Microelectrode Arrays and the Use of PEG-Functionalized Diblock Copolymer Coatings

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Experimental Section
2.1. Procedure for Coating Arrays with Block Copolymer
2.2. Procedure for Conducting Cyclic Voltammetry Experiments on a 12-K Array
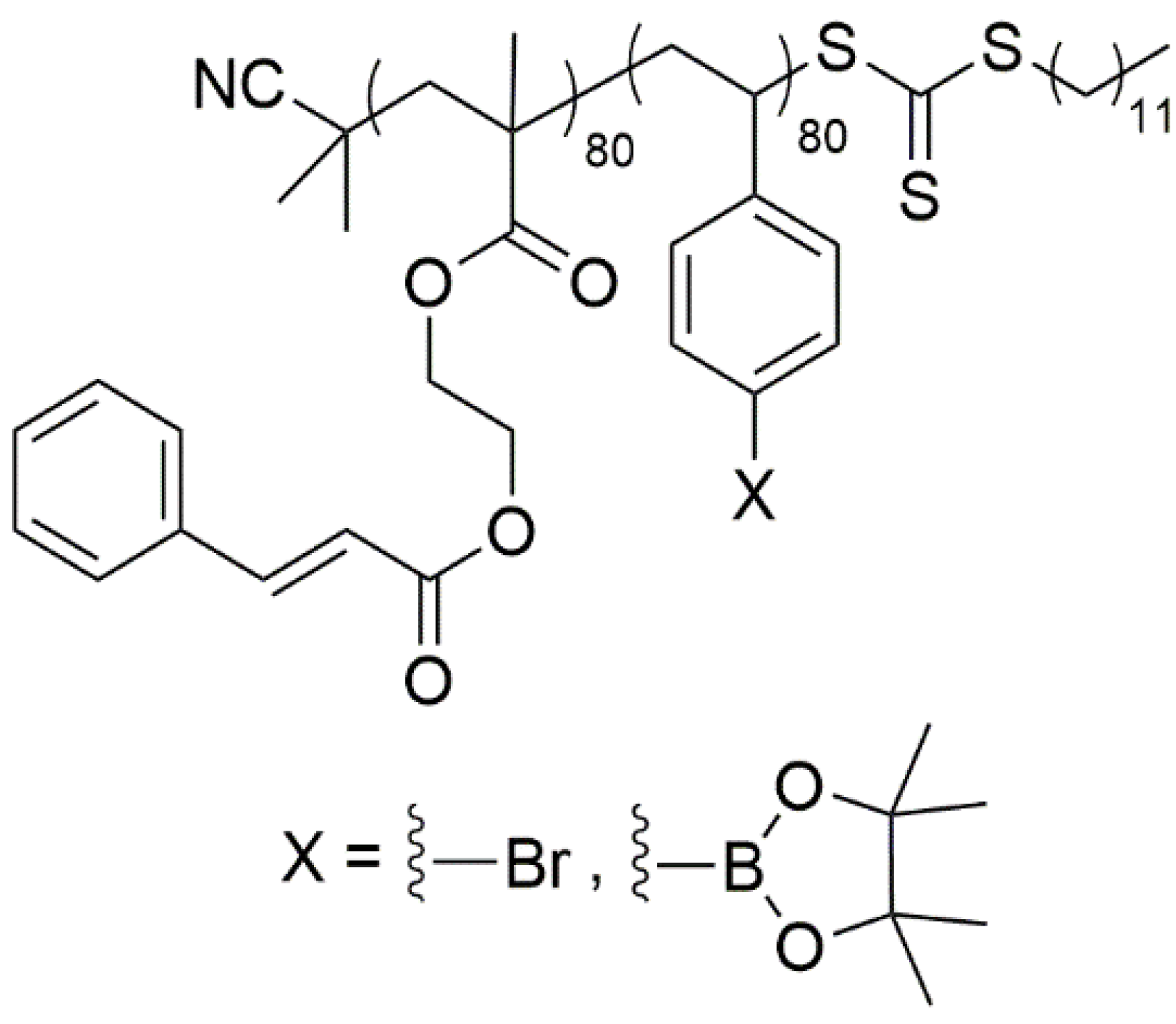
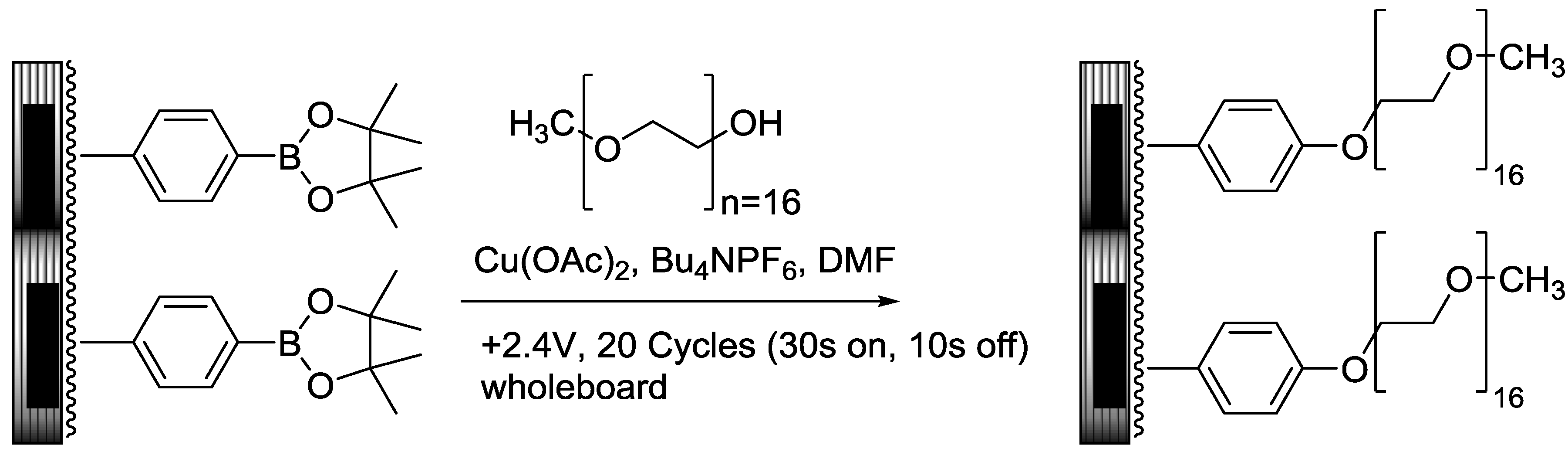
2.3. PEGylation of PCEMA-b-pBSt Polymer Using Chan-Lam Coupling on Arrays

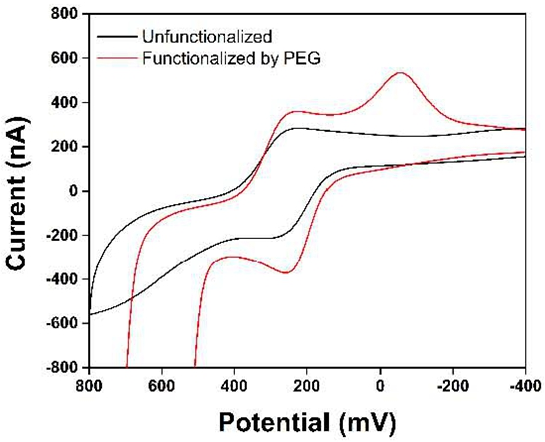
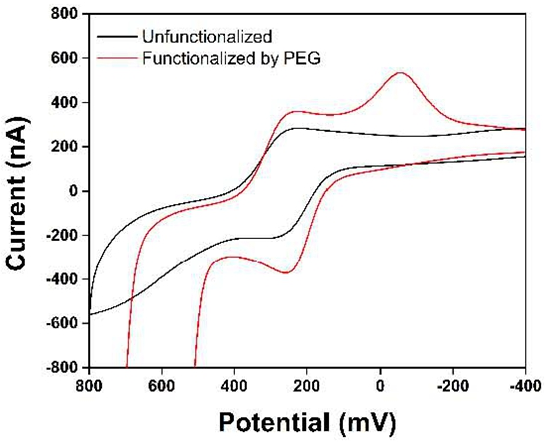
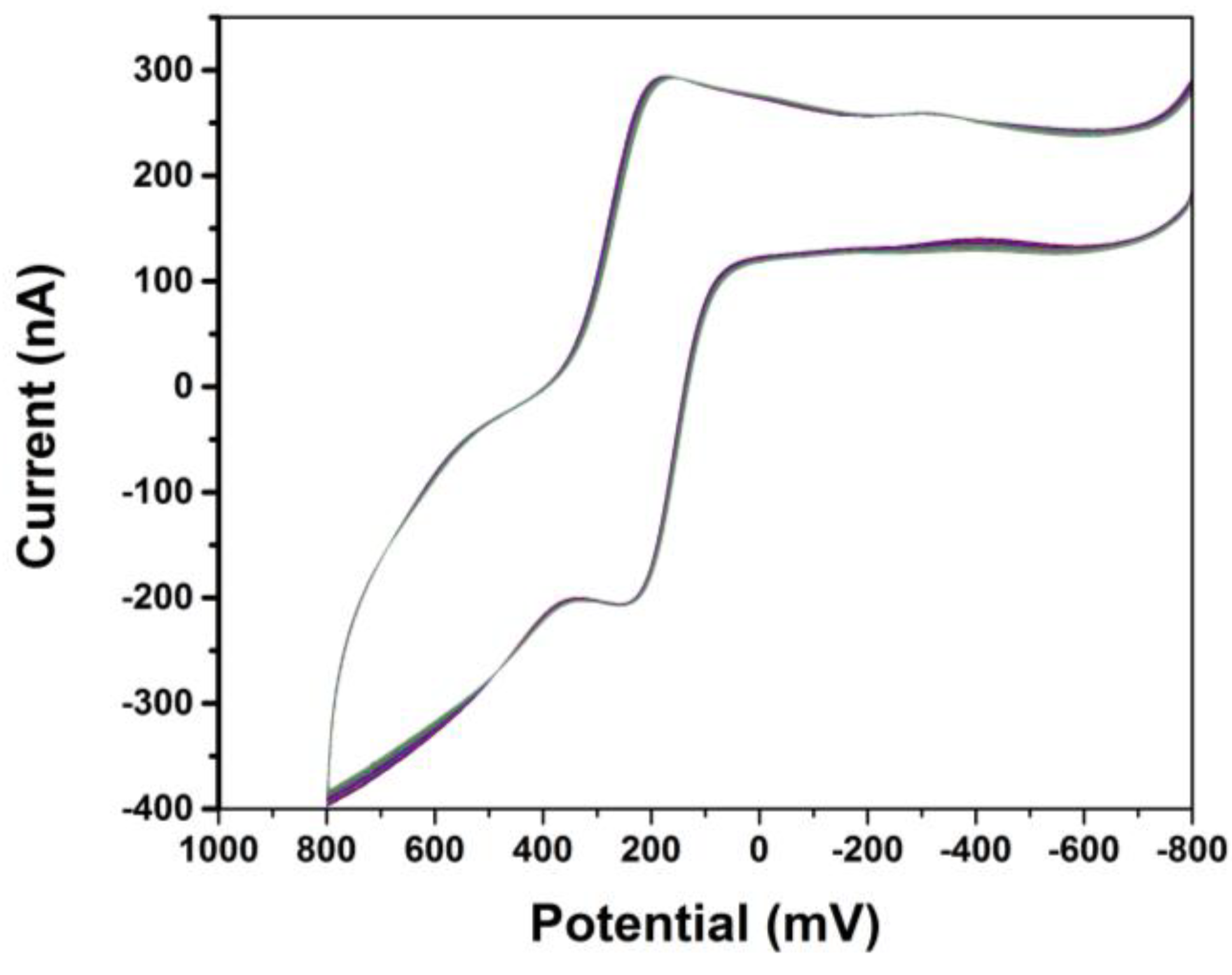
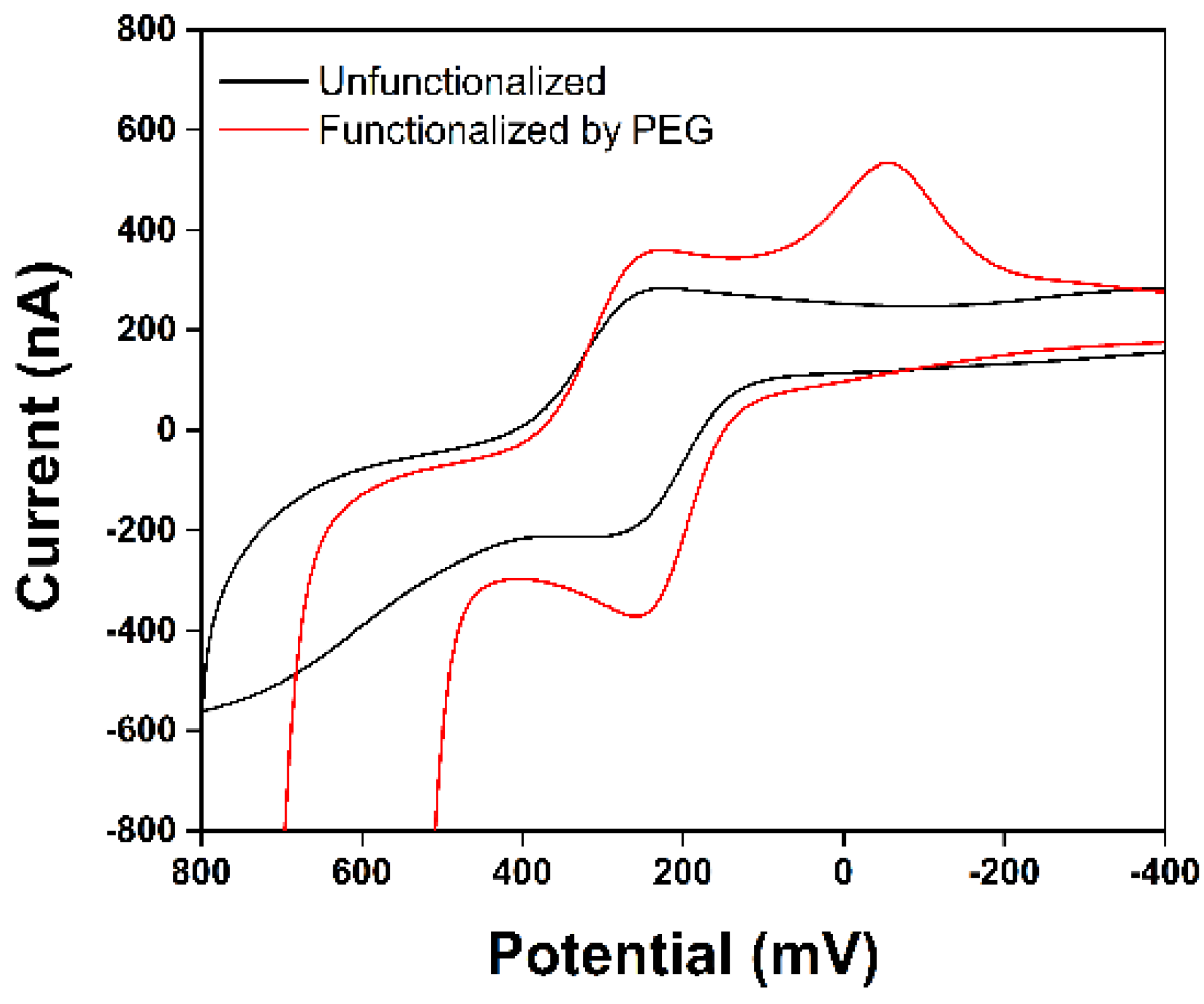
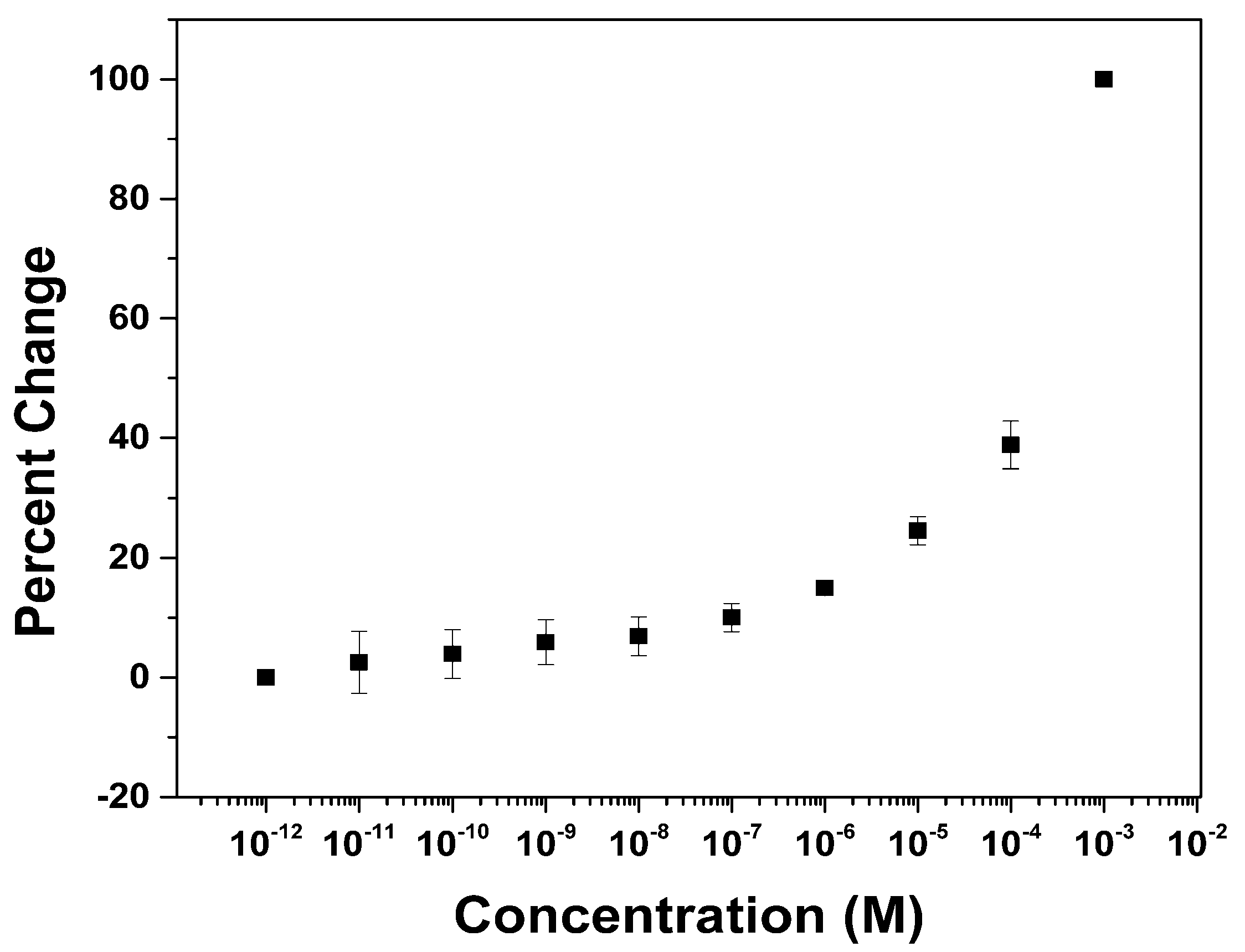
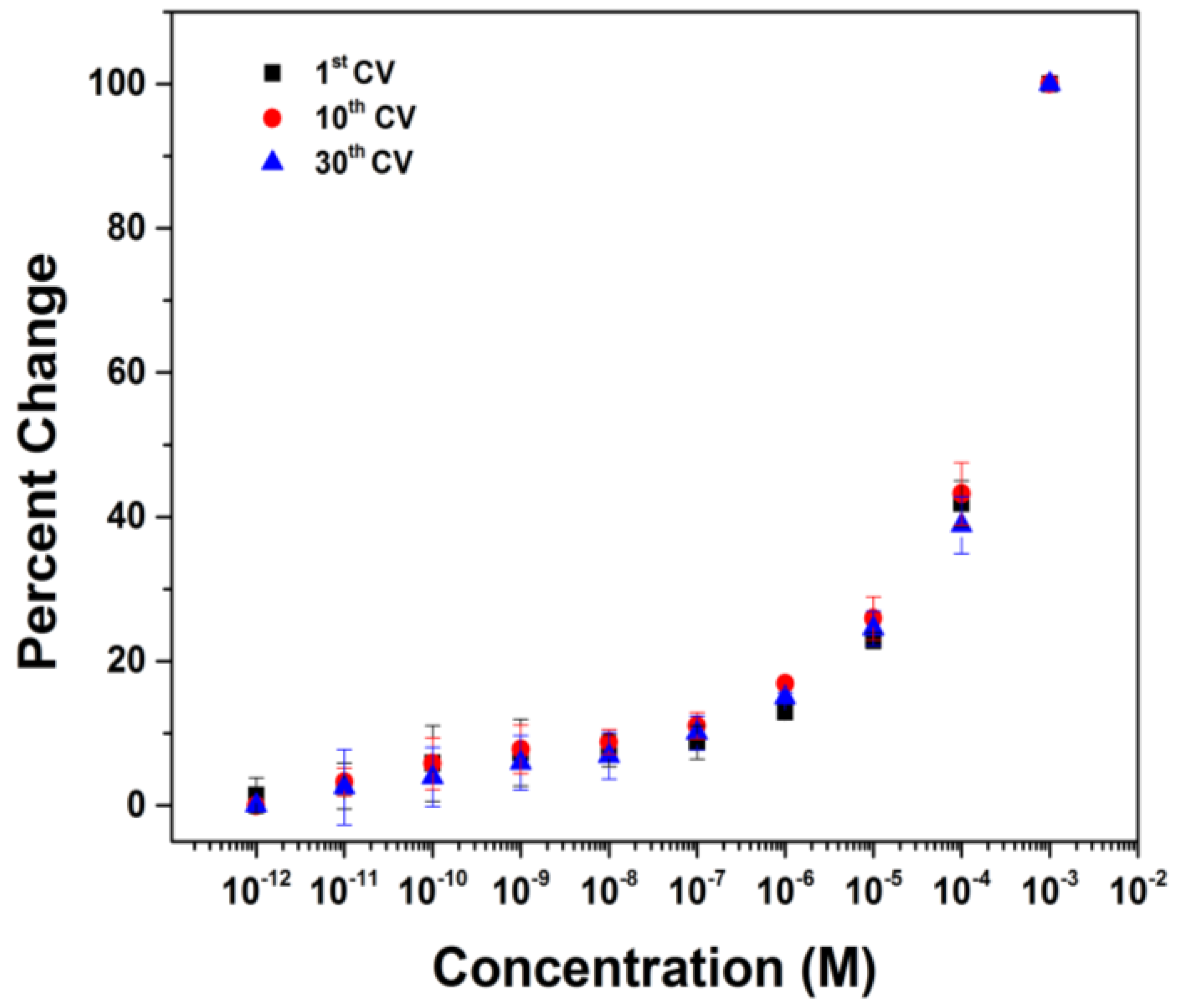
3. Results and Discussion





4. Conclusions
Acknowledgments
References and Notes
- For a description of the chips used here see “ Dill, K.; Montgomery, D.D.; Wang, W.; Tsai, J.C. Antigen Detection Using Microelectrode Array Microchips. Anal. Chim. Acta 2001, 444, 69–78. [Google Scholar] ”. 1 K chips: electrode diameter = 92 μm; Distance between the Pt-electrodes (rectangular cells) = 245.3 μm and 337.3 μm; 12 K slide: diameter = 44 µm; Distance between the Pt-electrodes (square cells) = 33 µm.
- Microelectrode arrays and the power supply for addressing them can be purchased from CustomArray, Inc.: 18916 North Creek Parkway, Suite 115, Bothell, WA 98011, USA. Available online: http://www.CustomArrayInc.com (accessed on 29 July 2014).
- Sullivan, M.G.; Utomo, H.; Fagan, P.J.; Ward, M.D. Automated electrochemical analysis with combinatorial electrode arrays. Anal. Chem. 1999, 71, 4369–4375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, H.; John, R. Development of a generic microelectrode array biosensing system. Anal. Chim. Acta 2000, 421, 175–187. [Google Scholar] [CrossRef]
- Hintsche, R.; Albers, J.; Bernt, H.; Eder, A. Multiplexing of microelectrode arrays in voltammetric measurements. Electroanalysis 2000, 12, 660–665. [Google Scholar] [CrossRef]
- Gardner, R.D.; Zhou, A.; Zufelt, N.A. Development of a microelectrode array sensing platform for combination electrochemical and spectrochemical aqueous ion testing. Sens. Actuator B Chem. 2009, 136, 177–185. [Google Scholar] [CrossRef]
- Beyer, M.; Nesterov, A.; Block, I.; König, K.; Felgenhauer, T.; Fernandez, S.; Leibe, K.; Torralba, G.; Hausmann, M.; Trunk, U.; Lindenstruth, V.; Bischoff, F.R.; Stadler, V.; Breitling, F. Combinatorial synthesis of peptide arrays onto a microchip. Science 2007, 318, 1888. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K.; Dinolfo, P.H.; Chidsey, C.E.D.; Collman, J.P. Selective functionalization of independently addressed microelectrodes by electrochemical activation and deactivation of a coupling catalyst. J. Am. Chem. Soc. 2006, 128, 1794–1795. [Google Scholar] [CrossRef] [PubMed]
- Wassum, K.M.; Tolosa, V.M.; Wang, J.; Walker, E.; Monbouquette, H.G.; Maidment, N.T. Silicon wafer-based platinum microelectrode array biosensor for near real-time measurement of glutamate in vivo. Sensors 2008, 8, 5023–5036. [Google Scholar] [CrossRef] [PubMed]
- Kerkoff, H.G.; Zhang, X.; Mailly, F.; Nouet, P.; Liu, H.; Richardson, A. A dependable microelectronic peptide synthesizer using electrode data. VLSI Design 2008, 4, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Nie, J.; Zhang, Y.; Shen, G.; Yu, R. Individually addressable microelectrode arrays fabricated with gold-coated pencil graphite particles for multiplexed and high sensitive impedance immunoassays. Biosens. Bioelectron. 2009, 25, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Yazvenko, N.; Wilmoth, J.; Cooper, J.; Lyon, W.; Danley, D. Use of a multiplexed CMOS microarray to optimize and compare oligonucleotide binding to DNA probes synthesized or immobilized on individual electrodes. Sensors 2010, 10, 7371–7385. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, Y.; Xia, P.; Luo, Y.; Rui, Q. Fabrication of TiO2 and metal nanoparticle-microelectrode arrays by photolithography and site-selective photocatalytic deposition. Anal. Chem. 2009, 81, 8249–8255. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.W.L.; Yousaf, M.N. Site-selective immobilization of ligands with control of density on electroactive microelectrode arrays. ChemPhysChem 2007, 8, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Graaf, M.D.; Moeller, K.D. The use of UV-cross-linkable di-block copolymers as functional reaction surfaces for microelectrode arrays. J. Electrochem. Soc. 2013, 160, G3020–G3029. [Google Scholar] [CrossRef]
- Moeller, K.D. Electrochemically generated organometallic reagents and site-selective synthesis on a microelectrode array. Organometallics 2014, in press. [Google Scholar]
- Bartels, J.; Lu, P.; Maurer, K.; Walker, A.V.; Moeller, K.D. Site-Selectively functionalizing microelectrode arrays: The use of Cu(I)-catalysts. Langmuir 2011, 27, 11199–11205. [Google Scholar] [CrossRef]
- Stuart-Fellet, M.; Bartels, J.L.; Bi, B.; Moeller, K.D. Site-selective chemistry and the attachment of peptides to the surface of a microelectrode array. In J. Am. Chem. Soc.; 2012; Volume 134, pp. 16891–16898. [Google Scholar]
- Tanabe, T.; Bi, B.; Hu, L.; Maurer, K.; Moeller, K.D. Building addressable libraries: Amino acid derived fluorescent linkers. Langmuir 2012, 28, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Gyurcsányi, R.E.; Jágerszki, G.; Kiss, G.; Tóth, K. Chemical imaging of biological systems with the scanning electrochemical microscope. Bioelectrochemistry 2004, 63, 207–215. [Google Scholar]
- Roberts, W.S.; Lonsdale, D.J.; Griffiths, J.; Higson, S.P.J. Advances in the application of scanning electrochemical microscopy to bioanalytical systems. Biosens. Bioelectron. 2007, 23, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Scanning Electrochemical Microscopy, 2nd ed.; Bard, A.J.; Mirkin, M.V. (Eds.) Marcel Dekker, Inc.: New York, NY, USA, 2001.
- Dykstra, P.H.; Roy, V.; Byrd, C.; Bentley, W.E.; Ghodssi, R. Microfluidic electrochemical sensor array for characterizing protein interactions with various functionalized surfaces. Anal. Chem. 2011, 83, 5920–5927. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Gonda, H.; Yano, K.; Kuroki, S.; Suzutani, T.; Katayama, Y. An electrochemical devise for the assay of the interaction between a dioxin receptor and its various ligands. Bioorg. Med. Chem. Lett. 2004, 14, 137–141. [Google Scholar] [CrossRef]
- Endo, A.; Hyashita, T. Electrochemical detection of ion and molecule recognition based on supramolecular assembly. J. Ion Exchange 2008, 19, 110–126. [Google Scholar] [CrossRef]
- Wirtz, M.; Martin, C.R. Nanotube membrane sensors. Resistive sensing and ion channel mimetics. Sens. Update 2002, 11, 35–64. [Google Scholar]
- Odashima, K.; Sugawara, M.; Umezawa, Y. Channel mimetic sensing membranes based on host-guest molecular recognition by synthetic receptors. ACS Symp. Series 1994, 561, 123–134. [Google Scholar]
- Yue, M.; Zhu, X.; Zheng, Y.; Hu, T.; Yang, L.; Wu, X. Amphotericin B ion channel mimetic sensor: A new type of potassium-selective sensor based on electrode-supported hybrid bilayer membranes. Electrochimica Acta 2012, 73, 78–85. [Google Scholar] [CrossRef]
- Malecka, K.; Grabowska, I.; Radecki, J.; Stachyra, A.; Góra-Sochacka, A.; Sirko, A.; Radecka, H. Voltammetric detection of a specific DNA sequence of avian influenza virus H5N1 using HS-ssDNA probe deposited onto gold electrode. Electroanalysis 2012, 24, 439–446. [Google Scholar] [CrossRef]
- Zhy, J.; Qin, Y.; Zhang, Y. Magnesium-selective ion-channel mimetic sensor with a traditional calcium ionophore. Anal. Chem. 2010, 82, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bakker, E. Ion channel mimetic chronopotentiometric polymeric membrane ion sensor for surface-confined protein detection. Langmuir 2009, 25, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Kurzatkowska, K.; Dolusic, E.; Dehaen, W.; Sieroń-Stoltny, K.; Sieroń, A.; Radecka, H. Gold electrode incorporating corrole as an ion-channel mimetic sensor for determination of dopamine. Anal. Chem. 2009, 81, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Komura, T.; Yamaguchi, T.; Kura, K.; Tanabe, J. Voltammetric and impedance study of the binding of ferrocene derivatives to a sulfonated calix[6]arene host in aqueous solutions. J. Electroanal. Chem. 2002, 523, 126–135. [Google Scholar] [CrossRef]
- Odashima, K.; Kotato, M.; Sugawara, M.; Umezawa, Y. The elusive hydroxyl radical. Measuring OH in the atmosphere. Anal. Chem. 1993, 65, 927–939. [Google Scholar]
- Chiari, M.; Cretich, M.; Damin, F.; Di Carlo, G.; Oldani, C. Advanced polymers for molecular recognitioin and sensing at the interface. J. Chromatogr. B 2008, 866, 89–103. [Google Scholar] [CrossRef]
- Piehler, J.; Brecht, A.; Valiokas, R.; Liedberg, B.; Gauglitz, G. A high-density poly(ethylene glycol) polymer brush for immobilization on glass-type surfaces. Biosens. Bioelectron. 2000, 15, 473–481. [Google Scholar] [CrossRef]
- Michel, R.; Pasche, S.; Textor, M.; Castner, D.G. The influence of PEG architecture on protein adsorption and conformation. Langmuir 2005, 21, 12337–12332. [Google Scholar]
- Sanjeeva Rao, K.; Wu, T.-S. Chan-Lam coupling reactions: Synthesis of heterocycles. Tetrahedron 2012, 68, 7735–7754. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Uppal, S.; Graaf, M.D.; Moeller, K.D. Microelectrode Arrays and the Use of PEG-Functionalized Diblock Copolymer Coatings. Biosensors 2014, 4, 318-328. https://doi.org/10.3390/bios4030318
Uppal S, Graaf MD, Moeller KD. Microelectrode Arrays and the Use of PEG-Functionalized Diblock Copolymer Coatings. Biosensors. 2014; 4(3):318-328. https://doi.org/10.3390/bios4030318
Chicago/Turabian StyleUppal, Sakshi, Matthew D. Graaf, and Kevin D. Moeller. 2014. "Microelectrode Arrays and the Use of PEG-Functionalized Diblock Copolymer Coatings" Biosensors 4, no. 3: 318-328. https://doi.org/10.3390/bios4030318
APA StyleUppal, S., Graaf, M. D., & Moeller, K. D. (2014). Microelectrode Arrays and the Use of PEG-Functionalized Diblock Copolymer Coatings. Biosensors, 4(3), 318-328. https://doi.org/10.3390/bios4030318