Using Complementary Acoustic and Optical Techniques for Quantitative Monitoring of Biomolecular Adsorption at Interfaces



Abstract
:1. Introduction
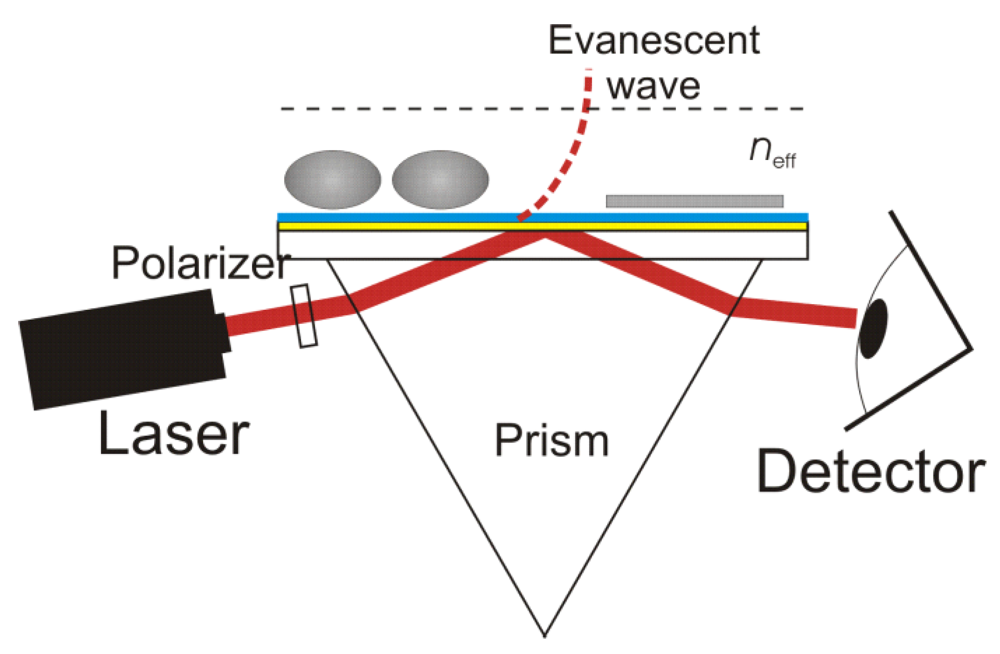
2. Surface Plasmon Resonance (SPR)


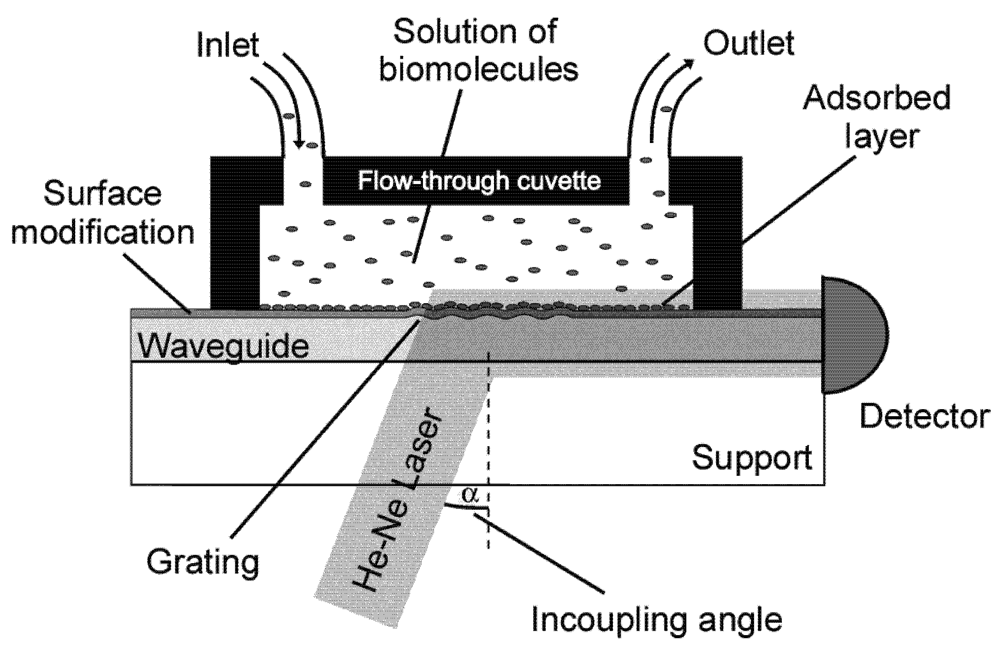
3. Optical Waveguide Lightmode Spectroscopy (OWLS)


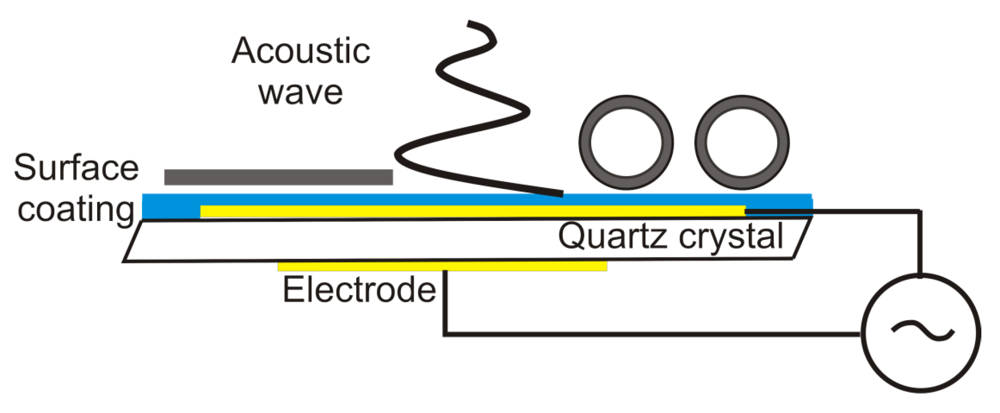
4. Quartz Crystal Microbalance (QCM)
 , where νq is the speed of shear waves in the crystal and tq is the thickness of the crystal. When the driving electric field oscillates at the fundamental frequency the mechanical amplitude of the crystal shear oscillation is increased more than hundred-fold. The width of the crystal resonance is very narrow, which gives an extremely well-defined resonant frequency and the ability to measure changes in the resonant frequency very precisely. Since the resonant frequency is determined by the total oscillating mass, which also includes all mass that is coupled to the surface, it can be used to measure the mass adsorbed on the surface in real time without need for any labels. A good approximation for the mass adsorbed to the sensor surfaces is the Sauerbrey relation [37]:
, where νq is the speed of shear waves in the crystal and tq is the thickness of the crystal. When the driving electric field oscillates at the fundamental frequency the mechanical amplitude of the crystal shear oscillation is increased more than hundred-fold. The width of the crystal resonance is very narrow, which gives an extremely well-defined resonant frequency and the ability to measure changes in the resonant frequency very precisely. Since the resonant frequency is determined by the total oscillating mass, which also includes all mass that is coupled to the surface, it can be used to measure the mass adsorbed on the surface in real time without need for any labels. A good approximation for the mass adsorbed to the sensor surfaces is the Sauerbrey relation [37]:


 , where η is the viscosity and ρ the density of the medium, i.e., typically water, and f the fundamental resonance frequency of the crystal [38]. For a 4.95 MHz crystal in water this yields δ ~ 250 nm for the fundamental resonance frequency, and as can be seen the extinction depth will decrease approximately as the inverse square root of the overtone number, n. Important to note is that the decay length is strongly dependent on the medium and an adsorbed film can significantly increase the decay length and therefore the probing depth of the acoustic sensor.
, where η is the viscosity and ρ the density of the medium, i.e., typically water, and f the fundamental resonance frequency of the crystal [38]. For a 4.95 MHz crystal in water this yields δ ~ 250 nm for the fundamental resonance frequency, and as can be seen the extinction depth will decrease approximately as the inverse square root of the overtone number, n. Important to note is that the decay length is strongly dependent on the medium and an adsorbed film can significantly increase the decay length and therefore the probing depth of the acoustic sensor. Hz−1 for a typical 4.95 MHz sensor crystal.
Hz−1 for a typical 4.95 MHz sensor crystal. 5. Comparing Acoustic and Optical Evanescent Techniques

6. An Overview of the Application of Complementary Sensor Techniques
7. Case Studies on Using Complementary Data Sets Obtained by Evanescent Optical and Acoustic Sensing Techniques
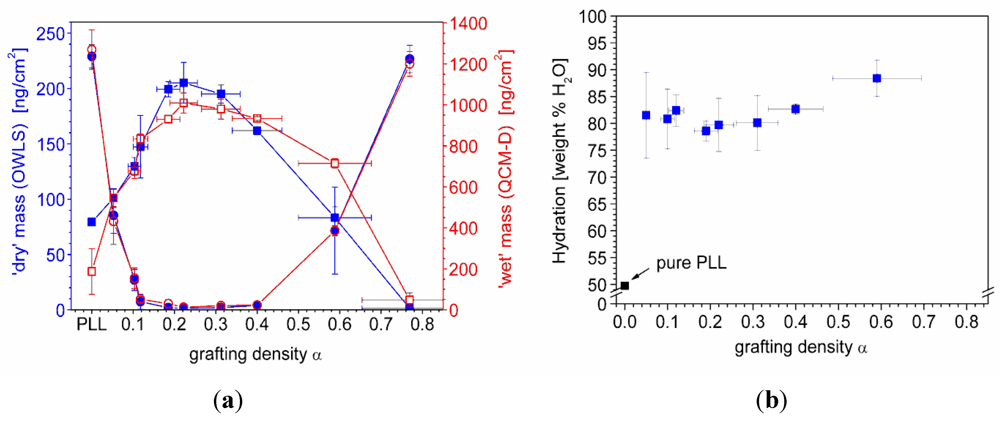
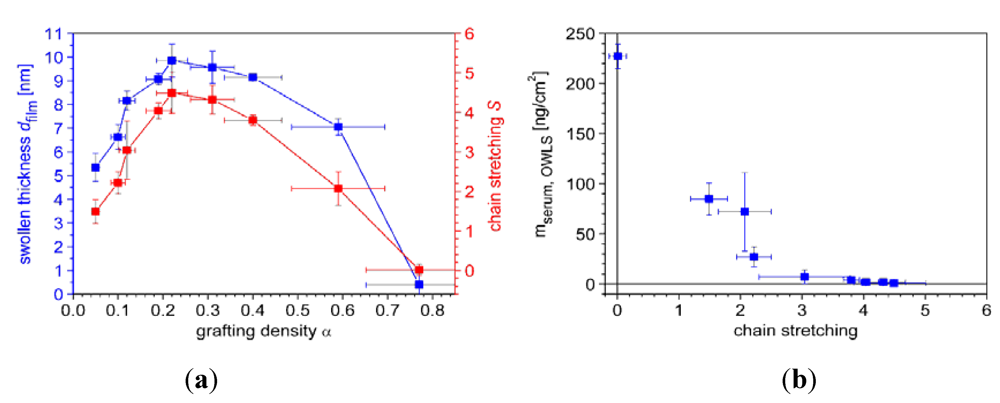
7.1. Case Study 1: Measuring Layer Thickness and Hydration by Complementary Evanescent Optical and Acoustic Techniques—A Case Study on PLL-g-PMOXA


 ) and serum proteins (
) and serum proteins (  ) as obtained from OWLS measurements (see Figure 5(a)) and ‘wet’ mass of adsorbed copolymer (
) as obtained from OWLS measurements (see Figure 5(a)) and ‘wet’ mass of adsorbed copolymer (  ) and serum proteins (
) and serum proteins (  ) as obtained from QCM-D measurements using Voigt modelling (see Figure 5(b)) for PLL-g-PMOXA graft copolymers of varying graft density. α = 0 corresponds to pure PLL. Copolymer and serum adsorbed masses were taken after rinsing with buffer after a stable value was reached; (b) Hydration in weight percent of water in the PLL-g-PMOXA adlayers calculated from the data in (a) according to Equation (4).
) and serum proteins ( ) as obtained from OWLS measurements (see Figure 5(a)) and ‘wet’ mass of adsorbed copolymer ( ) and serum proteins ( ) as obtained from QCM-D measurements using Voigt modelling (see Figure 5(b)) for PLL-g-PMOXA graft copolymers of varying graft density. α = 0 corresponds to pure PLL. Copolymer and serum adsorbed masses were taken after rinsing with buffer after a stable value was reached; (b) Hydration in weight percent of water in the PLL-g-PMOXA adlayers calculated from the data in (a) according to Equation (4).
) as obtained from QCM-D measurements using Voigt modelling (see Figure 5(b)) for PLL-g-PMOXA graft copolymers of varying graft density. α = 0 corresponds to pure PLL. Copolymer and serum adsorbed masses were taken after rinsing with buffer after a stable value was reached; (b) Hydration in weight percent of water in the PLL-g-PMOXA adlayers calculated from the data in (a) according to Equation (4).
) and serum proteins ( ) as obtained from OWLS measurements (see Figure 5(a)) and ‘wet’ mass of adsorbed copolymer ( ) and serum proteins ( ) as obtained from QCM-D measurements using Voigt modelling (see Figure 5(b)) for PLL-g-PMOXA graft copolymers of varying graft density. α = 0 corresponds to pure PLL. Copolymer and serum adsorbed masses were taken after rinsing with buffer after a stable value was reached; (b) Hydration in weight percent of water in the PLL-g-PMOXA adlayers calculated from the data in (a) according to Equation (4).








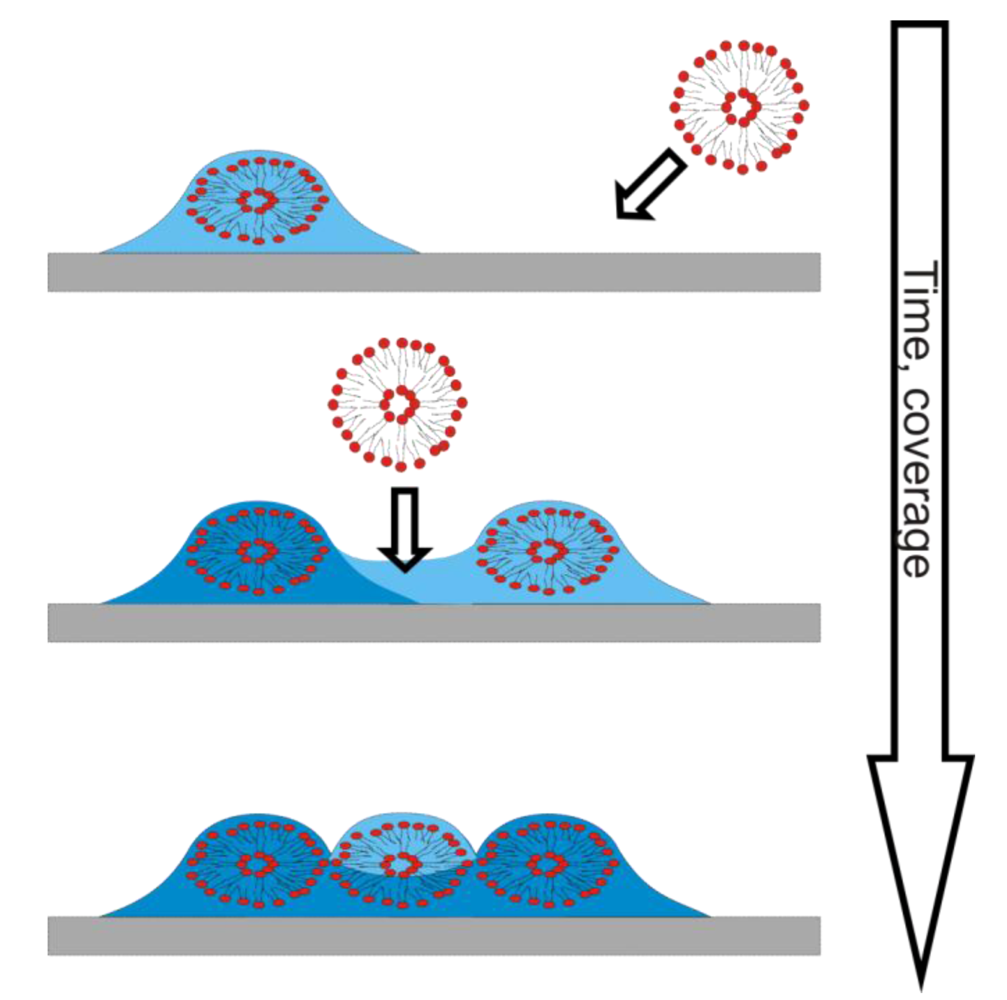
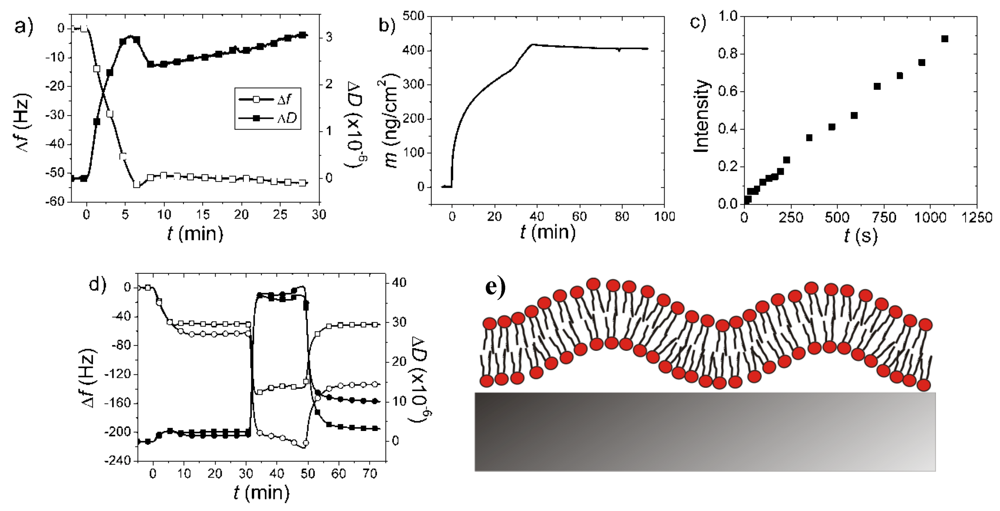
7.2. Case Study 2: How to Reveal Adsorption Kinetics of Biomolecular Systems Undergoing Structural Transformation—A Case Study on SLB Formation


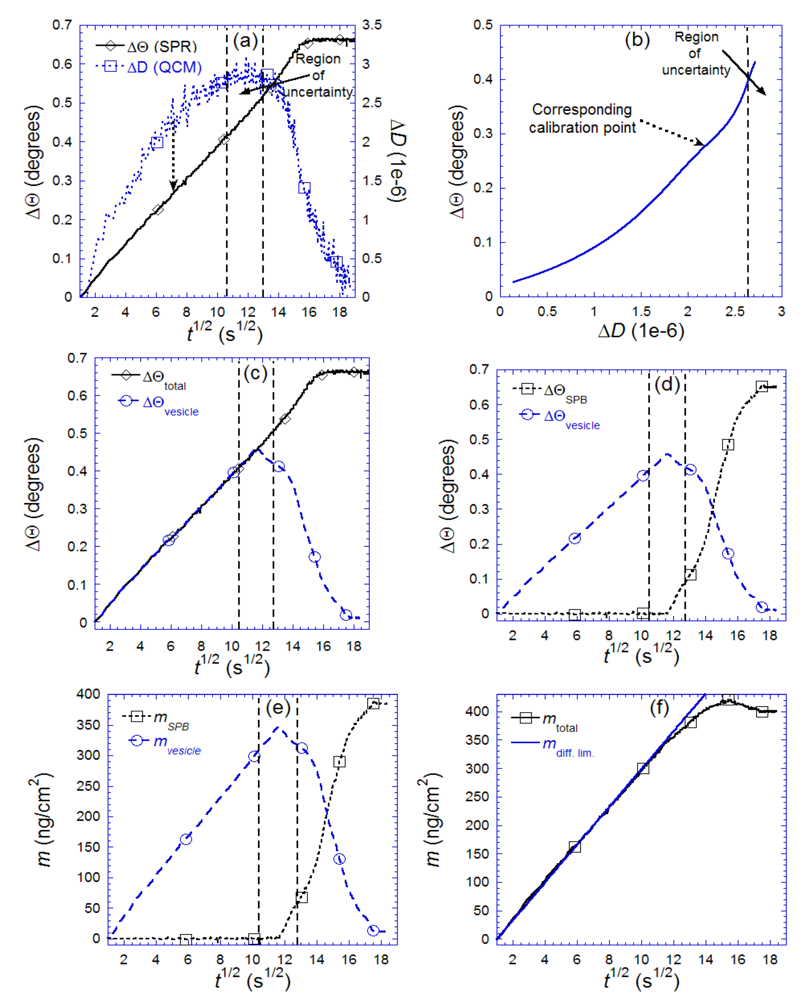
7.3. Case Study 3: Real-Time Modeling of Biomolecular Film Properties—A Case Study on SLB Self-Assembly and Protein Adsorption
 for (a) vesicle to bilayer formation; (b) streptavidin binding and 2D-crystallization on top of a biotinylated lipid bilayer. Shown is also the difference between the two measured masses, attributed to dynamically coupled water (mwater). The masses are calculated with the iterative method described in the text. Also shown in both plots are the expected adsorption rates for mass-transport limited adsorption (mdiff. lim.). Adapted with permission from Reimhult et al. [14]. Anal. Chem. 2004, 76, 7211–7220. Copyright 2004 American Chemical Society.
for (a) vesicle to bilayer formation; (b) streptavidin binding and 2D-crystallization on top of a biotinylated lipid bilayer. Shown is also the difference between the two measured masses, attributed to dynamically coupled water (mwater). The masses are calculated with the iterative method described in the text. Also shown in both plots are the expected adsorption rates for mass-transport limited adsorption (mdiff. lim.). Adapted with permission from Reimhult et al. [14]. Anal. Chem. 2004, 76, 7211–7220. Copyright 2004 American Chemical Society.
for (a) vesicle to bilayer formation; (b) streptavidin binding and 2D-crystallization on top of a biotinylated lipid bilayer. Shown is also the difference between the two measured masses, attributed to dynamically coupled water (mwater). The masses are calculated with the iterative method described in the text. Also shown in both plots are the expected adsorption rates for mass-transport limited adsorption (mdiff. lim.). Adapted with permission from Reimhult et al. [14]. Anal. Chem. 2004, 76, 7211–7220. Copyright 2004 American Chemical Society.
for (a) vesicle to bilayer formation; (b) streptavidin binding and 2D-crystallization on top of a biotinylated lipid bilayer. Shown is also the difference between the two measured masses, attributed to dynamically coupled water (mwater). The masses are calculated with the iterative method described in the text. Also shown in both plots are the expected adsorption rates for mass-transport limited adsorption (mdiff. lim.). Adapted with permission from Reimhult et al. [14]. Anal. Chem. 2004, 76, 7211–7220. Copyright 2004 American Chemical Society.
7.4. Case Study 4: The Dangers of Jumping to Conclusions Using Single Technique Kinetic Measurements—A Case Study on Bacterial Membrane Mimics

7.5. Case Study 5: Analysis of Molecular Ordering and the Influence of Optically Anisotropic Films—A Case Study on Birefringence Analysis of SLBs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| POPC SLB | POPC SLB (Ca2+) | POPC:POPS SLB (Ca2+) | DOPC SLB | DOPC SLB (Ca2+) | DOPC:DOPS SLB (Ca2+) | |
|---|---|---|---|---|---|---|
| n | 1.4788 (2.8e–3) | 1.4782 (1.2e–3) | 1.4711 (1.4e–3) | 1.456 (2.5e–3) | 1.4693 (4.2e–3) | 1.4904 (3.6e–3) |
| Birefringence | 0.02164 (5.3e–4) | 0.01960 (3.5e–4) | 0.01955 (7.1e–4) | 0.01586 (7.4e–4) | 0.0139 (2.1e–3) | 0.0250 (1.9e–3) |
| Thickness (nm) | 4.976 (9.9e–2) | 4.962 (4.3e–2) | 4.689 (5.5e–2) | 3.992 (8.4e–2) | 4.46 (1.4e–1) | 5.2855 (5.1e–3) |
8. Implications of Case Studies for Sensitivity and Single-Technique Kinetics Measurements
| Biofilm | SPR | QCM |
|---|---|---|
| Liposomes | 1,800 | 4,200 |
| SLB | 500 | 630 |
| Streptavidin | 240 | 590 |
| Single 30-mer DNA-strand | 42 | 380 |
| fc-hybridization of 30-mer DNA-strand | 22 | 330 |
-dependence under static conditions and a linear dependence under flow conditions [107], while with one rate-limiting step, kinetically limited adsorption yields a single-exponential time dependence [108]. However, when the type of adsorption is known, this information can be used as an Ansatz to understand the details of the adsorption process by looking at the deviation from the expected behavior in the QCM-D data.9. Summary
Acknowledgments
References
- Liedberg, B.; Nylander, C.; Lundström, I. Surface plasmon resonance for gas detection and biosensing. Sens. Actuat. 1983, 4, 299–304. [Google Scholar] [CrossRef]
- Pockrand, I.; Swalen, J.D.; Gordon, J.G.; Philpott, M.R. Surface plasmon spectroscopy of organic monolayer assemblies. Surf. Sci. 1978, 74, 237–244. [Google Scholar] [CrossRef]
- Swalen, J.D.; Gordon, J.G.; Philpott, M.R.; Brillante, A.; Pockrand, I.; Santo, R. Plasmon and polariton dispersion by direct optical observation. Am. J. Phys. 1980, 48, 669–672. [Google Scholar]
- Löfås, S.; Malmqvist, M.; Rönnberg, I.; Stenberg, E.; Liedberg, B.; Lundström, I. Bioanalysis with surface plasmon resonance. Sens. Actuat. B 1991, 5, 79–84. [Google Scholar] [CrossRef]
- Liedberg, B.; Nylander, C.; Lundström, I. Biosensing with surface plasmon resonance—How it all started. Biosens. Bioelectron. 1995, 10, i–ix. [Google Scholar]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors: Review. Sens. Actuat. B 1999, 54, 3–15. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the 1999 surface plasmon resonance biosensor literature. J. Mol. Recognit. 2000, 13, 388–407. [Google Scholar]
- Jung, L.S.; Campbell, C.T.; Chinowsky, T.M.; Mar, M.N.; Yee, S.S. Quantitative interpretation of the response of surface plasmon resonance sensors to adsorbed films. Langmuir 1998, 14, 5636–5648. [Google Scholar] [CrossRef]
- Kretschmann, E. Die Bestimmung optischer Konstanten von Metallen durch Anregung von Oberflächenplasmaschwingungen. Z. Phys. 1971, 241, 313–324. [Google Scholar]
- Liedberg, B.; Lundström, I.; Stenberg, E. Principles of biosensing with an extended coupling matrix and surface plasmon resonance. Sens. Actuat. B 1993, 11, 63–72. [Google Scholar]
- Biacore Life Sciences. Available online: http://www.biacore.com (accessed on 31 July 2012).
- Bailey, L.E.; Kambhampati, D.; Kanazawa, K.K.; Knoll, W.; Frank, C.W. Using surface plasmon resonance and the quartz crystal microbalance to monitor in situ the interfacial behavior of thin organic films. Langmuir 2002, 18, 479–489. [Google Scholar] [CrossRef]
- Baumgart, T.; Kreiter, M.; Lauer, H.; Naumann, R.; Jung, G.; Jonczyk, A.; Offenhausser, A.; Knoll, W. Fusion of small unilamellar vesicles onto laterally mixed self-assembled monolayers of thiolipopeptides. J. Colloid Interf. Sci. 2003, 258, 298–309. [Google Scholar]
- Reimhult, E.; Larsson, C.; Kasemo, B.; Hook, F. Simultaneous surface plasmon resonance and quartz crystal microbalance with dissipation monitoring measurements of biomolecular adsorption events involving structural transformations and variations in coupled water. Anal. Chem. 2004, 76, 7211–7220. [Google Scholar] [CrossRef]
- Reimhult, E.; Zach, M.; Hook, F.; Kasemo, B. A multitechnique study of liposome adsorption on Au and lipid bilayer formation on SiO2. Langmuir 2006, 22, 3313–3319. [Google Scholar]
- De Feijter, J.A.; Benjamins, J.; Veer, F.A. Ellipsometry as a tool to study the adsorption behavior of synthetic and biopolymers at the air-water interface. Biopolymers 1978, 17, 1759–1772. [Google Scholar]
- Ekgasit, S.; Yu, F.; Knoll, W. Displacement of molecules near a metal surface as seen by an SPR-SPFS biosensor. Langmuir 2005, 21, 4077–4082. [Google Scholar]
- Yu, F.; Persson, B.; Lofas, S.; Knoll, W. Attomolar sensitivity in bioassays based on surface plasmon fluorescence spectroscopy. J. Am. Chem. Soc. 2004, 126, 8902–8903. [Google Scholar]
- Neumann, T.; Johansson, M.L.; Kambhampati, D.; Knoll, W. Surface-plasmon fluorescence spectroscopy. Adv. Funct. Mater. 2002, 12, 575–586. [Google Scholar]
- Liebermann, T.; Knoll, W. Surface-plasmon field-enhanced fluorescence spectroscopy. Colloids Surf.-Physicochem. Eng. Aspects 2000, 171, 115–130. [Google Scholar] [CrossRef]
- Kurrat, R.; Textor, M.; Ramsden, J.J.; Boni, P.; Spencer, N.D. Instrumental improvements in optical waveguide light mode spectroscopy for the study of biomolecule adsorption. Rev. Sci. Instrum. 1997, 68, 2172–2176. [Google Scholar]
- Lukosz, W.; Tiefenthaler, K. Embossing technique for fabricating integrated optical components in hard inorganic waveguiding materials. Opt. Lett. 1983, 8, 537–539. [Google Scholar]
- Yoldas, B.E. Deposition and properties of optical oxide coatings from polymerized solutions. Appl. Opt. 1982, 21, 2960–2964. [Google Scholar]
- Ball, V.; Ramsden, J.J. Buffer dependence of refractive index increments of protein solutions. Biopolymers 1998, 46, 489–492. [Google Scholar]
- Voros, J.; Ramsden, J.J.; Csucs, G.; Szendro, I.; de Paul, S.M.; Textor, M.; Spencer, N.D. Optical grating coupler biosensors. Biomaterials 2002, 23, 3699–3710. [Google Scholar] [CrossRef]
- Rodenhausen, K.B.; Schubert, M. Virtual separation approach to study porous ultra-thin films by combined spectroscopic ellipsometry and quartz crystal microbalance methods. Thin Solid Films 2011, 519, 2772–2776. [Google Scholar]
- Zhylyak, G.; Ramoz-Perez, V.; Michael, L.; Hug, T.; Citterio, D.; Spichiger-Keller, U.E. Planar integrated optical waveguide used as a transducer to yield chemical information: Detection of the activity of proteolytic enzymes e.g., serine-proteases. Opt. Lasers Eng. 2005, 43, 603–617. [Google Scholar] [CrossRef]
- Halter, M.; Gabi, M.; Textor, M.; Voros, J.; Grandin, H.M. Enhanced optical waveguide light mode spectroscopy via detection of fluorophore absorbance. Rev. Sci. Instrum. 2006, 77. [Google Scholar]
- Duveneck, G.L.; Abel, A.P.; Bopp, M.A.; Kresbach, G.M.; Ehrat, M. Planar waveguides for ultra-high sensitivity of the analysis of nucleic acids. Anal. Chim. Acta 2002, 469, 49–61. [Google Scholar]
- Duveneck, G.L.; Bopp, M.A.; Ehrat, M.; Balet, L.P.; Haiml, M.; Keller, U.; Marowsky, G.; Soria, S. Two-photon fluorescence excitation of macroscopic areas on planar waveguides. Biosens. Bioelectron. 2003, 18, 503–510. [Google Scholar]
- Grandin, H.M.; Staedler, B.; Textor, M.; Voeroes, J. Waveguide excitation fluorescence microscopy: A new tool for sensing and imaging the biointerface. Biosens. Bioelectron. 2006, 21, 1476–1482. [Google Scholar] [CrossRef]
- Bally, M.; Halter, M.; Voros, J.; Grandin, H.M. Optical microarray biosensing techniques. Surf. Interface Anal. 2006, 38, 1442–1458. [Google Scholar]
- Picart, C.; Gergely, C.; Arntz, Y.; Voegel, J.-C.; Schaaf, P.; Cuisinier, F.J.G.; Senger, B. Measurement of film thickness up to several hundreds of nanometers using optical waveguide lightmode spectroscopy. Biosens. Bioelectron. 2004, 20, 553–561. [Google Scholar]
- Kozma, P.; Hamori, A.; Kurunczi, S.; Cottier, K.; Horvath, R. Grating coupled optical waveguide interferometer for label-free biosensing. Sens. Actuat. B 2011, 155, 446–450. [Google Scholar]
- Bearinger, J.P.; Voros, J.; Hubbell, J.A.; Textor, M. Electrochemical optical waveguide lightmode spectroscopy (EC-OWLS): A pilot study using evanescent-field optical sensing under voltage control to monitor polycationic polymer adsorption onto indium tin oxide (ITO)-coated waveguide chips. Biotechnol. Bioeng. 2003, 82, 465–473. [Google Scholar]
- Grieshaber, D.; MacKenzie, R.; Voros, J.; Reimhult, E. Electrochemical biosensors—Sensor principles and architectures. Sensors 2008, 8, 1400–1458. [Google Scholar]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dunner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. [Google Scholar]
- Rodahl, M.; Kasemo, B. On the measurement of thin liquid overlayers with the quartz-crystal microbalance. Sens. Actuat. A 1996, 54, 448–456. [Google Scholar]
- Johannsmann, D.; Mathauer, K.; Wegner, G.; Knoll, W. Viscoelastic properties of thin-films probed with a quartz-crystal resonator. Phys. Rev. B 1992, 46, 7808–7815. [Google Scholar]
- Rodahl, M.; Höök, F.; Krozer, A.; Brzezinski, P.; Kasemo, B. Quartz crystal microbalance setup for frequency and Q-factor measurements in gaseous and liquid environments. Rev. Sci. Instrum. 1995, 66, 3924–3930. [Google Scholar]
- Rodahl, M.; Höök, F.; Fredriksson, C.; Keller, C.A.; Krozer, A.; Brzezinski, P.; Voinova, M.; Kasemo, B. Simultaneous frequency and dissipation factor QCM measurements of biomolecular adsorption and cell adhesion. Faraday Discuss. 1997, 107, 229–246. [Google Scholar]
- Rodahl, M.; Kasemo, B. A simple setup to simultaneously measure the resonant frequency and the absolute dissipation factor of a quartz crystal microbalance. Rev. Sci. Instrum. 1996, 67, 3238–3241. [Google Scholar]
- Voinova, M.V.; Rodahl, M.; Jonson, M.; Kasemo, B. Viscoelastic acoustic response of layered polymer films at fluid-solid interfaces: Continuum mechanics approach. Phys. Scr. 1999, 59, 391–396. [Google Scholar]
- Hook, F.; Kasemo, B.; Nylander, T.; Fant, C.; Sott, K.; Elwing, H. Variations in coupled water, viscoelastic properties, and film thickness of a Mefp-1 protein film during adsorption and cross-linking: A quartz crystal microbalance with dissipation monitoring, ellipsometry, and surface plasmon resonance study. Anal. Chem. 2001, 73, 5796–5804. [Google Scholar] [CrossRef]
- Reviakine, I.; Johannsmann, D.; Richter, R.P. Hearing what you cannot see and visualizing what you hear: Interpreting quartz crystal microbalance data from solvated interfaces. Anal. Chem. 2011, 83, 8838–8848. [Google Scholar]
- Johannsmann, D.; Reviakine, I.; Richter, R.P. Dissipation in films of adsorbed nanospheres studied by Quartz Crystal Microbalance (QCM). Anal. Chem. 2009, 81, 8167–8176. [Google Scholar]
- Friedt, J.M.; Francis, L.; Reekmans, G.; de Palma, R.; Campitelli, A.; Sleytr, U.B. Simultaneous surface acoustic wave and surface plasmon resonance measurements: Electrodeposition and biological interactions monitoring. J. Appl. Phys. 2004, 95, 1677–1680. [Google Scholar]
- Zhou, C.; Friedt, J.-M.; Angelova, A.; Choi, K.-H.; Laureyn, W.; Frederix, F.; Francis, L.A.; Campitelli, A.; Engelborghs, Y.; Borghs, G. Human immunoglobulin adsorption investigated by means of quartz crystal microbalance dissipation, atomic force microscopy, surface acoustic wave, and surface plasmon resonance techniques. Langmuir 2004, 20, 5870–5878. [Google Scholar]
- Laschitsch, A.; Menges, B.; Johannsmann, D. Simultaneous determination of optical and acoustic thicknesses of protein layers using surface plasmon resonance spectroscopy and quartz crystal microweighing. Appl. Phys. Lett. 2000, 77, 2252–2254. [Google Scholar]
- Plunkett, M.A.; Wang, Z.; Rutland, M.W.; Johannsmann, D. Adsorption of pNIPAM layers on hydrophobic gold surfaces, measured in situ by QCM and SPR. Langmuir 2003, 19, 6837–6844. [Google Scholar] [CrossRef]
- Hook, F.; Voros, J.; Rodahl, M.; Kurrat, R.; Boni, P.; Ramsden, J.J.; Textor, M.; Spencer, N.D.; Tengvall, P.; Gold, J.; Kasemo, B. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids Surf. B: Biointerfaces 2002, 24, 155–170. [Google Scholar] [CrossRef]
- Larsson, C.; Rodahl, M.; Hook, F. Characterization of DNA immobilization and subsequent hybridization on a 2D arrangement of streptavidin on a biotin-modified lipid bilayer supported on SiO2. Anal. Chem. 2003, 75, 5080–5087. [Google Scholar]
- Stengel, G.; Hook, F.; Knoll, W. Viscoelastic modeling of template-directed DNA synthesis. Anal. Chem. 2005, 77, 3709–3714. [Google Scholar]
- Voeroes, J. The density and refractive index of adsorbing protein layers. Biophys. J. 2004, 87, 553–561. [Google Scholar]
- Rechendorff, K.; Hovgaard, M.B.; Foss, M.; Zhdanov, V.P.; Besenbacher, F. Enhancement of protein adsorption induced by surface roughness. Langmuir 2006, 22, 10885–10888. [Google Scholar]
- Evans-Nguyen, K.M.; Fuierer, R.R.; Fitchett, B.D.; Tolles, L.R.; Conboy, J.C.; Schoenfisch, M.H. Changes in adsorbed fibrinogen upon conversion to fibrin. Langmuir 2006, 22, 5115–5121. [Google Scholar]
- Malmstroem, J.; Agheli, H.; Kingshott, P.; Sutherland, D.S. Viscoelastic modeling of highly hydrated laminin layers at homogeneous and nanostructured surfaces: Quantification of protein layer properties using QCM-D and SPR. Langmuir 2007, 23, 9760–9768. [Google Scholar]
- Stevens, M.M.; Allen, S.; Sakata, J.K.; Davies, M.C.; Roberts, C.J.; Tendler, S.J.B.; Tirrell, D.A.; Williams, P.M. pH-Dependent behavior of surface-immobilized artificial leucine zipper proteins. Langmuir 2004, 20, 7747–7752. [Google Scholar]
- Su, X.; Wu, Y.-J.; Knoll, W. Comparison of surface plasmon resonance spectroscopy and quartz crystal microbalance techniques for studying DNA assembly and hybridization. Biosens. Bioelectron. 2005, 21, 719–726. [Google Scholar]
- Su, X.; Wu, Y.-J.; Robelek, R.; Knoll, W. Surface plasmon resonance spectroscopy and quartz crystal microbalance study of streptavidin film structure effects on biotinylated DNA assembly and target DNA hybridization. Langmuir 2005, 21, 348–353. [Google Scholar]
- Peh, W.Y.X.; Reimhult, E.; Teh, H.F.; Thomsen, J.S.; Su, X.D. Understanding ligand binding effects on the conformation of estrogen receptor alpha-DNA complexes: A combinational quartz crystal microbalance with dissipation and surface plasmon resonance study. Biophys. J. 2007, 92, 4415–4423. [Google Scholar]
- Richter, R.P.; Brisson, A.R. Following the formation of supported lipid bilayers on mica: A study combining AFM, QCM-D, and ellipsometry. Biophys. J. 2005, 88, 3422–3433. [Google Scholar] [CrossRef]
- Stalgren, J.J.R.; Eriksson, J.; Boschkova, K. A comparative study of surfactant adsorption on model surfaces using the quartz crystal microbalance and the ellipsometer. J. Colloid Interface Sci. 2002, 253, 190–195. [Google Scholar] [CrossRef]
- Benetti, E.M.; Reimhult, E.; de Bruin, J.; Zapotoczny, S.; Textor, M.; Vancso, G.J. Poly(methacrylic acid) grafts grown from designer surfaces: The effect of initiator coverage on polymerization kinetics, morphology, and properties. Macromolecules 2009, 42, 1640–1647. [Google Scholar] [CrossRef]
- Picart, C.; Lavalle, P.; Hubert, P.; Cuisinier, F.J.G.; Decher, G.; Schaaf, P.; Voegel, J.C. Buildup mechanism for poly(L-lysine)/hyaluronic acid films onto a solid surface. Langmuir 2001, 17, 7414–7424. [Google Scholar]
- Picart, C.; Mutterer, J.; Richert, L.; Luo, Y.; Prestwich, G.D.; Schaaf, P.; Voegel, J.C.; Lavalle, P. Molecular basis for the explanation of the exponential growth of polyelectrolyte multilayers. Proc. Natl. Acad. Sci. USA 2002, 99, 12531–12535. [Google Scholar]
- Halthur, T.J.; Elofsson, U.M. Multilayers of charged polypeptides as studied by in situ ellipsometry and quartz crystal microbalance with dissipation. Langmuir 2004, 20, 1739–1745. [Google Scholar] [CrossRef]
- Halthur, T.J.; Claesson, P.M.; Elofsson, U.M. Immobilization of enamel matrix derivate protein onto polypeptide multilayers: Comparative in situ measurements using ellipsometry, quartz crystal microbalance with dissipation, and dual-polarization interferometry. Langmuir 2006, 22, 11065–11071. [Google Scholar] [CrossRef]
- Ramos, J.J.I.; Stahl, S.; Richter, R.P.; Moya, S.E. Water content and buildup of poly(diallyldimethylammonium chloride)/poly(sodium 4-styrenesulfonate) and poly(allylamine hydrochloride)/poly(sodium 4-styrenesulfonate) polyelectrolyte multilayers studied by an in situ combination of a quartz crystal microbalance with dissipation monitoring and spectroscopic ellipsometry. Macromolecules 2010, 43, 9063–9070. [Google Scholar] [CrossRef]
- Bittrich, E.; Rodenhausen, K.B.; Eichhorn, K.J.; Hofmann, T.; Schubert, M.; Stamm, M.; Uhlmann, P. Protein adsorption on and swelling of polyelectrolyte brushes: A simultaneous ellipsometry-quartz crystal microbalance study. Biointerphases 2010, 5, 1–9. [Google Scholar]
- Lord, M.S.; Stenzel, M.H.; Simmons, A.; Milthorpe, B.K. Lysozyme interaction with poly(HEMA)-based hydrogel. Biomaterials 2006, 27, 1341–1345. [Google Scholar]
- Rodenhausen, K.B.; Kasputis, T.; Pannier, A.K.; Gerasimov, J.Y.; Lai, R.Y.; Solinsky, M.; Tiwald, T.E.; Wang, H.; Sarkar, A.; Hofmann, T.; et al. Combined optical and acoustical method for determination of thickness and porosity of transparent organic layers below the ultra-thin film limit. Rev. Sci. Instrum. 2011, 82, 103111:1–103111:10. [Google Scholar]
- Wang, G.; Rodahl, M.; Edvardsson, M.; Svedhem, S.; Ohlsson, G.; Hook, F.; Kasemo, B. A combined reflectometry and quartz crystal microbalance with dissipation setup for surface interaction studies. Rev. Sci. Instrum. 2008, 79, 075107:1–075107:7. [Google Scholar]
- Rodenhausen, K.B.; Guericke, M.; Sarkar, A.; Hofmann, T.; Ianno, N.; Schubert, M.; Tiwald, T.E.; Solinsky, M.; Wagner, M. Micelle-assisted bilayer formation of cetyltrimethylammonium bromide thin films studied with combinatorial spectroscopic ellipsometry and quartz crystal microbalance techniques. Thin Solid Films 2011, 519, 2821–2824. [Google Scholar]
- Rodenhausen, K.B.; Duensing, B.A.; Kasputis, T.; Pannier, A.K.; Hofmann, T.; Schubert, M.; Tiwald, T.E.; Solinsky, M.; Wagner, M. In situ monitoring of alkanethiol self-assembled monolayer chemisorption with combined spectroscopic ellipsometry and quartz crystal microbalance techniques. Thin Solid Films 2011, 519, 2817–2820. [Google Scholar] [CrossRef]
- Edvardsson, M.; Svedhem, S.; Wang, G.; Richter, R.; Rodahl, M.; Kasemo, B. QCM-D and reflectometry instrument: Applications to supported lipid structures and their biomolecular interactions. Anal. Chem. 2009, 81, 349–361. [Google Scholar]
- Kunze, A.; Svedhem, S.; Kasemo, B. Lipid transfer between charged supported lipid bilayers and oppositely charged vesicles. Langmuir 2009, 25, 5146–5158. [Google Scholar]
- Kunze, A.; Zhao, F.; Marel, A.K.; Svedhem, S.; Kasemo, B. Ion-mediated changes of supported lipid bilayers and their coupling to the substrate. A case of bilayer slip? Soft Matter 2011, 7, 8582–8591. [Google Scholar]
- Kenausis, G.L.; Voeroes, J.; Elbert, D.L.; Huang, N.; Hofer, R.; Ruiz-Taylor, L.; Textor, M.; Hubbell, J.A.; Spencer, N.D. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: Attachment mechanism and effects of polymer architecture on resistance to protein adsorption. J. Phys. Chem. B 2000, 104, 3298–3309. [Google Scholar]
- Huang, N.-P.; Michel, R.; Voros, J.; Textor, M.; Hofer, R.; Rossi, A.; Elbert, D.L.; Hubbell, J.A.; Spencer, N.D. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: Surface-analytical characterization and resistance to serum and fibrinogen adsorption. Langmuir 2001, 17, 489–498. [Google Scholar]
- Pasche, S.; de Paul, S.M.; Voeroes, J.; Spencer, N.D.; Textor, M. Poly(L-lysine)-graft-poly(ethylene glycol) assembled monolayers on Niobium Oxide surfaces: A quantitative study of the influence of polymer interfacial architecture on resistance to protein adsorption by ToF-SIMS and in situ OWLSu OWLS. Langmuir 2003, 19, 9216–9225. [Google Scholar]
- Pasche, S.; Textor, M.; Meagher, L.; Spencer, N.D.; Griesser, H.J. Relationship between interfacial forces measured by colloid-probe atomic force microscopy and protein resistance of poly(ethylene glycol)-grafted poly(L-lysine) adlayers on niobia surfaces. Langmuir 2005, 21, 6508–6520. [Google Scholar]
- Konradi, R.; Pidhatika, B.; Muehlebach, A.; Textor, M. Poly-2-methyl-2-oxazoline: A peptide-like polymer for protein-repellent surfaces. Langmuir 2008, 24, 613–616. [Google Scholar]
- Feuz, L.; Leermakers, F.A.M.; Textor, M.; Borisov, O. Bending rigidity and induced persistence length of molecular bottle brushes: A self-consistent-field theory. Macromolecules 2005, 38, 8891–8901. [Google Scholar]
- Merrill, E.W. Distinctions and correspondences among surfaces contacting blood. Ann. N. Y. Acad. Sci. 1987, 516, 196–203. [Google Scholar]
- Chapman, R.G.; Ostuni, E.; Liang, M.N.; Meluleni, G.; Kim, E.; Yan, L.; Pier, G.; Warren, H.S.; Whitesides, G.M. Polymeric thin films that resist the adsorption of proteins and the adhesion of bacteria. Langmuir 2001, 17, 1225–1233. [Google Scholar]
- Keller, C.A.; Kasemo, B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 1998, 75, 1397–1402. [Google Scholar]
- Richter, R.P.; Berat, R.; Brisson, A.R. Formation of solid-supported lipid bilayers: An integrated view. Langmuir 2006, 22, 3497–3505. [Google Scholar]
- Rossetti, F.F.; Bally, M.; Michel, R.; Textor, M.; Reviakine, I. Interactions between titanium dioxide and phosphatidyl serine-containing liposomes: Formation and patterning of supported phospholipid bilayers on the surface of a medically relevant material. Langmuir 2005, 21, 6443–6450. [Google Scholar]
- Reimhult, E.; Hook, F.; Kasemo, B. Intact vesicle adsorption and supported biomembrane formation from vesicles in solution: Influence of surface chemistry, vesicle size, temperature, and osmotic pressure. Langmuir 2003, 19, 1681–1691. [Google Scholar] [CrossRef]
- Baumann, M.K.; Amstad, E.; Mashaghi, A.; Textor, M.; Reimhult, E. Characterization of supported lipid bilayers incorporating the phosphoinositides phosphatidylinositol 4,5-biphosphate and phosphoinositol-3,4,5-triphosphate by complementary techniques. Biointerphases 2010, 5, 114–119. [Google Scholar]
- Kaufmann, S.; Papastavrou, G.; Kumar, K.; Textor, M.; Reimhult, E. A detailed investigation of the formation kinetics and layer structure of poly(ethylene glycol) tether supported lipid bilayers. Soft Matter 2009, 5, 2804–2814. [Google Scholar]
- Salamon, Z.; Tollin, G. Optical anisotropy in lipid bilayer membranes: Coupled plasmon-waveguide resonance measurements of molecular orientation, polarizability, and shape. Biophys. J. 2001, 80, 1557–1567. [Google Scholar] [CrossRef]
- Keller, C.A.; Glasmästar, K.; Zhdanov, V.P.; Kasemo, B. Formation of supported membranes from vesicles. Phys. Rev. Lett. 2000, 84, 5443–5446. [Google Scholar]
- Mashaghi, A.; Swann, B.; Popplewell, J.; Textor, M.; Reimhult, E. Optical anisotropy of supported lipid structures probed by waveguide spectroscopy and its application to study of supported lipid bilayer formation kinetics. Anal. Chem. 2008, 80, 3666–3676. [Google Scholar]
- Cuypers, P.A.; Corsel, J.W.; Janssen, M.P.; Kop, J.M.M.; Hermens, W.T.; Hemker, H. Adsorption of prothrombin to phosphatidylserine by ellipsometry. J. Biol. Chem. 1983, 258, 2426–2431. [Google Scholar]
- Tellechea, E.; Johannsmann, D.; Steinmetz, N.F.; Richter, R.P.; Reviakine, I. Model-independent analysis of QCM data on colloidal particle adsorption. Langmuir 2009, 25, 5177–5184. [Google Scholar]
- Merz, C.; Knoll, W.; Textor, M.; Reimhult, E. Formation of supported bacterial lipid membrane mimics. Biointerphases 2008, 3, FA41–FA50. [Google Scholar]
- Jonsson, M.P.; Jonsson, P.; Dahlin, A.B.; Hook, F. Supported lipid bilayer formation and lipid-membrane-mediated biorecognition reactions studied with a new nanoplasmonic sensor template. Nano Lett. 2007, 7, 3462–3468. [Google Scholar]
- Cross, G.H.; Reeves, A.; Brand, S.; Swann, M.J.; Peel, L.L.; Freeman, N.J.; Lu, J.R. The metrics of surface adsorbed small molecules on the Young’s fringe dual-slab waveguide interferometer. J. Phys. D-Appl. Phys. 2004, 37, 74–80. [Google Scholar] [CrossRef]
- Cross, G.H.; Reeves, A.A.; Brand, S.; Popplewell, J.F.; Peel, L.L.; Swann, M.J.; Freeman, N.J. A new quantitative optical biosensor for protein characterisation. Biosens. Bioelectron. 2003, 19, 383–390. [Google Scholar]
- Cross, G.H.; Ren, Y.T.; Freeman, N.J. Young’s fringes from vertically integrated slab waveguides: Applications to humidity sensing. J. Appl. Phys. 1999, 86, 6483–6488. [Google Scholar] [CrossRef] [Green Version]
- Swann, M.J.; Peel, L.L.; Carrington, S.; Freeman, N.J. Dual-polarization interferometry: An analytical technique to measure changes in protein structure in real time, to determine the stoichiometry of binding events, and to differentiate between specific and nonspecific interactions. Anal. Biochem. 2004, 329, 190–198. [Google Scholar]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta-Rev. Biomembr. 2000, 1469, 159–195. [Google Scholar] [CrossRef]
- Biesalski, M.; Ruhe, J. Swelling of a polyelectrolyte brush in humid air. Langmuir 2000, 16, 1943–1950. [Google Scholar]
- Ruhe, J.; Ballauff, M.; Biesalski, M.; Dziezok, P.; Grohn, F.; Johannsmann, D.; Houbenov, N.; Hugenberg, N.; Konradi, R.; Minko, S.; et al. Polyelectrolyte brushes. In Polyelectrolytes with Defined Molecular Architecture I; Schmidt, M., Ed.; Springer-Verlag: Berlin, Germany, 2004; Volume 165, pp. 79–150. [Google Scholar]
- Wojciechowski, P.W. Interfacial Phenomena and Bioproducts; Brash, J.L., Wojciechowski, P.W., Eds.; CRC Press: New York, NY, USA, 1996; p. 209. [Google Scholar]
- Kreuzer, H.J.; Gortel, Z.W. Physisorption Kinetics; Springer Verlag: Berlin, Germany, 1986. [Google Scholar]
- Huang, C.J.; Dostalek, J.; Sessitsch, A.; Knoll, W. Long-range surface plasmon-enhanced fluorescence spectroscopy biosensor for ultrasensitive detection of E. coli O157:H7. Anal. Chem. 2011, 83, 674–677. [Google Scholar]
- Bally, M.; Gunnarsson, A.; Svensson, L.; Larson, G.; Zhdanov, V.P.; Hook, F. Interaction of single viruslike particles with vesicles containing glycosphingolipids. Phys. Rev. Lett. 2001, 107, 188103:1–188103:5. [Google Scholar]
- Dostalek, J.; Knoll, W. Biosensors based on surface plasmon-enhanced fluorescence spectroscopy. Biointerphases 2008, 3, FD12–FD22. [Google Scholar]
- Naumann, R.; Schiller, S.M.; Giess, F.; Grohe, B.; Hartman, K.B.; Karcher, I.; Koper, I.; Lubben, J.; Vasilev, K.; Knoll, W. Tethered lipid Bilayers on ultraflat gold surfaces. Langmuir 2003, 19, 5435–5443. [Google Scholar]
- Sugihara, K.; Delai, M.; Szendro, I.; Guillaume-Gentil, O.; Voros, J.; Zambelli, T. Simultaneous OWLS and EIS monitoring of supported lipid bilayers with the pore forming peptide melittin. Sens. Actuat. B 2012, 161, 600–606. [Google Scholar]
- Ferner-Ortner-Bleckmann, J.; Schrems, A.; Ilk, N.; Egelseer, E.M.; Sleytr, U.B.; Schuster, B. Multitechnique study on a recombinantly produced Bacillus halodurans laccase and an S-layer/laccase fusion protein. Biointerphases 2011, 6, 63–72. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Konradi, R.; Textor, M.; Reimhult, E. Using Complementary Acoustic and Optical Techniques for Quantitative Monitoring of Biomolecular Adsorption at Interfaces. Biosensors 2012, 2, 341-376. https://doi.org/10.3390/bios2040341
Konradi R, Textor M, Reimhult E. Using Complementary Acoustic and Optical Techniques for Quantitative Monitoring of Biomolecular Adsorption at Interfaces. Biosensors. 2012; 2(4):341-376. https://doi.org/10.3390/bios2040341
Chicago/Turabian StyleKonradi, Rupert, Marcus Textor, and Erik Reimhult. 2012. "Using Complementary Acoustic and Optical Techniques for Quantitative Monitoring of Biomolecular Adsorption at Interfaces" Biosensors 2, no. 4: 341-376. https://doi.org/10.3390/bios2040341
APA StyleKonradi, R., Textor, M., & Reimhult, E. (2012). Using Complementary Acoustic and Optical Techniques for Quantitative Monitoring of Biomolecular Adsorption at Interfaces. Biosensors, 2(4), 341-376. https://doi.org/10.3390/bios2040341