Powder and Solutions with Different Concentrations. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2024, 309, 123814. [Google Scholar] [CrossRef]
- Deriabina, A.; Prutskij, T.; Morales Ochoa, H.D.; Gonzalez Jimenez, E.; Deriabin, S. Comparative Analysis of Fluorescence Emission in Myricetin, Kaempferol, and Quercetin Powders and Solutions. Int. J. Mol. Sci. 2024, 25, 2558. [Google Scholar] [CrossRef]
- Deriabina, A.; Prutskij, T.; Castillo Trejo, L.; Sanchez Gutierrez, M.P.; Gonzalez Jimenez, E. Experimental and Theoretical Study of Fluorescent Properties of Morin. Molecules 2022, 27, 4965. [Google Scholar] [CrossRef]
- Ji, F.; Wu, Z.; Wang, M.; Guo, Y.; Wang, C.; Wang, S.; Zhao, G. New Insights into the Excited State Intramolecular Proton Transfer (ESIPT) Competition Mechanism for Different Intramolecular Hydrogen Bonds of Kaempferol and Quercetin in Solution. J. Lumin. 2022, 248, 118914. [Google Scholar] [CrossRef]
- Sahu, A.K.; Mishra, A.K. Photophysical Behavior of Plant Flavonols Galangin, Kaempferol, Quercetin, and Myricetin in Homogeneous Media and the DMPC Model Membrane: Unveiling the Influence of the B-Ring Hydroxylation of Flavonols. J. Phys. Chem. B 2022, 126, 2863–2875. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhao, J.; Zheng, R.; Wang, Y.; Lv, J. A DFT/TDDFT Investigation of the Excited State Proton Transfer Reaction of Fisetin Chromophore. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2015, 151, 368–374. [Google Scholar] [CrossRef]
- Amat, A.; Clementi, C.; De Angelis, F.; Sgamellotti, A.; Fantacci, S. Absorption and Emission of the Apigenin and Luteolin Flavonoids: A TDDFT Investigation. J. Phys. Chem. A 2009, 113, 15118–15126. [Google Scholar] [CrossRef]
- Carrillo-Martinez, E.J.; Flores-Hernández, F.Y.; Salazar-Montes, A.M.; Nario-Chaidez, H.F.; Hernández-Ortega, L.D. Quercetin, a Flavonoid with Great Pharmacological Capacity. Molecules 2024, 29, 1000. [Google Scholar] [CrossRef] [PubMed]
- Shaker, L.M.; Al-Amiery, A.A.; Abed, T.K.; Al-Azzawi, W.K.; Kadhum, A.A.H.; Sulaiman, G.M.; Mohammed, H.A.; Khan, M.; Khan, R.A. An Overview of the Density Functional Theory on Antioxidant Bioactivity Predictive Feasibilities: Insights from Natural Antioxidant Products. J. Mol. Struct. 2024, 1301, 137393. [Google Scholar] [CrossRef]
- Vasisht, K.; Chadha, K.; Karan, M.; Bhalla, Y.; Jena, A.K.; Chadha, R. Enhancing Biopharmaceutical Parameters of Bioflavonoid Quercetin by Cocrystallization. CrystEngComm 2016, 18, 1403–1415. [Google Scholar] [CrossRef]
- Domagała, S.; Munshi, P.; Ahmed, M.; Guillot, B.; Jelsch, C. Structural Analysis and Multipole Modelling of Quercetin Monohydrate—A Quantitative and Comparative Study. Acta Crystallogr. B Struct. Sci. 2011, 67, 63–78. [Google Scholar] [CrossRef]
- Rossi, M.; Rickles, L.F.; Halpin, W.A. The Crystal and Molecular Structure of Quercetin: A Biologically Active and Naturally Occurring Flavonoid. Bioorgan. Chem. 1986, 14, 55–69. [Google Scholar] [CrossRef]
- Jin, G.Z.; Yamagata, Y.; Tomita, K. Structure of Quercetin Dihydrate. Acta Crystallogr. C Cryst. Struct. Commun. 1990, 46, 310–313. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. In Proceedings of the Horizons of Quantum Chemistry; Fukui, K., Pullman, B., Eds.; Springer: Dordrecht, The Netherlands, 1980; pp. 5–15. [Google Scholar]
- Parr, R.G.; Weitao, Y. Density-Functional Theory. In Density-Functional Theory; Oxford University Press: London, UK, 1995. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor Chem Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Laine, M.; Barbosa, N.A.; Wieczorek, R.; Melnikov, M.Y.; Filarowski, A. Calculations of BODIPY Dyes in the Ground and Excited States Using the M06-2X and PBE0 Functionals. J. Mol. Model. 2016, 22, 260. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- CRC Handbook of Chemistry and Physics, 84th Edition Edited by David R. Lide. J. Am. Chem. Soc. 2004, 126, 1586. [CrossRef]
- Cossi, M.; Barone, V. Solvent Effect on Vertical Electronic Transitions by the Polarizable Continuum Model. J. Chem. Phys. 2000, 112, 2427–2435. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, J.; Xie, Y. Improvement Strategies for the Oral Bioavailability of Poorly Water-Soluble Flavonoids: An Overview. Int. J. Pharm. 2019, 570, 118642. [Google Scholar] [CrossRef]
- Tommasini, S.; Raneri, D.; Ficarra, R.; Calabrò, M.L.; Stancanelli, R.; Ficarra, P. Improvement in Solubility and Dissolution Rate of Flavonoids by Complexation with β-Cyclodextrin. J. Pharm. Biomed. Anal. 2004, 35, 379–387. [Google Scholar] [CrossRef]
- Smith, A.J.; Kavuru, P.; Wojtas, L.; Zaworotko, M.J.; Shytle, R.D. Cocrystals of Quercetin with Improved Solubility and Oral Bioavailability. Mol. Pharm. 2011, 8, 1867–1876. [Google Scholar] [CrossRef]
- Olejniczak, S.; Potrzebowski, M.J. Solid State NMR Studies and Density Functional Theory (DFT) Calculations of Conformers of Quercetin. Org. Biomol. Chem. 2004, 2, 2315. [Google Scholar] [CrossRef]
- Dubey, R.; Desiraju, G.R. Combinatorial Selection of Molecular Conformations and Supramolecular Synthons in Quercetin Cocrystal Landscapes: A Route to Ternary Solids. IUCrJ 2015, 2, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kavuru, P.; Aboarayes, D.; Arora, K.K.; Clarke, H.D.; Kennedy, A.; Marshall, L.; Ong, T.T.; Perman, J.; Pujari, T.; Wojtas, Ł.; et al. Hierarchy of Supramolecular Synthons: Persistent Hydrogen Bonds Between Carboxylates and Weakly Acidic Hydroxyl Moieties in Cocrystals of Zwitterions. Cryst. Growth Des. 2010, 10, 3568–3584. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Zhang, X.; Wu, S.; Gong, J. Exploring the Influence of Crystal Packing on the Optical-Physical Property of Quercetin-Based Binary and Ternary Solid Forms. Chin. Chem. Lett. 2023, 34, 107983. [Google Scholar] [CrossRef]
- Ahmedova, A.; Paradowska, K.; Wawer, I. 1H, 13C MAS NMR and DFT GIAO Study of Quercetin and Its Complex with Al(III) in Solid State. J. Inorg. Biochem. 2012, 110, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Wawer, I.; Zielinska, A. 13C CP/MAS NMR Studies of Flavonoids. Magn. Reson. Chem. 2001, 39, 374–380. [Google Scholar] [CrossRef]
- Sowa, M.; Ślepokura, K.; Matczak-Jon, E. Improving Solubility of Fisetin by Cocrystallization. CrystEngComm 2014, 16, 10592–10601. [Google Scholar] [CrossRef]
- Cox, P.J.; Kumarasamy, Y.; Nahar, L.; Sarker, S.D.; Shoeb, M. Luteolin. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, o975–o977. [Google Scholar] [CrossRef]
- He, H.; Huang, Y.; Zhang, Q.; Wang, J.-R.; Mei, X. Zwitterionic Cocrystals of Flavonoids and Proline: Solid-State Characterization, Pharmaceutical Properties, and Pharmacokinetic Performance. Cryst. Growth Des. 2016, 16, 2348–2356. [Google Scholar] [CrossRef]

| ES0 (a.u) | ES1 (a.u) | ES1 − ES0 (eV) | λab (nm) | fab | μS0 (D) | ES1* (a.u) | ES0* (a.u) | ES1* − ES0* (eV) | λem (nm) | fem | μS1 (D) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S0 Opt | S0 Opt | S1 Opt | S1 Opt | |||||||||
| Fisetin | ||||||||||||
| F0100S enol | −1028.61944 | −1028.47277 | 3.9911 | 310.65 | 0.6232 | 1.92 | −1028.48200 | −1028.61049 | 3.4962 | 354.63 | 0.6733 | 2.35 |
| F0100S keto | −1028.59672 | −1028.49262 | 2.8325 | 437.72 | 0.5277 | 3.68 | −1028.49846 | −1028.59158 | 2.5341 | 489.28 | 0.5148 | 3.15 |
| F0000S enol | −1028.61943 | −1028.47404 | 3.9563 | 313.38 | 0.6245 | 2.06 | −1028.48303 | −1028.61063 | 3.4722 | 357.08 | 0.6709 | 1.92 |
| F0000S keto | −1028.59616 | −1028.49317 | 2.8025 | 442.41 | 0.5292 | 4.80 | −1028.49880 | −1028.59120 | 2.5145 | 493.08 | 0.5154 | 4.31 |
| Luteolin | ||||||||||||
| L0000A enol | −1028.62994 | −1028.47479 | 4.2217 | 293.68 | 0.4830 | 4.72 | - | - | - | - | - | - |
| L0000A keto | - | - | - | - | - | - | −1028.51678 | −1028.59237 | 2.0570 | 602.75 | 0.0541 | 7.06 |
| L0101A enol | −1028.62165 | −1028.46553 | 4.2484 | 291.84 | 0.3954 | 5.45 | - | - | - | - | - | - |
| L0101A keto | - | - | - | - | - | - | −1028.50814 | −1028.58125 | 1.9895 | 623.19 | 0.0399 | 9.77 |
| Quercetin | ||||||||||||
| Q00001A enol | −1103.82539 | −1103.68525 | 3.8134 | 325.17 | 0.6137 | 1.24 | −1103.69358 | −1103.81652 | 3.3470 | 370.48 | 0.6121 | 1.77 |
| Q00001A keto O3 | - | - | - | - | - | - | −1103.69792 | −1103.78879 | 2.4728 | 501.46 | 0.5086 | 3.32 |
| Q00001A keto O5 | - | - | - | - | - | - | −1103.70799 | −1103.78516 | 2.0999 | 590.50 | 0.1009 | 5.12 |
| ES0 (a.u) | ES1 (a.u) | ES1 − ES0 (eV) | λab (nm) | fab | μS0 (D) | ES1* (a.u) | ES0* (a.u) | ES1* − ES0* (eV) | λem (nm) | fem | μS1 (D) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S0 Opt | S0 Opt | S1 Opt | S1 Opt | |||||||||
| METHANOL | ||||||||||||
| Fisetin | ||||||||||||
| F0100S enol | −1028.63734 | −1028.49589 | 3.8492 | 322.11 | 0.7242 | 3.15 | −1028.50771 | −1028.62438 | 3.1745 | 390.56 | 1.0327 | 4.12 |
| F0100S keto | −1028.61643 | −1028.50454 | 3.0445 | 407.24 | 0.6319 | 5.55 | −1028.51289 | −1028.60894 | 2.6135 | 474.40 | 0.8204 | 4.62 |
| F0000S enol | −1028.63717 | −1028.49566 | 3.8508 | 321.97 | 0.724 | 2.76 | −1028.50759 | −1028.62445 | 3.1800 | 389.89 | 1.0336 | 2.01 |
| F0000S keto | −1028.61600 | −1028.50446 | 3.0351 | 408.50 | 0.6325 | 6.99 | −1028.51285 | −1028.60862 | 2.6062 | 475.73 | 0.8202 | 5.97 |
| Luteolin | ||||||||||||
| L0000A enol | −1028.64902 | −1028.50110 | 4.0252 | 308.02 | 0.0003 | 6.67 | - | - | - | - | - | - |
| L0000A keto | - | - | - | - | - | - | −1028.5402 | −1028.624 | 2.2788 | 544.08 | 0.3191 | 8.77 |
| L0101A enol | −1028.64580 | −1028.49820 | 4.0164 | 308.70 | 0.6991 | 7.51 | - | - | - | - | - | - |
| L0101A keto | - | - | - | - | - | - | −1028.5356 | −1028.6142 | 2.1388 | 579.69 | 0.3000 | 12.03 |
| Quercetin | ||||||||||||
| Q00001A enol | −1103.84661 | −1103.70812 | 3.7684 | 329.02 | 0.7071 | 2.03 | −1103.71784 | −1103.83612 | 3.2185 | 385.23 | 1.0064 | 3.11 |
| Q00001A keto O3 | −1103.81936 | −1103.70920 | 2.9975 | 413.63 | 0.6337 | 5.75 | −1103.71748 | −1103.81215 | 2.5761 | 481.29 | 0.7960 | 5.05 |
| Q00001A keto O5 | - | - | - | - | - | - | −1103.73257 | −1103.81851 | 2.3384 | 530.22 | 0.4758 | 5.75 |
| PROPYLENE GLYCOL | ||||||||||||
| Fisetin | ||||||||||||
| F0100S enol | −1028.63716 | −1028.50829 | 3.5069 | 353.54 | 0.7478 | 3.14 | −1028.50746 | −1028.62550 | 3.2121 | 385.99 | 1.0294 | 4.11 |
| F0100S keto | −1028.61623 | −1028.50724 | 2.9657 | 418.06 | 0.651 | 5.53 | −1028.51275 | −1028.60949 | 2.632 | 471.07 | 0.8177 | 4.61 |
| F0000S enol | −1028.63699 | −1028.50793 | 3.5120 | 353.03 | 0.7475 | 2.75 | −1028.50734 | −1028.62556 | 3.217 | 385.41 | 1.0302 | 2.01 |
| F0000S keto | −1028.61580 | −1028.50714 | 2.9568 | 419.32 | 0.6514 | 6.97 | −1028.51271 | −1028.60915 | 2.6244 | 472.44 | 0.8176 | 5.95 |
| Luteolin | ||||||||||||
| L0000A enol | −1028.64884 | −1028.51396 | 3.6700 | 337.83 | 0.7272 | 6.65 | - | - | - | - | - | - |
| L0000A keto | - | - | - | - | - | - | −1028.54001 | −1028.62368 | 2.2767 | 544.58 | 0.3142 | 8.76 |
| L0101A enol | −1028.64502 | −1028.51140 | 3.6360 | 341.00 | 0.7481 | 7.51 | −1028.51463 | −1028.63366 | 3.2389 | 382.80 | 1.0091 | 10.28 |
| L0101A keto | - | - | - | - | - | - | −1028.53530 | −1028.61932 | 2.2863 | 542.30 | 0.0688 | 12.03 |
| Quercetin | ||||||||||||
| Q00001A enol | −1103.84650 | −1103.71263 | 3.6426 | 340.38 | 0.7296 | 2.0305 | −1103.71760 | −1103.83593 | 3.2198 | 385.07 | 1.0030 | 3.10 |
| Q00001A keto O3 | −1103.81910 | −1103.71084 | 2.9459 | 420.88 | 0.6532 | 5.7260 | −1103.71727 | −1103.81243 | 2.5892 | 478.85 | 0.7938 | 5.02 |
| Q00001A keto O5 | - | - | - | - | - | - | −1103.73235 | −1103.81821 | 2.3365 | 530.64 | 0.4704 | 5.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deriabina, A.; Prutskij, T.; Morales Ochoa, H.D.; Delgado Curiel, E.; Palacios Corte, V. Comparative Study of Fluorescence Emission of Fisetin, Luteolin and Quercetin Powders and Solutions: Further Evidence of the ESIPT Process. Biosensors 2024, 14, 413. https://doi.org/10.3390/bios14090413
Deriabina A, Prutskij T, Morales Ochoa HD, Delgado Curiel E, Palacios Corte V. Comparative Study of Fluorescence Emission of Fisetin, Luteolin and Quercetin Powders and Solutions: Further Evidence of the ESIPT Process. Biosensors. 2024; 14(9):413. https://doi.org/10.3390/bios14090413
Chicago/Turabian StyleDeriabina, Alexandra, Tatiana Prutskij, Hector Daniel Morales Ochoa, Esteban Delgado Curiel, and Veranda Palacios Corte. 2024. "Comparative Study of Fluorescence Emission of Fisetin, Luteolin and Quercetin Powders and Solutions: Further Evidence of the ESIPT Process" Biosensors 14, no. 9: 413. https://doi.org/10.3390/bios14090413
APA StyleDeriabina, A., Prutskij, T., Morales Ochoa, H. D., Delgado Curiel, E., & Palacios Corte, V. (2024). Comparative Study of Fluorescence Emission of Fisetin, Luteolin and Quercetin Powders and Solutions: Further Evidence of the ESIPT Process. Biosensors, 14(9), 413. https://doi.org/10.3390/bios14090413