FRET Microscopy in Yeast


Abstract
:1. Introduction
2. Mapping the Organization of Yeast Protein Complexes by FRET
3. Analyzing Biochemistry and Biophysics of Yeast by FRET Biosensors
4. FRET Microscopy Techniques for Yeast Models
5. Fluorescent Proteins and Imaging Tips for FRET Microscopy in Yeast
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Förster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 6, 166–175. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Terjung, S.; Belyaev, J. Fluorescent Imaging Techniques. FRET and Complementary Methods. In Optical Probes in Biology, 1st ed.; Zhang, J., Mehta, S., Schultz, C., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 33–70. [Google Scholar]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced fluorescence microscopy techniques-FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. Fluorescence Energy Transfer as a Spectroscopic Ruler. Annu. Rev. Biochem. 2003, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, A.J.P.; Pérez-Medina, C.; Meijerink, A.; Mulder, W.J.M. Investigating supramolecular systems using Förster resonance energy transfer. Chem. Soc. Rev. 2018, 47, 7027–7044. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Kremers, G.-J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szöllősi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Jovin, T.M. Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 2006, 10, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Damelin, M.; Silver, P.A. Mapping interactions between nuclear transport factors in living cells reveals pathways through the nuclear pore complex. Mol. Cell 2000, 5, 133–140. [Google Scholar] [CrossRef]
- Damelin, M.; Silver, P.A. In Situ Analysis of Spatial Relationships between Proteins of the Nuclear Pore Complex. Biophys. J. 2002, 83, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Joglekar, A.; Chen, R.; Lawrimore, J. A Sensitized Emission Based Calibration of FRET Efficiency for Probing the Architecture of Macromolecular Machines. Cell. Mol. Bioeng. 2013, 6, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudhan, P.; Felzer-Kim, I.; Gurunathan, K.; Joglekar, A.P. Assembling the protein architecture of the budding yeast kinetochore-microtubule attachment using FRET. Curr. Biol. 2014, 24, 1437–1446. [Google Scholar] [CrossRef]
- Guo, W.; Kumar, S.; Görlitz, F.; Garcia, E.; Alexandrov, Y.; Munro, I.; Kelly, D.J.; Warren, S.; Thorpe, P.; Dunsby, C.; et al. Automated Fluorescence Lifetime Imaging High-Content Analysis of Förster Resonance Energy Transfer between Endogenously Labeled Kinetochore Proteins in Live Budding Yeast Cells. SLAS Technol. 2019, 24, 308–320. [Google Scholar] [CrossRef]
- Flory, M.R.; Carson, A.R.; Muller, E.G.; Aebersold, R. An SMC-domain protein in fission yeast links telomeres to the meiotic centrosome. Mol. Cell 2004, 16, 619–630. [Google Scholar] [CrossRef]
- Muller, E.G.D.; Snydsman, B.E.; Novik, I.; Hailey, D.W.; Gestaut, D.R.; Niemann, C.A.; Toole, E.T.O.; Giddings, T.H.; Sundin, B.A.; Davis, T.N. The Organization of the Core Proteins of the Yeast Spindle Pole Body. Mol. Cell. Biol. 2005, 16, 3341–3352. [Google Scholar] [CrossRef] [Green Version]
- Kollman, J.M.; Zelter, A.; Muller, E.G.D.; Fox, B.; Rice, L.M.; Davis, T.N.; Agard, D.A. The Structure of the gamma-Tubulin Small Complex: Implications of Its Architecture nad Flexibility for Microtubule Nucleation. Mol. Cell. Biol. 2008, 19, 207–215. [Google Scholar] [CrossRef]
- Gryaznova, Y.; Caydasi, A.; Malengo, G.; Sourjik, V.; Pereira, G. A FRET-based study reveals site-specific regulation of spindle position checkpoint proteins at yeast centrosomes. Elife 2016, 5, 1–27. [Google Scholar] [CrossRef]
- Viswanath, S.; Bonomi, M.; Kim, S.J.; Klenchin, V.A.; Taylor, K.C.; Yabut, K.C.; Umbreit, N.T.; Van Epps, H.A.; Meehl, J.; Jones, M.H.; et al. The molecular architecture of the yeast spindle pole body core determined by Bayesian integrative modeling. Mol. Biol. Cell 2017, 28, 3298–3314. [Google Scholar] [CrossRef] [Green Version]
- McDonald, N.A.; Lind, A.L.; Smith, S.E.; Li, R.; Gould, K.L. Nanoscale architecture of the Schizosaccharomyces pombe contractile ring. Elife 2017, 6, 1–23. [Google Scholar] [CrossRef]
- Skruzny, M.; Pohl, E.; Gnoth, S.; Malengo, G.; Sourjik, V. The protein architecture of the endocytic coat analyzed by FRET. BioRxiv 2018. [Google Scholar] [CrossRef]
- Warren, D.T.; Andrews, P.D.; Gourlay, C.W.; Ayscough, K.R. Sla1p couples the yeast endocytic machinery to proteins regulating actin dynamics. J. Cell Sci. 2002, 115, 1703–1715. [Google Scholar] [PubMed]
- Mc Intyre, J.; Muller, E.G.D.; Weitzer, S.; Snydsman, B.E.; Davis, T.N.; Uhlmann, F. In vivo analysis of cohesin architecture using FRET in the budding yeast Saccharomyces cerevisiae. EMBO J. 2007, 26, 3783–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaumik, S.R.; Raha, T.; Aiello, D.P.; Green, M.R. In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev. 2004, 18, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Chen, J.; Takasuka, T.E.; Jacobi, J.L.; Kaufman, P.D.; Irudayaraj, J.M.K.; Kirchmaier, A.L. Proliferating Cell Nuclear Antigen (PCNA) is required for cell cycle-regulated silent chromatin on replicated and nonreplicated genes. J. Biol. Chem. 2010, 285, 35142–35154. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Seeber, A.; Sack, R.; Tsai-Pflugfelder, M.; Bhullar, B.; Vlaming, H.; van Leeuwen, F.; Guénolé, A.; van Attikum, H.; Srivas, R.; et al. Yeast PP4 interacts with ATR homolog Ddc2-Mec1 and regulates checkpoint signaling. Mol. Cell 2015, 57, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Overton, M.C.; Blumer, K.J. G-protein-coupled receptors function as oligomers in vivo. Curr. Biol. 2000, 10, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Jansma, D.B.; Miller, D.; Friesen, J.D. Protein interaction quantified in vivo by spectrally resolved fluorescence. Biochem. J. 2005, 277, 265–277. [Google Scholar]
- Stoneman, M.R.; Paprocki, J.D.; Biener, G.; Yokoi, K.; Shevade, A.; Kuchin, S.; Raicu, V. Quaternary structure of the yeast pheromone receptor Ste2 in living cells. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 1456–1464. [Google Scholar] [CrossRef]
- Singh, A.; Severance, S.; Kaur, N.; Wiltsie, W.; Kosman, D.J. Assembly, activation, and trafficking of the Fet3p·Ftr1p high affinity iron permease complex in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 13355–13364. [Google Scholar] [CrossRef]
- Sinani, D.; Adle, D.J.; Kim, H.; Lee, J. Distinct mechanisms for Ctr1-mediated copper and cisplatin transport. J. Biol. Chem. 2007, 282, 26775–26785. [Google Scholar] [CrossRef] [PubMed]
- Strachotová, D.; Holoubek, A.; Kučerová, H.; Benda, A.; Humpolíčková, J.; Váchová, L.; Palková, Z. Ato protein interactions in yeast plasma membrane revealed by fluorescence lifetime imaging (FLIM). Biochim. Biophys. Acta-Biomembr. 2012, 1818, 2126–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dye, B.T.; Schell, K.; Miller, D.J.; Ahlquist, P. Detecting protein-protein interaction in live yeast by flow cytometry. Cytom. Part A 2005, 63, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Tabke, K.; Albertmelcher, A.; Vitavska, O.; Huss, M.; Schmitz, H.; Wieczorek, H. Reversible disassembly of the yeast V-ATPase revisited under in vivo conditions. Biochem. J. 2014, 462, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Barberis, M.; Scolari, S.; Klaus, C.; Herrmann, A.; Klipp, E. Unraveling interactions of cell cycle-regulating proteins Sic1 and B-type cyclins in living yeast cells: A FLIM-FRET approach. FASEB J. 2011, 26, 546–554. [Google Scholar] [CrossRef]
- Slaughter, B.D.; Schwartz, J.W.; Li, R. Mapping dynamic protein interactions in MAP kinase signaling using live-cell fluorescence fluctuation spectroscopy and imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 20320–20325. [Google Scholar] [CrossRef] [Green Version]
- Mallik, S.; Yang, W.; Norstrom, E.M.; Mastrianni, J.A. Live cell fluorescence resonance energy transfer predicts an altered molecular association of heterologous PrPSc with PrPC. J. Biol. Chem. 2010, 285, 8967–8975. [Google Scholar] [CrossRef]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A.P. Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 2013, 7, 469–476. [Google Scholar] [CrossRef]
- Saleh, A.A.; Bhadra, A.K.; Roy, I. Cytotoxicity of mutant huntingtin fragment in yeast can be modulated by the expression level of wild type huntingtin fragment. ACS Chem. Neurosci. 2014, 5, 205–215. [Google Scholar] [CrossRef]
- Sergeeva, A.V.; Sopova, J.V.; Belashova, T.A.; Siniukova, V.A.; Chirinskaite, A.V.; Galkin, A.P.; Zadorsky, S.P.; Sopova, J.V.; Belashova, T.A.; Siniukova, V.A. Amyloid properties of the yeast cell wall protein Toh1 and its interaction with prion proteins Rnq1 and Sup35. Prion 2019, 13, 21–32. [Google Scholar] [CrossRef]
- Damelin, M.; Silver, P. Analysis of Protein Interactions In Vivo with Fluorescence Resonance Energy Transfer (FRET). Cold Spring Harb. Protoc. 2006. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.R. Analysis of in vivo targets of transcriptional activators by fluorescence resonance energy transfer. Methods 2006, 40, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Overton, M.C.; Blumer, K.J. Use of fluorescence resonance energy transfer to analyze oligomerization of G-protein-coupled receptors expressed in yeast. Methods 2002, 27, 324–332. [Google Scholar] [CrossRef]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent Indicators for Ca2 + Based on Green Fluorescent Proteins and Calmodulin. Nature 1997, 388, 882–887. [Google Scholar]
- Broussard, J.A.; Green, K.J. Research Techniques Made Simple: Methodology and Applications of Förster Resonance Energy Transfer (FRET) Microscopy. J. Investig. Dermatol. 2017, 137, e185–e191. [Google Scholar] [CrossRef]
- Hochreiter, B.; Garcia, A.; Schmid, J. Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences. Sensors 2015, 15, 26281–26314. [Google Scholar] [CrossRef] [Green Version]
- Marx, V. Probes: FRET sensor design and optimization. Nat. Methods 2017, 14, 949–953. [Google Scholar] [CrossRef]
- Bermejo, C.; Ewald, J.C.; Lanquar, V.; Jones, A.M.; Frommer, W.B. In vivo biochemistry: Quantifying ion and metabolite levels in individual cells or cultures of yeast. Biochem. J. 2011, 438, 1–10. [Google Scholar] [CrossRef]
- Oku, M.; Hoseki, J.; Ichiki, Y.; Sakai, Y. A fluorescence resonance energy transfer (FRET)-based redox sensor reveals physiological role of thioredoxin in the yeast Saccharomyces cerevisiae. FEBS Lett. 2013, 587, 793–798. [Google Scholar] [CrossRef]
- Bermejo, C.; Haerizadeh, F.; Sadoine, M.S.C.; Chermak, D.; Frommer, W.B. Differential regulation of glucose transport activity in yeast by specific cAMP signatures. Biochem. J. 2013, 452, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Hu, Y.M.; Corkins, M.E.; Palmer, A.E.; Bird, A.J. Zinc transporters belonging to the Cation Diffusion Facilitator (CDF) family have complementary roles in transporting zinc out of the cytosol. PLoS Genet. 2018, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Qiao, W.; Mooney, M.; Bird, A.J.; Winge, D.R.; Eide, D.J. Zinc binding to a regulatory zinc-sensing domain monitored in vivo by using FRET. Proc. Natl. Acad. Sci. USA 2006, 103, 8674–8679. [Google Scholar] [CrossRef] [PubMed]
- Wegner, S.V.; Arslan, H.; Sunbul, M.; Yin, J.; He, C. Dynamic Copper(I) Imaging in Mammalian Cells with a Genetically Encoded Fluorescence Copper(I) Sensor. J. Am. Chem. Soc. 2010, 132, 2567–2569. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Oku, M.; Akeyama, N.; Itoyama, A.; Yurimoto, H.; Kuge, S.; Fujiki, Y.; Sakai, Y. A Novel Fluorescent Sensor Protein for Visualization of Redox States in the Cytoplasm and in Peroxisomes. Mol. Cell. Biol. 2010, 30, 3758–3766. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.-H.; Frommer, W.B. Fluorescent sensors for activity and regulation of the nitrate transceptor CHL1/NRT1.1 and oligopeptide transporters. Elife 2014, 3, 1–21. [Google Scholar] [CrossRef]
- Jones, A.M.; Danielson, J.Å.H.; Manojkumar, S.N.; Lanquar, V.; Grossmann, G.; Frommer, W.B. Abscisic acid dynamics in roots detected with genetically encoded FRET sensors. Elife 2014, 2014, 1–30. [Google Scholar] [CrossRef]
- Khan, T.; Kandola, T.S.; Wu, J.; Venkatesan, S.; Ketter, E.; Lange, J.J.; Rodríguez Gama, A.; Box, A.; Unruh, J.R.; Cook, M.; et al. Quantifying Nucleation In Vivo Reveals the Physical Basis of Prion-like Phase Behavior. Mol. Cell 2018, 71, 155–168. [Google Scholar] [CrossRef]
- Guerrero, J.L.; O’Malley, M.A.; Daugherty, P.S. Intracellular FRET-based Screen for Redesigning the Specificity of Secreted Proteases. ACS Chem. Biol. 2016, 11, 961–970. [Google Scholar] [CrossRef]
- Shin, I.; Ray, J.; Gupta, V.; Ilgu, M.; Beasley, J.; Bendickson, L.; Mehanovic, S.; Kraus, G.A.; Nilsen-Hamilton, M. Live-cell imaging of Pol II promoter activity to monitor gene expression with RNA IMAGEtag reporters. Nucleic Acids Res. 2014, 42, e90. [Google Scholar] [CrossRef]
- Conlon, P.; Gelin-Licht, R.; Ganesan, A.; Zhang, J.; Levchenko, A. Single-cell dynamics and variability of MAPK activity in a yeast differentiation pathway. Proc. Natl. Acad. Sci. USA 2016, 113, E5896–E5905. [Google Scholar] [CrossRef] [Green Version]
- Colombo, S.; Broggi, S.; Collini, M.; D’Alfonso, L.; Chirico, G.; Martegani, E. Detection of cAMP and of PKA activity in Saccharomyces cerevisiae single cells using Fluorescence Resonance Energy Transfer (FRET) probes. Biochem. Biophys. Res. Commun. 2017, 487, 594–599. [Google Scholar] [CrossRef]
- Suzuki, A.; Badger, B.L.; Haase, J.; Ohashi, T.; Erickson, H.P.; Salmon, E.D.; Bloom, K. How the kinetochore couples microtubule force and centromere stretch to move chromosomes. Nat. Cell Biol. 2016, 18, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Freikamp, A.; Cost, A.-L.; Grashoff, C. The Piconewton Force Awakens: Quantifying Mechanics in Cells. Trends Cell Biol. 2016, 26, 838–847. [Google Scholar] [CrossRef]
- Skruzny, M.; Brach, T.; Ciuffa, R.; Rybina, S.; Wachsmuth, M.; Kaksonen, M. Molecular basis for coupling the plasma membrane to the actin cytoskeleton during clathrin-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2012, 109, E2533–E2542. [Google Scholar] [CrossRef]
- Abella, M.; Andruck, L.; Malengo, G.; Skruzny, M. Mechanobiology of endocytic vesicle formation analyzed by FRET force sensors. Manuscript in preparation.
- Fehr, M.; Frommer, W.B.; Lalonde, S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. USA 2002, 99, 9846–9851. [Google Scholar] [CrossRef] [Green Version]
- Ha, J.S.; Song, J.J.; Lee, Y.M.; Kim, S.J.; Shin, C.S.; Lee, S.G. Design and Application of Highly Responsive FRET Biosensors for Sugar Detection in Living Yeast. Appl. Environ. Microbiol. 2007, 73, 7408–7414. [Google Scholar] [CrossRef]
- Bermejo, C.; Haerizadeh, F.; Takanaga, H.; Chermak, D.; Frommer, W.B. Dynamic analysis of cytosolic glucose and ATP levels in yeast using optical sensors. Biochem. J. 2010, 432, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, C.; Haerizadeh, F.; Takanaga, H.; Chermak, D.; Frommer, W.B. Optical sensors for measuring dynamic changes of cytosolic metabolite levels in yeast. Nat. Protoc. 2011, 6, 1806–1817. [Google Scholar] [CrossRef]
- Peroza, E.A.; Ewald, J.C.; Parakkal, G.; Skotheim, J.M.; Zamboni, N. A genetically encoded Förster resonance energy transfer sensor for monitoring in vivo trehalose-6-phosphate dynamics. Anal. Biochem. 2015, 474, 1–7. [Google Scholar] [CrossRef]
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA 2009, 106, 75–78. [Google Scholar] [CrossRef]
- Papagiannakis, A.; Niebel, B.; Wit, E.C.; Heinemann, M. Autonomous Metabolic Oscillations Robustly Gate the Early and Late Cell Cycle. Mol. Cell 2017, 65, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; Ota, K.; Ito, T. Circular permutation of ligand-binding module improves dynamic range of genetically encoded FRET-based nanosensor. Protein Sci. 2009, 18, 2518–2527. [Google Scholar] [CrossRef] [Green Version]
- Ameen, S.; Ahmad, M.; Mohsin, M.; Qureshi, M.I.; Ibrahim, M.M.; Abdin, M.Z.; Ahmad, A. Designing, construction and characterization of genetically encoded FRET-based nanosensor for real time monitoring of lysine flux in living cells. J. Nanobiotechnol. 2016, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Eichhof, I.; Ernst, J.F. Oxygen-independent FbFP: Fluorescent sentinel and oxygen sensor component in Saccharomyces cerevisiae and Candida albicans. Fungal Genet. Biol. 2016, 92, 14–25. [Google Scholar] [CrossRef]
- Padilla-Parra, S.; Tramier, M. FRET microscopy in the living cell: Different approaches, strengths and weaknesses. Bioessays 2012, 34, 369–376. [Google Scholar] [CrossRef]
- Pietraszewska-Bogiel, A.; Gadella, T.W. FRET microscopy: From principle to routine technology in cell biology. J. Microsc. 2010, 241, 111–118. [Google Scholar] [CrossRef]
- Ma, L.; Yang, F.; Zheng, J. Application of fluorescence resonance energy transfer in protein studies. J. Mol. Struct. 2014, 1077, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Rombola, C.; Jyothikumar, V.; Periasamy, A. Förster Resonance Energy Transfer Microscopy and Spectroscopy for Localizing Protein–Protein Interactions in Living Cells. Cytom. A 2013, 83, 780–793. [Google Scholar] [CrossRef]
- Becker, W. Fluorescence lifetime imaging-techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef]
- Ebrecht, R.; Don Paul, C.; Wouters, F.S. Fluorescence lifetime imaging microscopy in the medical sciences. Protoplasma 2014, 251, 293–305. [Google Scholar] [CrossRef]
- Snell, N.E.; Rao, V.P.; Seckinger, K.M.; Liang, J.; Leser, J.; Mancini, A.E.; Rizzo, M.A. Homotransfer FRET reporters for live cell imaging. Biosensors 2018, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wallrabe, H.; Seo, S.-A.; Periasamy, A. FRET Microscopy in 2010: The Legacy of Theodor Förster on the 100th Anniversary of his Birth. ChemPhysChem 2010, 12, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Mund, M.; van der Beek, J.A.; Deschamps, J.; Dmitrieff, S.; Hoess, P.; Monster, J.L.; Picco, A.; Nédélec, F.; Kaksonen, M.; Ries, J. Systematic Nanoscale Analysis of Endocytosis Links Efficient Vesicle Formation to Patterned Actin Nucleation. Cell 2018, 174, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A guide to fluorescent protein FRET pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef]
- Day, R.N.; Davidson, M.W. Fluorescent proteins for FRET microscopy: Monitoring protein interactions in living cells. Bioessays 2012, 34, 341–350. [Google Scholar] [CrossRef]
- Botman, D.; de Groot, D.H.; Schmidt, P.; Goedhart, J.; Teusink, B. In vivo characterisation of fluorescent proteins in budding yeast. Sci. Rep. 2019, 9, 2234. [Google Scholar] [CrossRef] [Green Version]
- Lambert, T.J. FPbase: A community-editable fluorescent protein database. Nat. Methods 2019, 16, 277–278. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751–759. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Meurer, M.; Ho, C.-T.; Besenbeck, B.; Fuller, J.; Lemberg, M.K.; Bukau, B.; Mogk, A.; Knop, M. Incomplete proteasomal degradation of green fluorescent proteins in the context of tandem fluorescent protein timers. Mol. Biol. Cell 2016, 27, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Mastop, M.; Bindels, D.S.; Shaner, N.C.; Postma, M.; Gadella, T.W.J.; Goedhart, J. Characterization of a spectrally diverse set of fluorescent proteins as FRET acceptors for mTurquoise2. Sci. Rep. 2017, 7, 11999. [Google Scholar] [CrossRef] [PubMed]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2016, 14, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Magiera, M.M.; Rathfelder, N.; Taxis, C.; Reber, S.; Maekawa, H.; Moreno-Borchart, A.; Doenges, G.; Schwob, E.; Schiebel, E.; et al. A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004, 21, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Khmelinskii, A.; Meurer, M.; Duishoev, N.; Delhomme, N.; Knop, M. Seamless gene tagging by endonuclease-driven homologous recombination. PLoS ONE 2011, 6, e23794. [Google Scholar] [CrossRef] [PubMed]
- Young, C.L.; Raden, D.L.; Caplan, J.L.; Czymmek, K.J.; Robinson, A.S. Cassette series designed for live-cell imaging of proteins and high-resolution techniques in yeast. Yeast 2012, 29, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Lim, W.A.; Thorn, K.S. Improved Blue, Green, and Red Fluorescent Protein Tagging Vectors for S. cerevisiae. PLoS ONE 2013, 8, e67902. [Google Scholar] [CrossRef] [PubMed]
- Higuchi-Sanabria, R.; Garcia, E.J.; Tomoiaga, D.; Munteanu, E.L.; Feinstein, P.; Pon, L.A. Characterization of Fluorescent Proteins for Three- and Four-Color Live-Cell Imaging in S. cerevisiae. PLoS ONE 2016, 11, e0146120. [Google Scholar] [CrossRef]
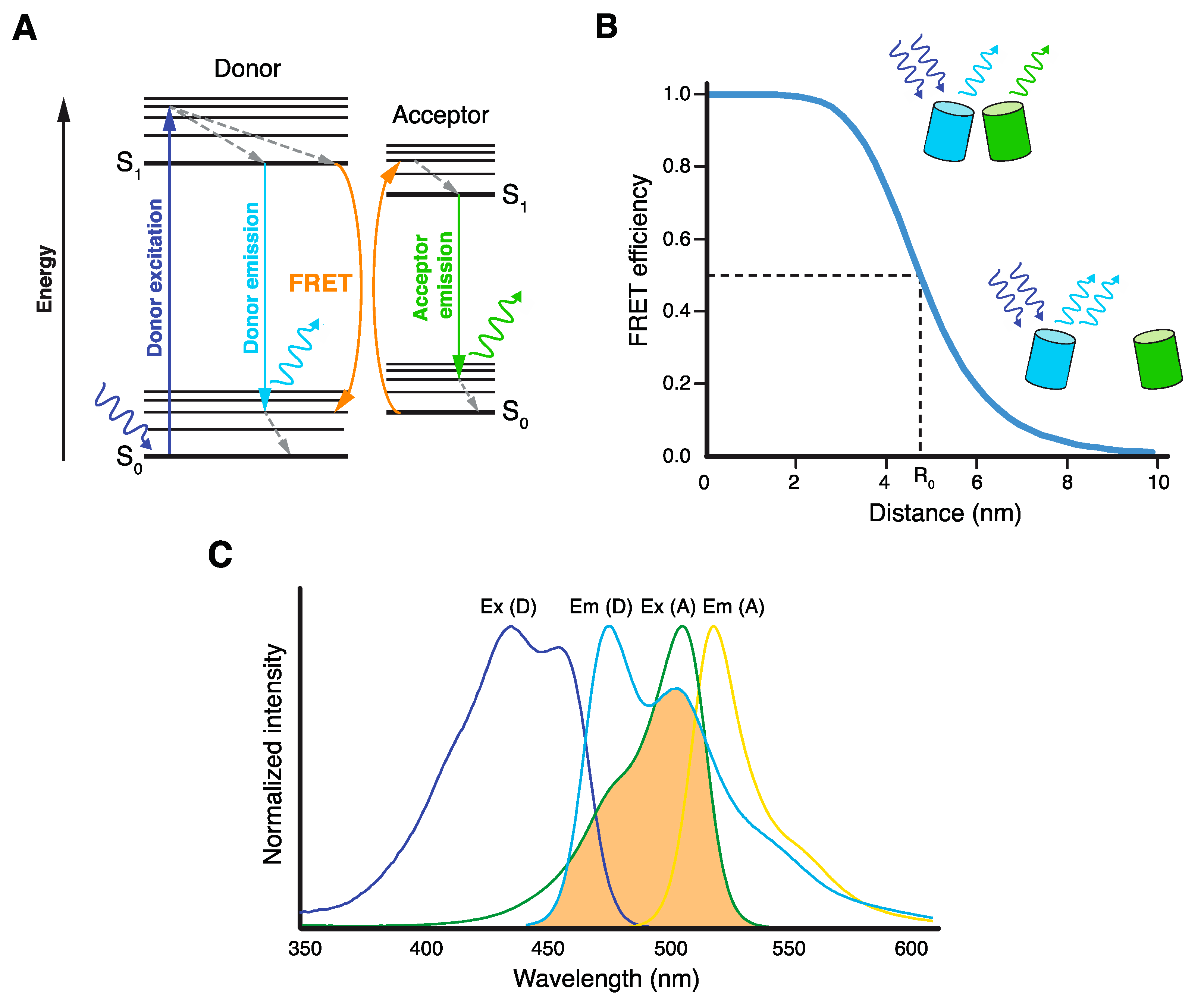




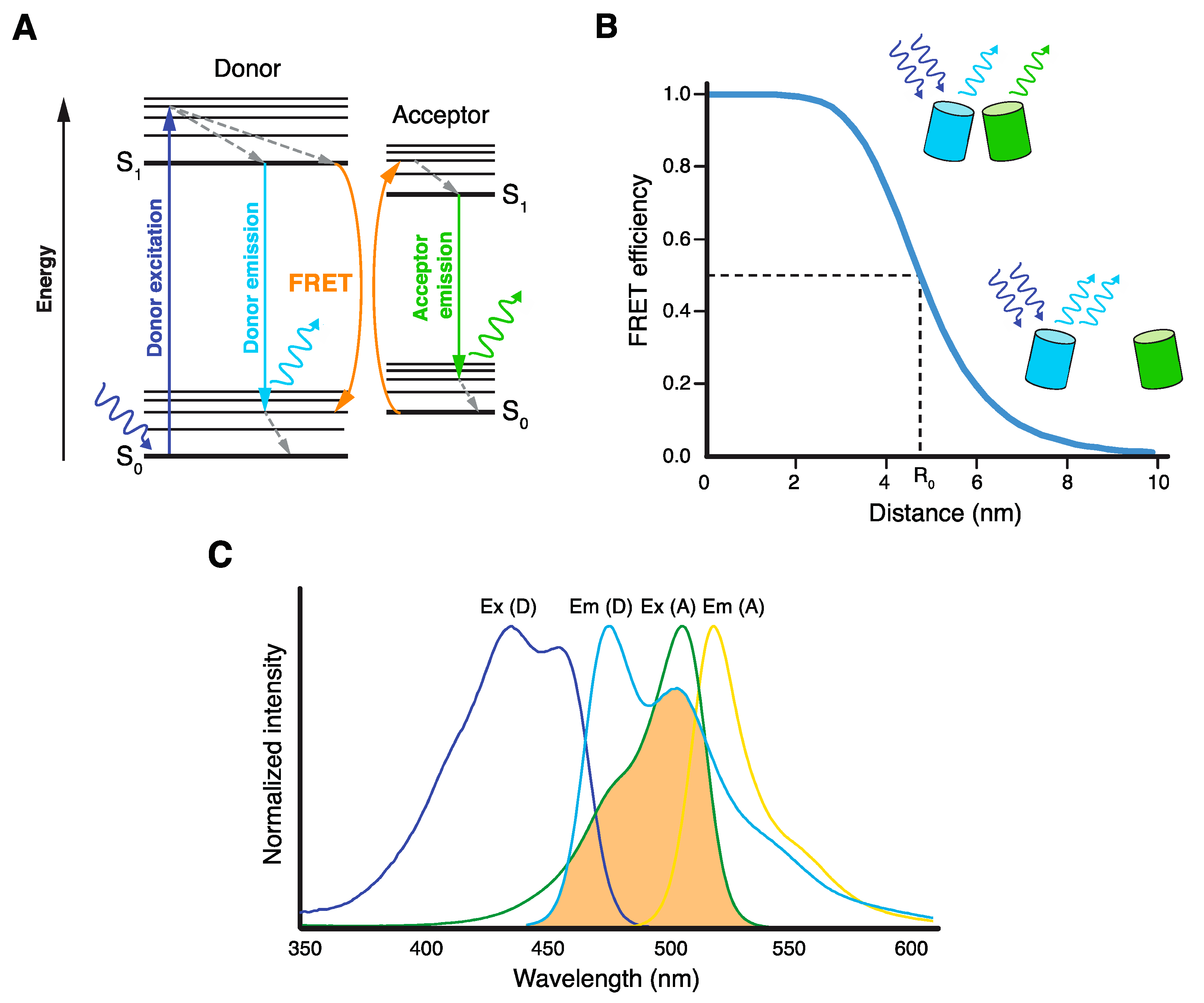{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Complex | FRET Technique 1,2 | FRET Donor–Acceptor 1,2 | References |
|---|---|---|---|
| Nuclear pore complex (NPC) | sensitized emission | CFP-YFP | [11,12,42] |
| Spindle pole body (SPB) | sensitized emission acceptor photobleaching | CFP-YFP mTq2-YFP | [16,17,18,19,20] |
| Kinetochore | sensitized emission FLIM | GFP-mCherry mTq2-YFP | [13,14,15] |
| Contractile ring | acceptor photobleaching | GFP/mNG-mCherry | [21] |
| Endocytic coat | acceptor photobleaching | GFP-mCherry mTq-mNG mNG-mScarlet | [22] |
| Cohesin | sensitized emission | CFP-YFP | [24] |
| SAGA-Gal4 transcription factor | acceptor photobleaching spectral FRET | CFP-YFP | [25,43] |
| PCNA-SAS-I complex | FLIM | CFP-YFP | [26] |
| ATR complex Dcp2-Mec1- PP4 phosphatase Psy2-Php3 | sensitized emission | GFP-RFP | [27] |
| Ste2 oligomerization | spectral FRET | CFP/GFP-YFP | [28,29,30,44] |
| Fet3-Ftr1 iron permease | spectral FRET | CFP-YFP | [31] |
| Ctr1 transporter oligomerization and copper binding | spectral FRET | CFP-YFP | [32] |
| Ato1-Ato2 proteins | acceptor photobleaching, FLIM | GFP-tdimer2 CFP-Venus | [33] |
| V-ATPase disassembly | sensitized emission | CFP-YFP | [34] |
| Tom70 oligomerization | sensitized emission | CFP-YFP | [35] |
| CDK inhibitor Sic1-cyclins | FLIM | mCerulean-YFP | [36] |
| Ste5-Fus3 interaction | acceptor photobleaching | GFP-mStrawberry | [37] |
| Prp prion aggregation | donor photobleaching | CFP-YFP | [38] |
| Prp-amyloid β interaction | acceptor photobleaching | CFP-YFP | [39] |
| HTT huntingtin aggregation | acceptor photobleaching | CFP-Venus | [40] |
| Toh1 aggregation with Rnq1 and Sup35 prion proteins | acceptor photobleaching | CFP-YFP | [41] |
| Studied Analyte/Process | Sensor Name (Sensor Origin) | FRET Donor–Acceptor (FRET Method) 1 | References |
|---|---|---|---|
| Maltose | FLIPmal (MBP) | CFP-YFP (spectral FRET) | [67,68] |
| Glucose (Galactose) | FLIPglu sensors (MglD) | CFP-Venus | [69,70] |
| Trehalose-6P | T6P-TRACKs (TreR) | CFP-Venus | [71] |
| ATP | AT1.03 sensors (ε subunit of FoF1-ATP synthase) | CFP-Venus | [69,72,73] |
| Histidine | FLIP-cpHisJ194 (HisJ) | CFP-Venus | [74] |
| Lysine | FLIPK (LAO) | CFP-YFP | [75] |
| Zinc ion | ZF1/2, ZF3/4, ZapCY1/2 (Zap1) | CFP-YFP/Citrine | [52,53] |
| Redox state | Redoxfluor (Yap1) | Cerulean-Citrine | [50,55] |
| Oxygen | YFOS (FbFP) | FbFP-YFP (spectral FRET) | [76] |
| Nitrate Oligopeptides | NiTrac sensors, PepTrac sensors | mCerulean-Aphrodite (spectral FRET) | [56] |
| Abscisic acid | ABACUS1 sensors | Cerulean-Citrine | [57] |
| Prion proteins nucleation | AmFRET | mEos3.1 (FACS) | [58] |
| Amyloid β cleavage by evolved protease | PrECISE | CyPet-Ypet (FACS) | [59] |
| PolII promoter activity | IMAGEtags (RNA aptamers) | Cy3-Cy5 (sensitized emission) | [60] |
| MAPK signaling pathway | yEKAREV | CFP-YPet | [61] |
| cAMP/PKA signaling pathway | Epac2-camps (Epac2) AKAR3 | CFP-YFP CFP-cpVenus | [62] |
| Force for chromosome segregation | Ndc80 tension sensor | CFP-YPet | [63] |
| Force for endocytic vesicle formation | molecular tension sensors in Sla2 protein | mTq2-mNG | [64,66] |
| FRET Technique | Advantages | Disadvantages |
|---|---|---|
| acceptor photobleaching | easy to set up and calculate | endpoint assay (time-lapse measurements only indirectly) |
| sensitized emission (including ratiometric FRET) | easy to set up time-lapse measurements | acquisition of controls necessary for FRET calculation (not for ratiometric FRET) |
| FRET-FLIM | pools of FRET-involved/absent molecules discernable time-lapse measurements | complex setup and data analysis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skruzny, M.; Pohl, E.; Abella, M. FRET Microscopy in Yeast. Biosensors 2019, 9, 122. https://doi.org/10.3390/bios9040122
Skruzny M, Pohl E, Abella M. FRET Microscopy in Yeast. Biosensors. 2019; 9(4):122. https://doi.org/10.3390/bios9040122
Chicago/Turabian StyleSkruzny, Michal, Emma Pohl, and Marc Abella. 2019. "FRET Microscopy in Yeast" Biosensors 9, no. 4: 122. https://doi.org/10.3390/bios9040122
APA StyleSkruzny, M., Pohl, E., & Abella, M. (2019). FRET Microscopy in Yeast. Biosensors, 9(4), 122. https://doi.org/10.3390/bios9040122