A DNA-Based Assay for Digoxin Detection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Construction of the Assay for Overnight Detection
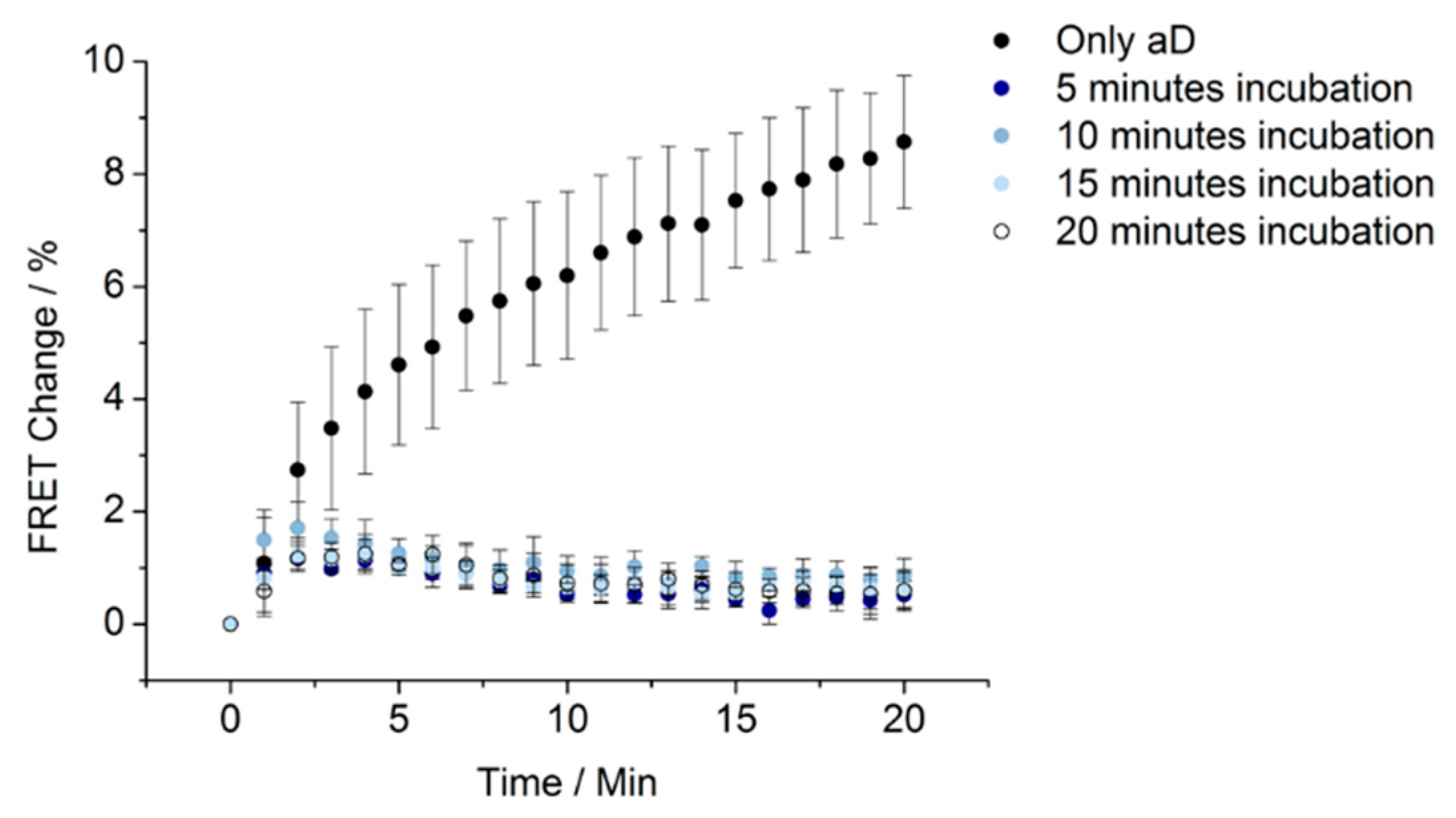
2.3. Construction of the Assay for 30-Minute Detection
2.4. FRET Experiments
2.5. Blood Samples
2.6. DNA Sequences
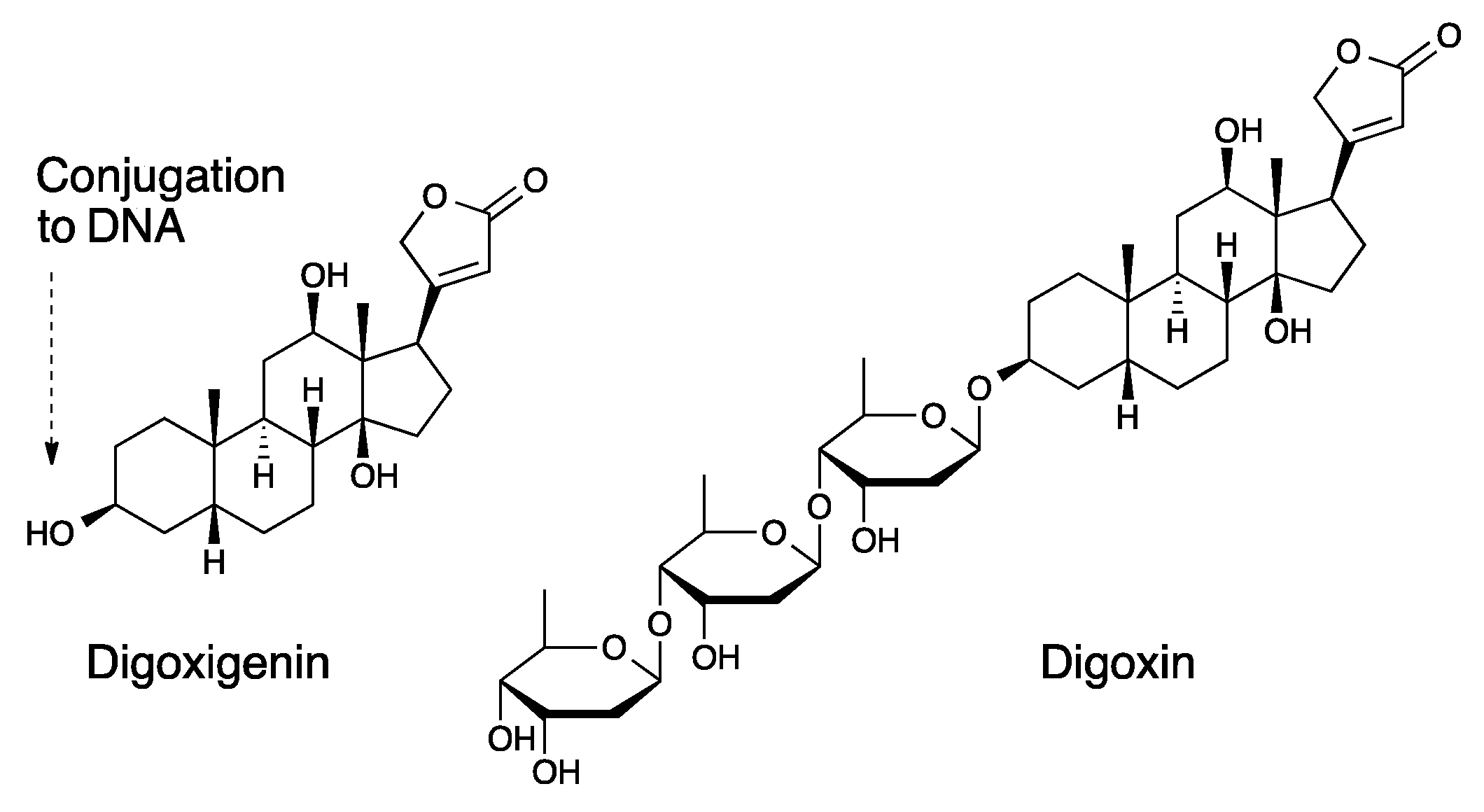
2.7. DNA-Ligand/Fluorophore Conjugation by NHS Ester-Amine Reaction
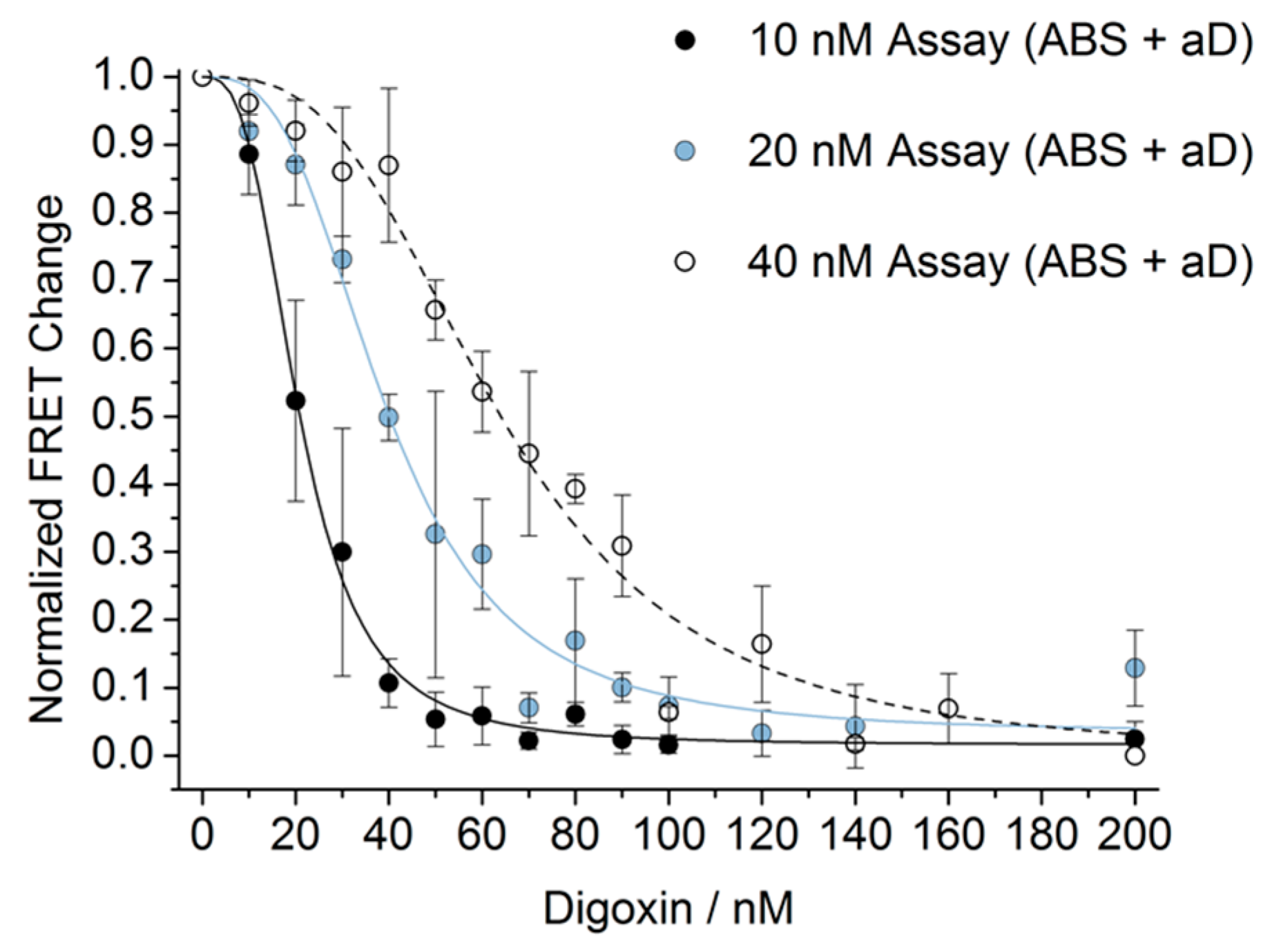
2.8. Calculation of Limit of Detection (LOD)
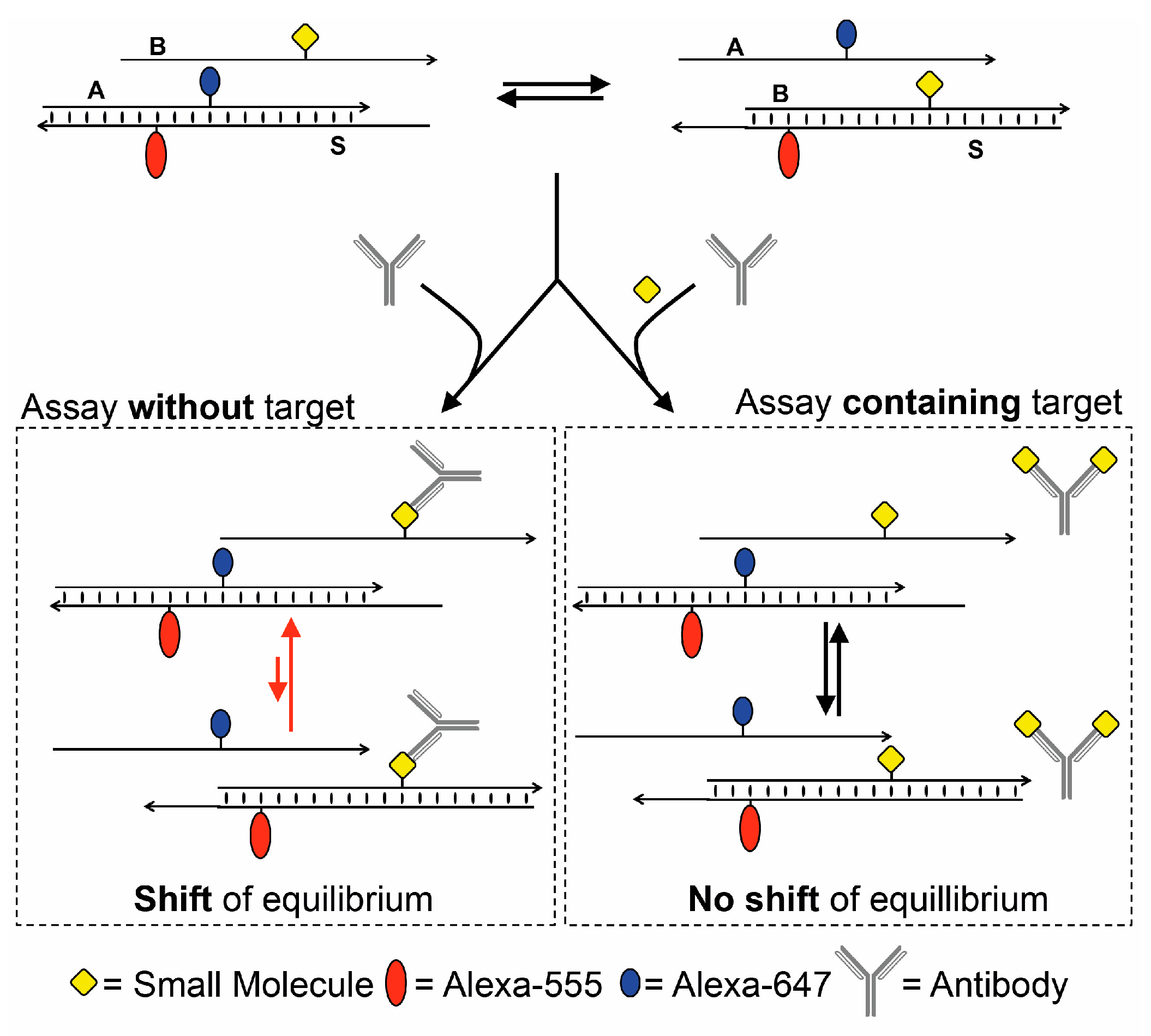
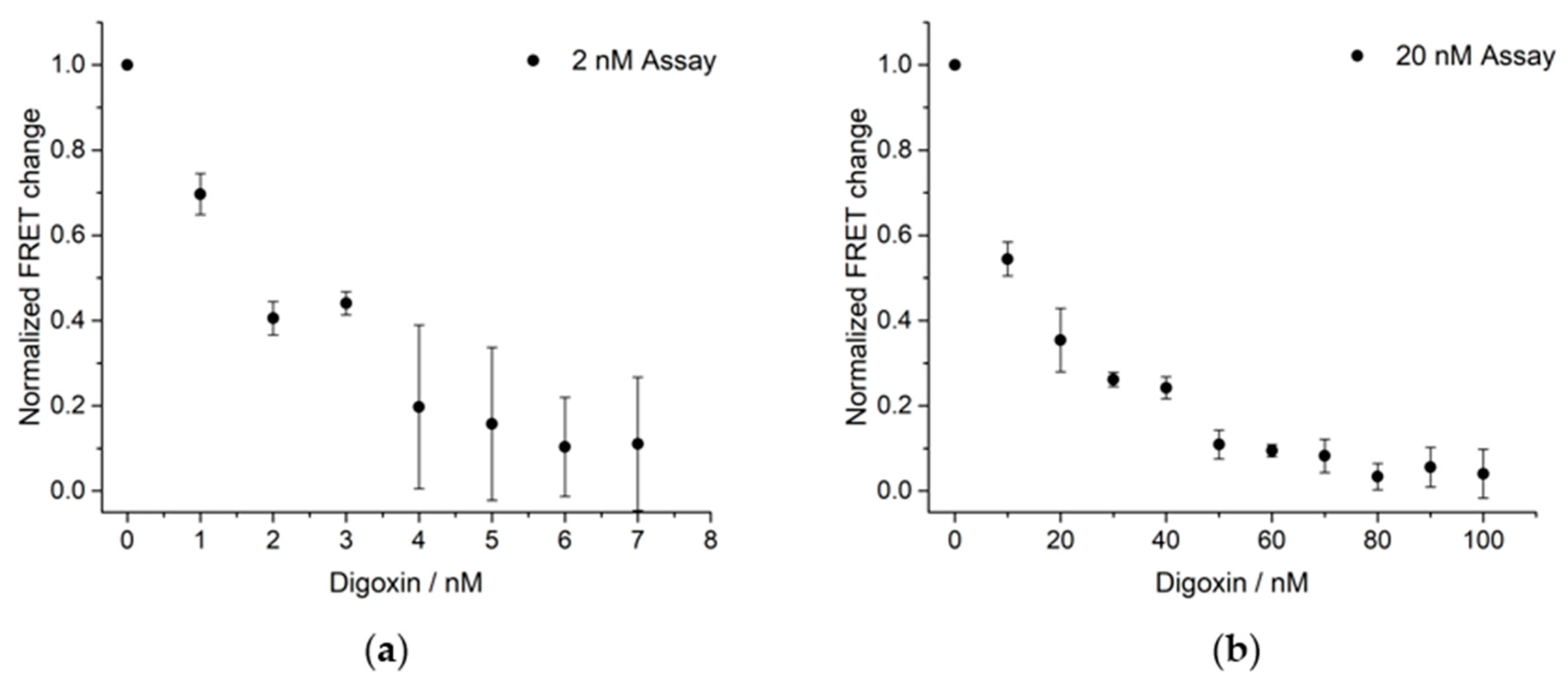
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, Z.; Wang, S. Recent development in application of high performance liquid chromatography-tandem mass spectrometry in therapeutic drug monitoring of immunosuppressants. J. Immunol. Methods 2008, 336, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Vogeser, M.; Seger, C. A decade of HPLC–MS/MS in the routine clinical laboratory—Goals for further developments. Clin. Biochem. 2008, 41, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Gerbitz, K.D.; Casetta, B.; Bauer, M.F. Rapid Determination of Total Homocysteine in Blood Spots by Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry. Clin. Chem. 2000, 46, 122–123. [Google Scholar] [PubMed]
- Lequin, R.M. Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, F.; Dever, B.; Li, X.-F.; Le, X.C. DNA-Mediated Homogeneous Binding Assays for Nucleic Acids and Proteins. Chem. Rev. 2013, 113, 2812–2841. [Google Scholar] [CrossRef] [PubMed]
- Nutiu, R.; Li, Y. Structure-Switching Signaling Aptamers. J. Am. Chem. Soc. 2003, 125, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Sassolas, A.; Blum, L.J.J.; Leca-Bouvier, B.D. Homogeneous assays using aptamers. Analyst 2011, 136, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, T.; Guo, X.; Lu, Z. Exonuclease III protection assay with FRET probe for detecting DNA-binding proteins. Nucleic Acids Res. 2005, 33, e23. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.-H.; Chan, D.S.; He, H.-Z.; Cheng, Z.; Yang, H.; Ma, D.-L. Luminescent detection of DNA-binding proteins. Nucleic Acids Res. 2012, 40, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Ohashi, H.; Iijima, I.; Ihara, M.; Takagi, H.; Hohsaka, T.; Ueda, H. “Quenchbodies”: Quench-Based Antibody Probes That Show Antigen-Dependent Fluorescence. J. Am. Chem. Soc. 2011, 133, 17386–17394. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Jeong, H.-J.; Arakawa, D.; Dong, J.; Ohashi, H.; Kaigome, R.; Saiki, F.; Yamane, K.; Takagi, H.; Ueda, H. Ultra Q-bodies: Quench-based antibody probes that utilize dye-dye interactions with enhanced antigen-dependent fluorescence. Sci. Rep. 2014, 4, 4640. [Google Scholar] [CrossRef] [PubMed]
- Heyduk, E.; Dummit, B.; Chang, Y.-H.; Heyduk, T. Molecular Pincers: Antibody-Based Homogeneous Protein Sensors. Anal. Chem. 2008, 80, 5152–5159. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011, 39, e102. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H.; Wang, Z.; Li, X.; Li, X.-F.; Le, X.C. Dynamic DNA Assemblies Mediated by Binding-Induced DNA Strand Displacement. J. Am. Chem. Soc. 2013, 135, 2443–2446. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H.; Lai, C.; Li, X.-F.; Le, C.X. A Molecular Translator that Acts by Binding-Induced DNA Strand Displacement for a Homogeneous Protein Assay. Angew. Chem. Int. Ed. 2012, 51, 9317–9320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, F.; Dever, B.; Wang, C.; Li, X.-F.; Le, X.C. Assembling DNA through Affinity Binding to Achieve Ultrasensitive Protein Detection. Angew. Chem. Int. Ed. 2013, 52, 10698–10705. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Dixon, W.; Ji, H.; Koong, A.C.; Mindrinos, M.; Davis, R.W. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nat. Methods 2007, 4, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Gustafsdottir, S.M.; Schlingemann, J.; Rada-Iglesias, A.; Schallmeiner, E.; Kamali-Moghaddam, M.; Wadelius, C.; Landegren, U. In vitro analysis of DNA–protein interactions by proximity ligation. Proc. Natl. Acad. Sci. USA 2007, 104, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Durner, J. Clinical Chemistry: Challenges for Analytical Chemistry and the Nanosciences from Medicine. Angew. Chem. Int. Ed. 2010, 49, 1026–1051. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Van Veldhuisen, D.J.; Colucci, W.S. Contemporary Use of Digoxin in the Management of Cardiovascular Disorders. Circulation 2006, 113, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Chen, S.X.; Yin, P. Optimizing the specificity of nucleic acid hybridization. Nat. Chem. 2012, 4, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hejesen, C.; Kjelstrup, M.B.; Birkedal, V.; Gothelf, K.V. A DNA-Mediated Homogeneous Binding Assay for Proteins and Small Molecules. J. Am. Chem. Soc. 2014, 136, 11115–11120. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys.-Berlin 1948, 2, 55–75. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Dewan, J.C.; Seeman, N.C. A DNA decamer with a sticky end: The crystal structure of d-CGACGATCGT. J. Mol. Biol. 1997, 267, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Yurke, B.; Turberfield, A.J.; Mills, A.P.; Simmel, F.C.; Neumann, J.L. A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Winfree, E. Control of DNA Strand Displacement Kinetics Using Toehold Exchange. J. Am. Chem. Soc. 2009, 131, 17303–17314. [Google Scholar] [CrossRef] [PubMed]
- Bhuckory, S.; Lefebvre, L.; Qiu, X.; Wegner, K.D.; Hildebrandt, N. Evaluating Quantum Dot Performance in Homogeneous FRET Immunoassays for Prostate Specific Antigen. Sensors 2016, 2, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C. The digoxigenin:anti-digoxigenin (DIG) technology—A survey on the concept and realization of a novel bioanalytical indicator system. Mol. Cell. Probes 1991, 5, 161–205. [Google Scholar] [CrossRef]
- Duhme, D.W.; Greenblatt, D.J.; Koch-Weser, J. Reduction of Digoxin Toxicity Associated with Measurement of Serum Levels: A Report from the Boston Collaborative Drug Surveillance Program. Ann. Intern. Med. 1974, 80, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, K.V.; Zhang, Z.; Kjelstrup, M.B.; Hejesen, C. Detection of Non-Nucleic Acid Analytes Using Strand Displacement Exchange Reactions. Patent WO2014041024, 20 March 2014. [Google Scholar]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kjelstrup, M.V.; Nielsen, L.D.F.; Hansen-Bruhn, M.; Gothelf, K.V. A DNA-Based Assay for Digoxin Detection. Biosensors 2018, 8, 19. https://doi.org/10.3390/bios8010019
Kjelstrup MV, Nielsen LDF, Hansen-Bruhn M, Gothelf KV. A DNA-Based Assay for Digoxin Detection. Biosensors. 2018; 8(1):19. https://doi.org/10.3390/bios8010019
Chicago/Turabian StyleKjelstrup, Michael V., Line D. F. Nielsen, Malthe Hansen-Bruhn, and Kurt V. Gothelf. 2018. "A DNA-Based Assay for Digoxin Detection" Biosensors 8, no. 1: 19. https://doi.org/10.3390/bios8010019
APA StyleKjelstrup, M. V., Nielsen, L. D. F., Hansen-Bruhn, M., & Gothelf, K. V. (2018). A DNA-Based Assay for Digoxin Detection. Biosensors, 8(1), 19. https://doi.org/10.3390/bios8010019