
Potential Anticancer Lipoxygenase Inhibitors from the Red Sea-Derived Brown Algae Sargassum cinereum: An In-Silico-Supported In-Vitro Study


 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
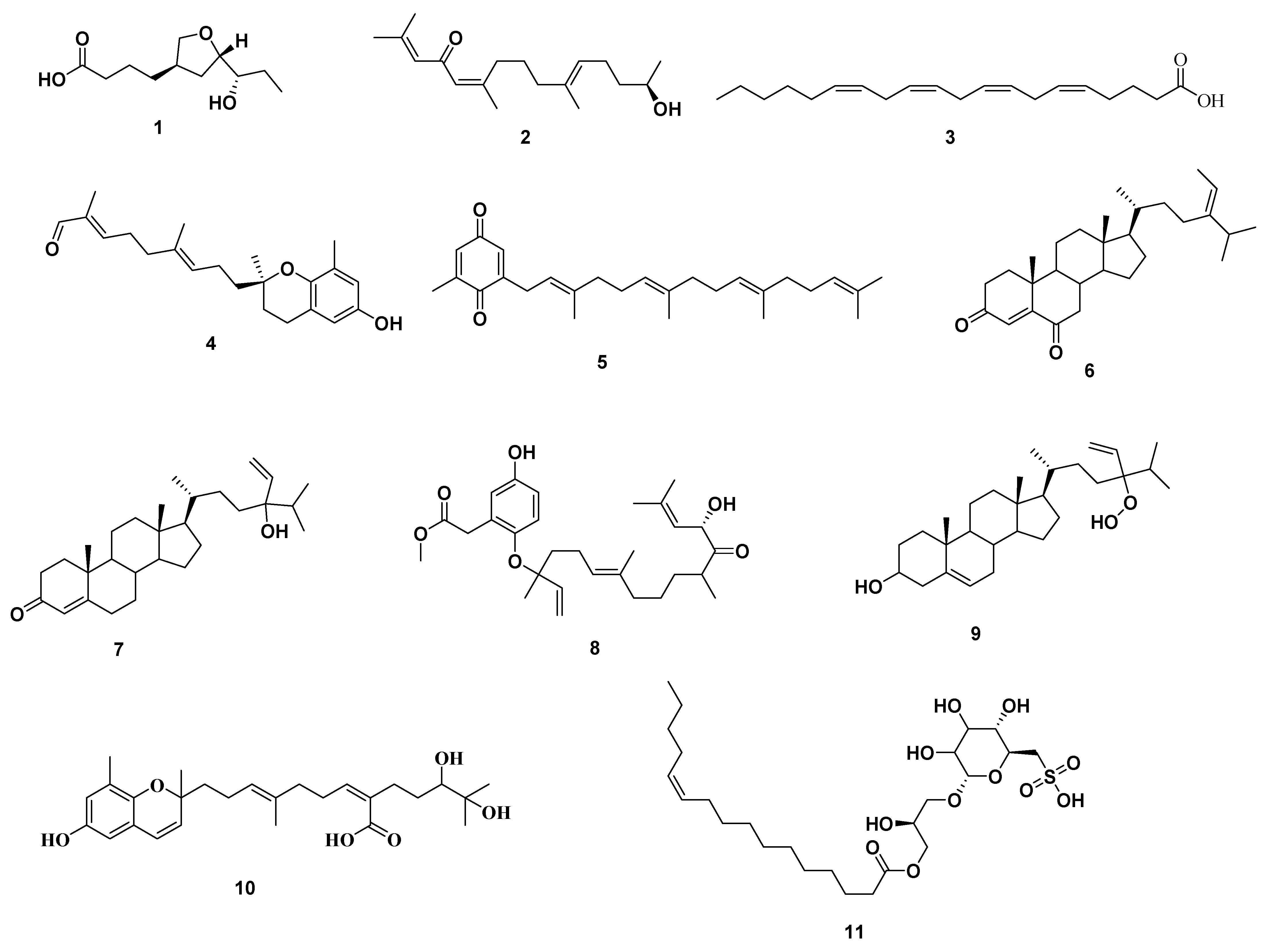
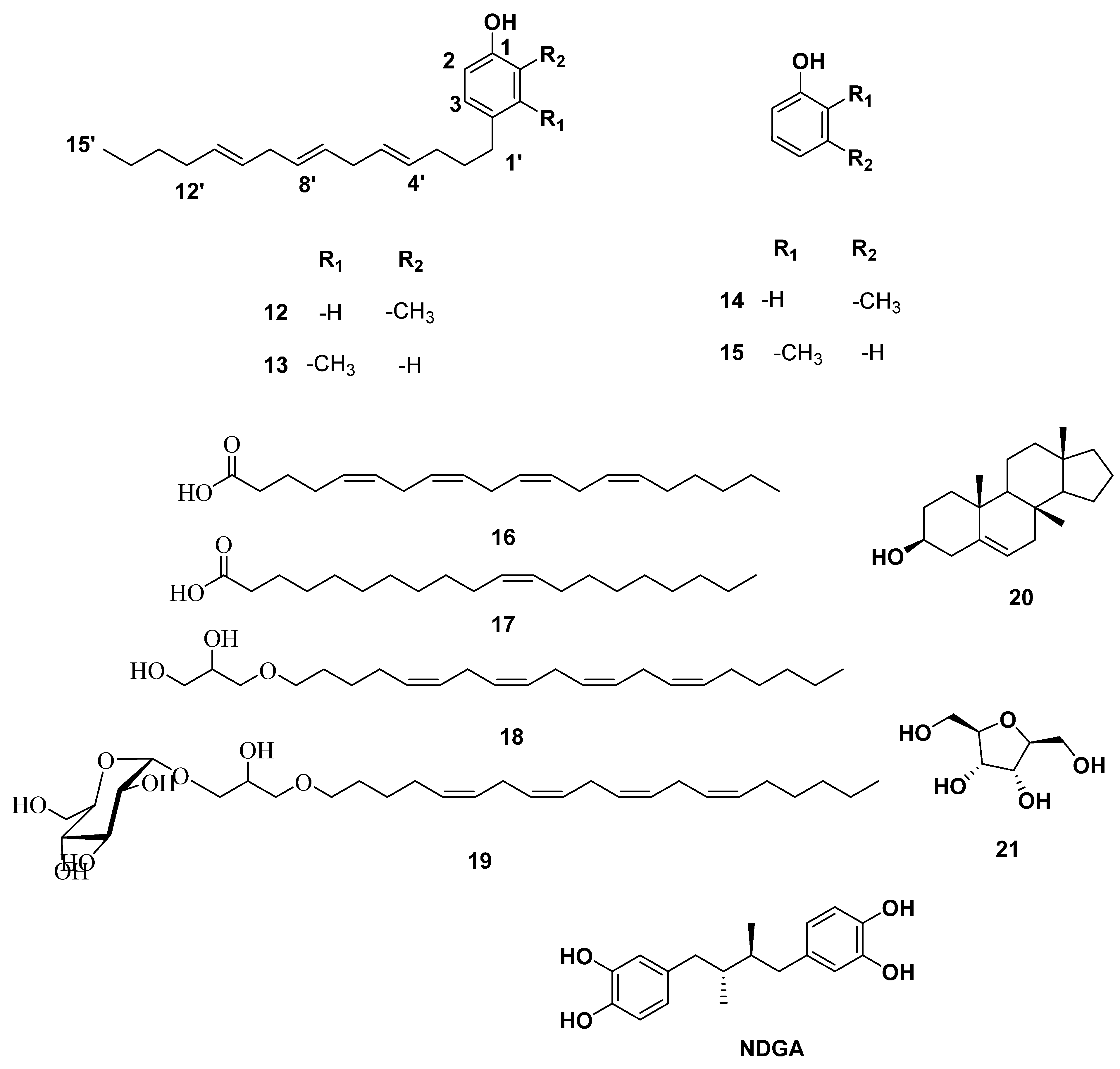
2.1. Chemical Dereplication of S. cinereum
2.2. Phytochemical Investigation of S. cinereum
2.3. Antiproliferative Activity of the Isolated Metabolites
2.4. Virtual Screening-based Target Identification
2.5. LOX Inhibition Assay
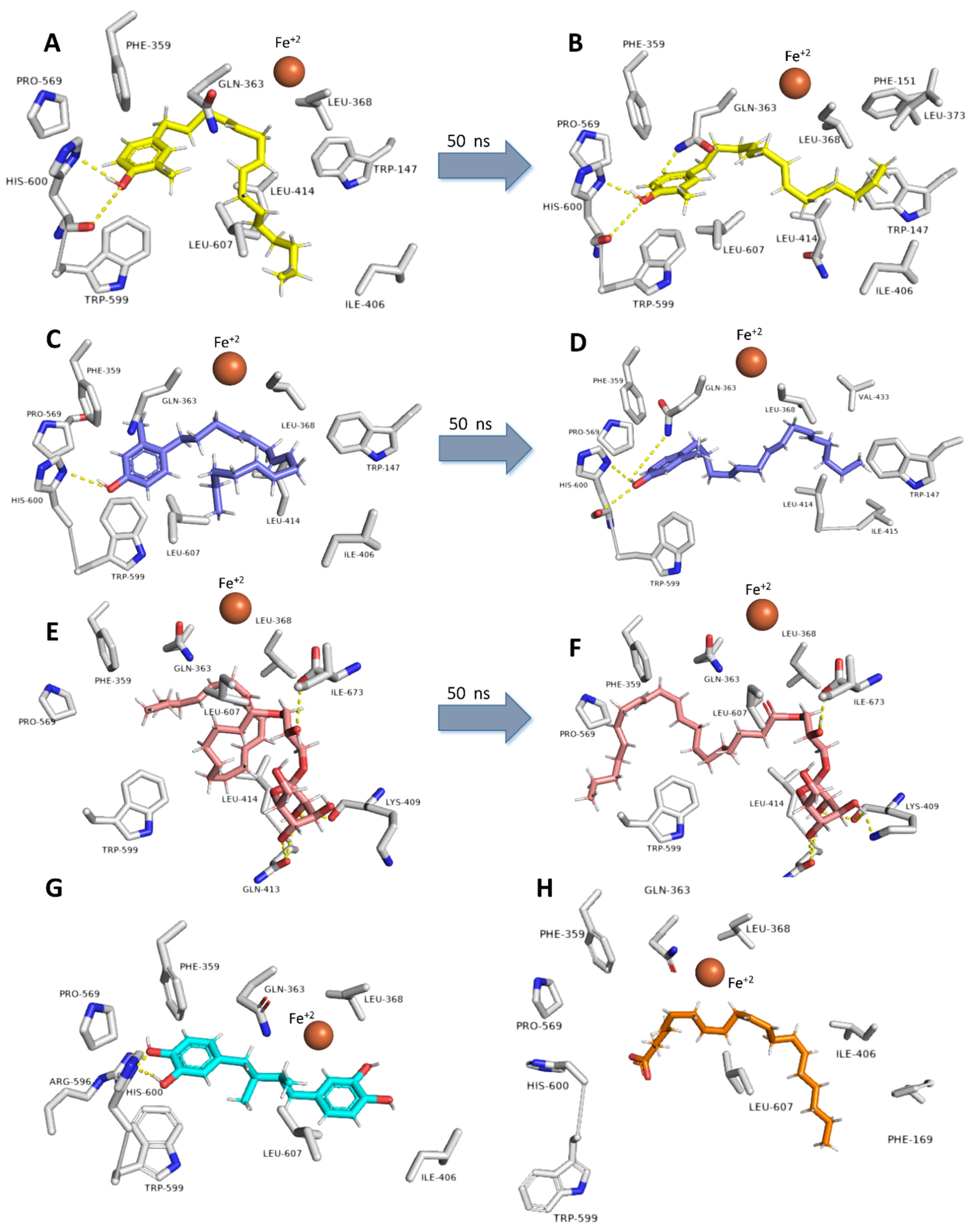
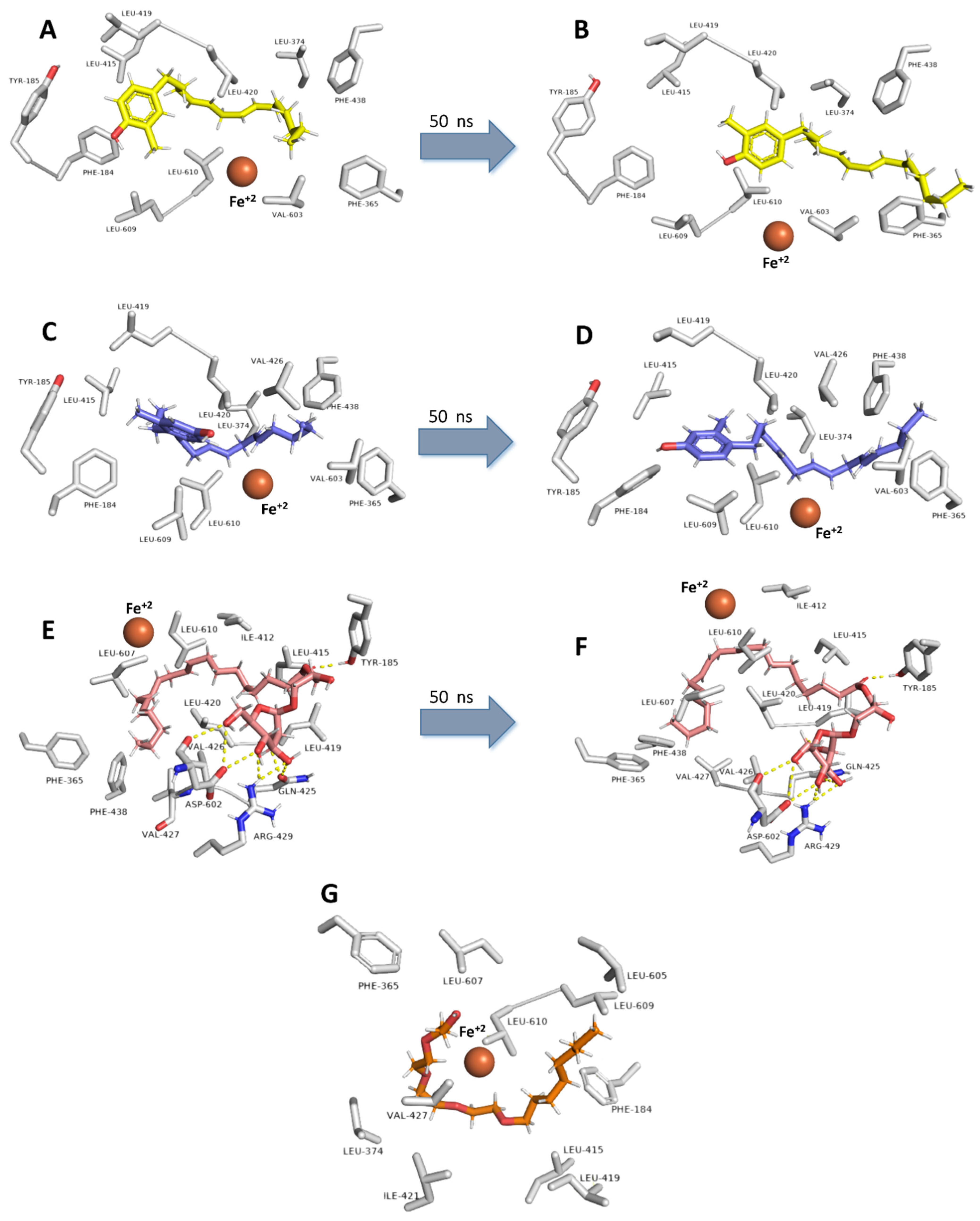
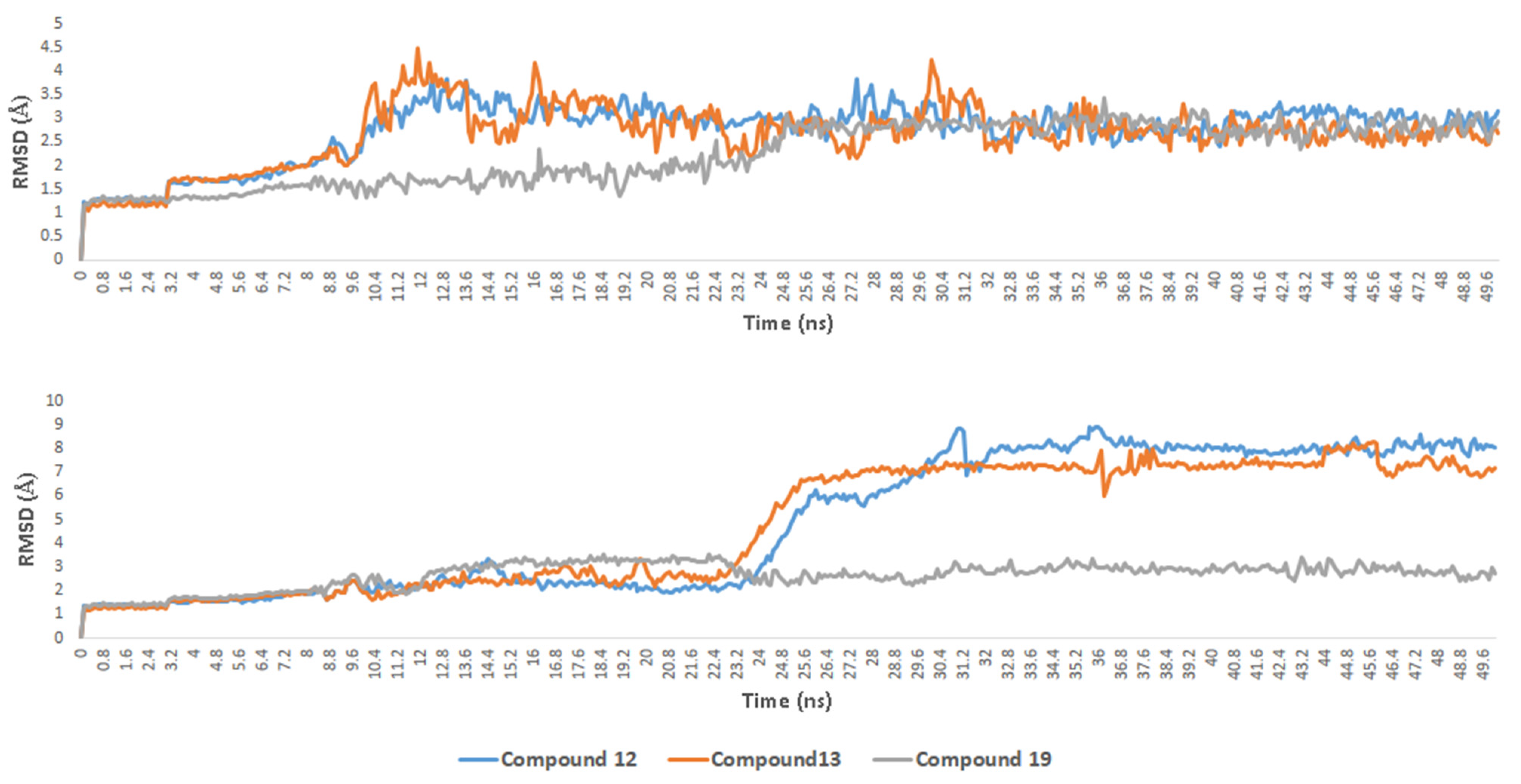
2.6. Molecular Docking and Dynamic Simulation
3. Materials and Methods
3.1. Algae Material
3.2. Chemicals and Reagents
3.3. Spectral Analyses
3.4. Extraction and Fractionation of Algae Material
3.5. Metabolomic Analysis Procedure
3.6. Isolation and Purification of Major Compounds
3.7. Antiproliferative Assay
3.8. Lipoxygenase (LOX) Inhibition Assay
3.9. Docking Study
3.10. Molecular Dynamic Simulation
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guiry, M. AlgaeBase. World-Wide Electronic Publication, National University of Ireland, Galway. 2010. Available online: https://ci.nii.ac.jp/naid/10028197296/ (accessed on 4 December 2020).
- Yip, Z.T.; Quek, R.Z.; Huang, D. Historical biogeography of the widespread macroalga Sargassum (Fucales, Phaeophyceae). J. Phycol. 2020, 56, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Yip, Z.T.; Quek, R.Z.; Low, J.K.; Wilson, B.; Bauman, A.G.; Chou, L.M.; Todd, P.A.; Huang, D. Diversity and phylogeny of Sargassum (Fucales, Phaeophyceae) in Singapore. Phytotaxa 2018, 369, 200–210. [Google Scholar] [CrossRef]
- Rushdi, M.I.; Abdel-Rahman, I.A.; Saber, H.; Attia, E.Z.; Abdelraheem, W.M.; Madkour, H.A.; Hassan, H.M.; Elmaidomy, A.H.; Abdelmohsen, U.R. Pharmacological and natural products diversity of the brown algae genus Sargassum. RSC Adv. 2020, 10, 24951–24972. [Google Scholar] [CrossRef]
- Yende, S.R.; Harle, U.N.; Chaugule, B.B. Therapeutic potential and health benefits of Sargassum species. Pharmacogn. Rev. 2014, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhang, L.; Zhang, J.; Liu, M.; Wei, L.; Shen, T.; Ma, C.; Wang, Y.; Chen, Y.; Zhu, D. 15-Lipoxygenase-1/15-hydroxyeicosatetraenoic acid promotes hepatocellular cancer cells growth through protein kinase B and heat shock protein 90 complex activation. Int. J. Biochem. Cell Biol. 2013, 45, 1031–1041. [Google Scholar] [CrossRef]
- Steele, V.E.; Holmes, C.A.; Hawk, E.T.; Kopelovich, L.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C.; Kelloff, G.J. Lipoxygenase inhibitors as potential cancer chemopreventives. Cancer Epidemiol. Prev. Biomark. 1999, 8, 467–483. [Google Scholar]
- Orafaie, A.; Matin, M.M.; Sadeghian, H. The importance of 15-lipoxygenase inhibitors in cancer treatment. Cancer Metastasis Rev. 2018, 37, 397–408. [Google Scholar] [CrossRef]
- Gilbert, N.C.; Gerstmeier, J.; Schexnaydre, E.E.; Börner, F.; Garscha, U.; Neau, D.B.; Werz, O.; Newcomer, M.E. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat. Chem. Biol. 2020, 16, 783–790. [Google Scholar] [CrossRef]
- Enomoto, M.; Kuwahara, S. Enantioselective Synthesis and Stereochemical Revision of Communiols A–C, Antibacterial 2, 4-Disubstituted Tetrahydrofurans from the Coprophilous Fungus Podospora communis. Biosci. Biotechnol. Biochem. 2008, 72, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Takada, N.; Watanabe, R.; Suenaga, K.; Yamada, K.; Uemura, D. Isolation and structures of hedaols A, B, and C, new bisnorditerpenes from a Japanese brown alga. J. Nat. Prod. 2001, 64, 653–655. [Google Scholar] [CrossRef]
- Yoon, W.-J.; Heo, S.-J.; Han, S.-C.; Lee, H.-J.; Kang, G.-J.; Kang, H.-K.; Hyun, J.-W.; Koh, Y.-S.; Yoo, E.-S. Anti-inflammatory effect of sargachromanol G isolated from Sargassum siliquastrum in RAW 264.7 cells. Arch. Pharmacal. Res. 2012, 35, 1421–1430. [Google Scholar] [CrossRef]
- Gerasimenko, N.; Logvinov, S. Seasonal composition of lipids, fatty acids pigments in the brown alga Sargassum pallidum: The potential for health. Open J. Mar. Sci. 2016, 6, 498. [Google Scholar] [CrossRef]
- Iwashima, M.; Mori, J.; Ting, X.; Matsunaga, T.; Hayashi, K.; Shinoda, D.; Saito, H.; Sankawa, U.; Hayashi, T. Antioxidant and antiviral activities of plastoquinones from the brown alga Sargassum micracanthum, and a new chromene derivative converted from the plastoquinones. Biol. Pharm. Bull. 2005, 28, 374–377. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.-F.; Yi, Y.-H.; Yao, X.-S.; Xu, Q.-Z.; Zhang, S.-Y.; Lin, H.-W. Bioactive steroids from the brown alga Sargassum carpophyllum. J. Asian Nat. Prod. Res. 2002, 4, 95–101. [Google Scholar] [CrossRef]
- Ayyad, S.-E.N.; Sowellim, S.Z.; El-Hosini, M.S.; Abo-Atia, A. The structural determination of a new steroidal metabolite from the brown alga Sargassum asperifolium. Zeitschrift Naturforschung C 2003, 58, 333–336. [Google Scholar] [CrossRef]
- Jung, M.; Jang, K.H.; Kim, B.; Lee, B.H.; Choi, B.W.; Oh, K.-B.; Shin, J. Meroditerpenoids from the brown alga Sargassum siliquastrum. J. Nat. Prod. 2008, 71, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Park, K.E.; Nam, T.J. Isolation of a new chromene from the brown alga Sargassum thunbergii. Bull. Korean Chem. Soc. 2007, 28, 1831. [Google Scholar]
- Cui, Z.; Li, Y.-S.; Liu, H.-B.; Yuan, D.; Lu, B.-R. Sulfoglycolipid from the marine brown alga Sargassum Hemiphyllum. J. Asian Nat. Prod. Res. 2001, 3, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, P.; Kunhi, A.; Divakar, S. New metabolic pathway for o-cresol degradation by Pseudomonas sp. CP4 as evidenced by 1H NMR spectroscopic studies. World J. Microbiol. Biotechnol. 2001, 17, 371–377. [Google Scholar] [CrossRef]
- Bogan, L.E., Jr.; Wolk, S.K. Synthesis of and assignment of carbon-13 NMR resonances to m-cresol novolak dimers. Macromolecules 1992, 25, 161–165. [Google Scholar] [CrossRef]
- Tawfike, A.; Attia, E.Z.; Desoukey, S.Y.; Hajjar, D.; Makki, A.A.; Schupp, P.J.; Edrada-Ebel, R.; Abdelmohsen, U.R. New bioactive metabolites from the elicited marine sponge-derived bacterium Actinokineospora spheciospongiae sp. nov. AMB Express 2019, 9, 12. [Google Scholar] [CrossRef]
- Wu, W.; Hasumi, K.; Peng, H.; Hu, X.; Wang, X.; Bao, B. Fibrinolytic compounds isolated from a brown alga, Sargassum fulvellum. Mar. Drugs 2009, 7, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stappen, I.; Höfinghoff, J.; Buchbauer, G.; Wolschann, P. Structure-activity relationships of sandalwood odorants: Synthesis of a new campholene derivative. Nat. Prod. Commun. 2010, 5, 1934578×1000500902. [Google Scholar] [CrossRef] [Green Version]
- Francesconi, K.A.; Edmonds, J.S.; Stick, R.V.; Skelton, B.W.; White, A.H. Arsenic-containing ribosides from the brown alga Sargassum lacerifolium: X-ray molecular structure of 2-amino-3-[5′-deoxy-5′-(dimethylarsinoyl) ribosyloxy] propane-1-sulphonic acid. J. Chem. Soc. Perkin Trans. 1 1991, 2707–2716. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.-M.; Deng, J.-J.; Yuan, G.-J.; Yang, F.; Guo, H.-T.; Xiang, M.; Ge, W.; Wu, Y.-G. 5-Lipoxygenase contributes to the progression of hepatocellular carcinoma. Mol. Med. Rep. 2011, 4, 1195–1200. [Google Scholar] [CrossRef] [Green Version]
- Hussey, H.; Tisdale, M. Inhibition of tumour growth by lipoxygenase inhibitors. Br. J. Cancer 1996, 74, 683–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avis, I.; Hong, S.H.; Martínez, A.; Moody, T.; Choi, Y.H.; Trepel, J.; Das, R.; Jett, M.; Mulshine, J.L. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001, 15, 2007–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishayee, K.; Khuda-Bukhsh, A.R. 5-lipoxygenase antagonist therapy: A new approach towards targeted cancer chemotherapy. Acta Biochim. Biophys. Sin. 2013, 45, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC 50-to-K i: A web-based tool for converting IC 50 to K i values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, M.R.F.; Stahl, E. Thin-Layer Chromatography: A Laboratory Handbook; Springer Science and Business Media: Berlin/Heidelberg, Germany, 2013; Available online: https://books.google.com.eg/books?hl=en&lr=&id=06v7CAAAQBAJ&oi=fnd&pg=PA1&dq=32.%09Ashworth,+M.R.F.%3B+Stahl,+E.+Thin-layer+chromatography:+a+laboratory+handbook%3B+Springer+Science+and+Business+Media:+2013&ots=UUi3D7RewN&sig=bkWmSFTqhRRmJ5lXwXr0rm7YcQA&redir_esc=y#v=onepage&q=32.%09Ashworth%2C%20M.R.F.%3B%20Stahl%2C%20E.%20Thin-layer%20chromatography%3A%20a%20laboratory%20handbook%3B%20Springer%20Science%20and%20Business%20Media%3A%202013&f=false (accessed on 9 November 2020).
- Abdelmohsen, U.R.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication strategies for targeted isolation of new antitrypanosomal actinosporins A and B from a marine sponge associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220–1244. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Yamamoto, S. Mammalian lipoxygenases: Molecular structures and functions. Biochim. Biophys. Acta Lipids Lipid Metab. 1992, 1128, 117–131. [Google Scholar] [CrossRef]
- Mitra, S.; Bartlett, S.G.; Newcomer, M.E. Identification of the substrate access portal of 5-lipoxygenase. Biochemistry 2015, 54, 6333–6342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. Available online: https://www.csb.pitt.edu/BBSI/2007/lectures/VMD.pdf (accessed on 15 January 2021). [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Jiang, W.; Lee, H.S.; Roux, B.t.; Im, W. CHARMM-GUI Ligand Binder for Absolute Binding Free Energy Calculations and Its Application. J. Chem. Inf. Model. 2013, 53, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, J.; Skalic, M.; Martinez-Rosell, G.; De Fabritiis, G. K deep: Protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]


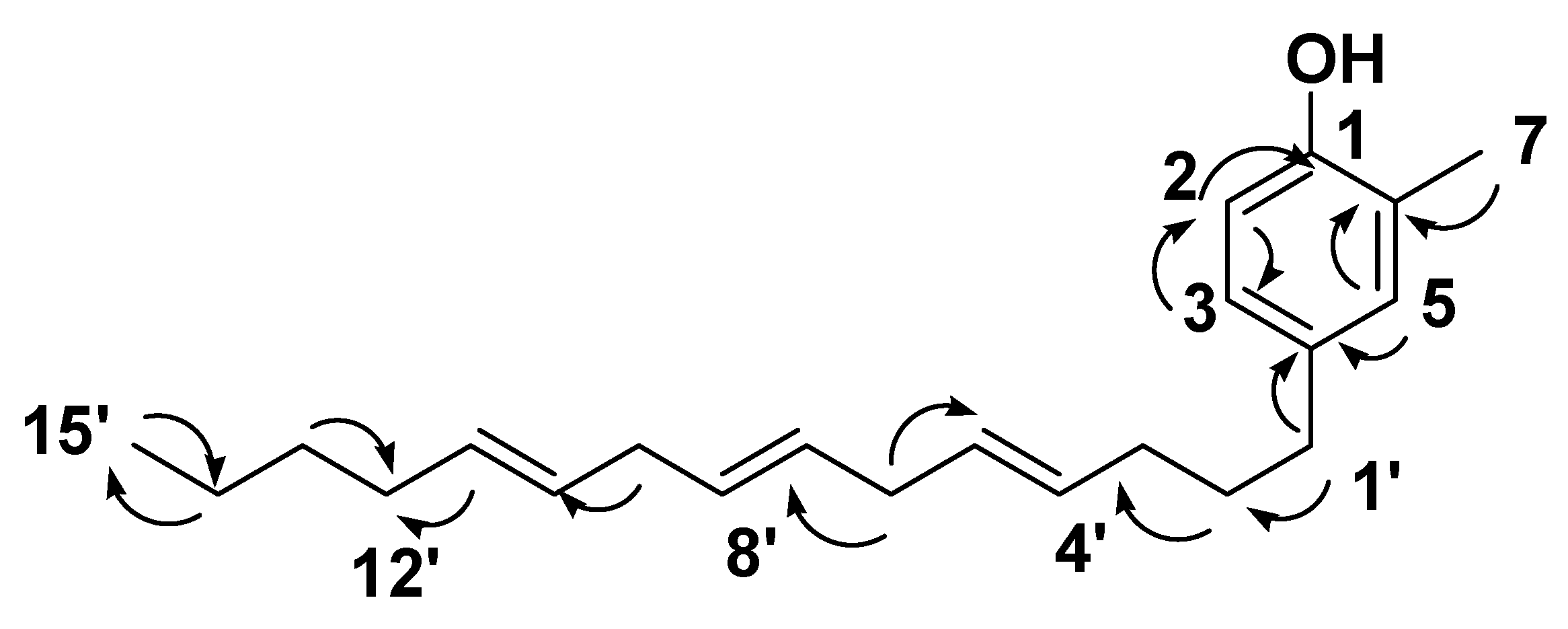
 ) correlations of compound 12.
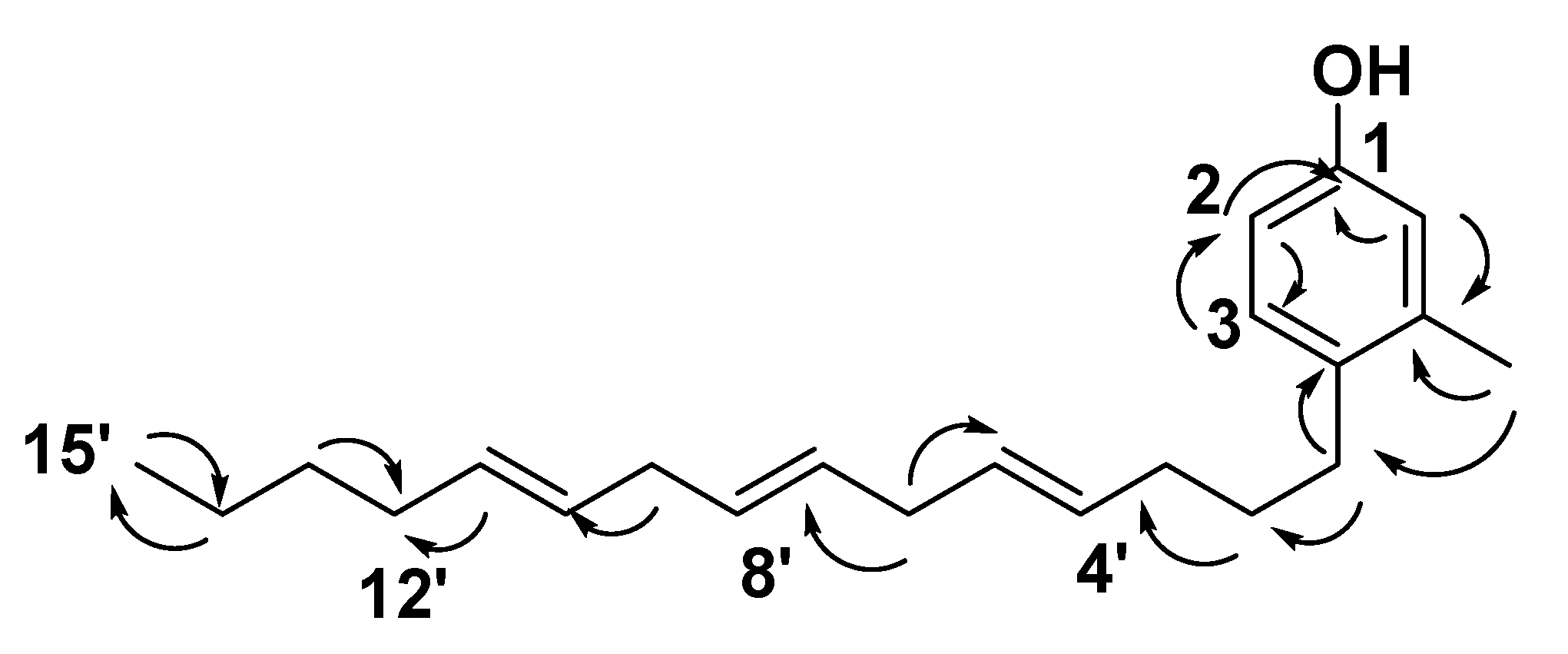
) correlations of compound 12. ) correlations of compound 13.
) correlations of compound 13.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 12 | 13 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 153.8, qC | 153.8, qC | ||
| 2 | 123.1, CH | 7.13, d (8.0) | 123.1, CH | 7.13, d (8.0) |
| 3 | 123.6, CH | 6.98, d (8.0) | 123.6, CH | 6.98, d (8.0) |
| 4 | 134.5, qC | 134.5, qC | ||
| 5 | 116.0, CH | 6.68, s | 140.6, qC | |
| 6 | 140.6, qC | 116.0, CH | 6.68, s | |
| 7 | 29.8, CH3 | 1.34, s | 31.9, CH3 | 1.23, s |
| 1′ | 33.4, CH2 | 2.26, m | 33.7, CH2 | 2.26, m |
| 2′ | 20.3, CH2 | 2.03, overlapped | 20.5, CH2 | 2.03, overlapped |
| 3′ | 27.1, CH2 | 2.01, overlapped | 27.1, CH2 | 2.01, overlapped |
| 4′ | 127.9, CH | 5.31–5.35, m | 127.9, CH | 5.31–5.35, m |
| 5′ | 128.8, CH | 5.31–5.35, m | 128.8, CH | 5.31–5.35, m |
| 6′ | 25.5, CH2 | 2.78, overlapped | 25.5, CH2 | 2.78, overlapped |
| 7′ | 128.0, CH | 5.31–5.35, m | 128.1, CH | 5.31–5.35, m |
| 8′ | 128.3, CH | 5.31–5.35, m | 128.4, CH | 5.31–5.35, m |
| 9′ | 25.6, CH2 | 2.78, overlapped | 25.6, CH2 | 2.78, overlapped |
| 10′ | 128.2, CH | 5.31–5.35, m | 128.3, CH | 5.31–5.35, m |
| 11′ | 129.4, CH | 5.31–5.35, m | 129.4, CH | 5.31–5.35, m |
| 12′ | 24.9, CH2 | 1.52, overlapped | 24.9, CH2 | 1.52, overlapped |
| 13′ | 28.9, CH2 | 1.24, overlapped | 28.9, CH2 | 1.24, overlapped |
| 14′ | 22.4, CH2 | 1.25, overlapped | 22.4, CH2 | 1.25, overlapped |
| 15′ | 14.5, CH3 | 0.89, t (6.6) | 14.3, CH3 | 0.85, t (6.6) |
| IC50 (µM) | |||
|---|---|---|---|
| Code | HepG2 | MCF-7 | Caco-2 |
| 12 | 14.5 ± 0.8 * | 17.6 ± 0.9 * | 18.2 ± 0.7 * |
| 13 | 13.1 ± 1.1 * | 12.7 ± 1.3 * | 11.2 ± 0.6 * |
| 14 | >50 | >50 | >50 |
| 15 | >50 | >50 | >50 |
| 16 | >50 | >50 | >50 |
| 17 | >50 | >50 | >50 |
| 18 | >50 | >50 | >50 |
| 19 | 18.5 ± 1.4 * | 21.6 ± 1.3* | 15.7 ± 0.9 * |
| 20 | >50 | >50 | >50 |
| 21 | >50 | >50 | >50 |
| Doxorubicin | 4.2 ± 0.3 | 3.8 ± 0.2 | 3.4 ± 0.1 |
| Compound | ΔGVina * | ΔGFEP ** | ΔGKDEEP *** | Ki# | IC50 # | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5-LOX | 15-LOX | 5-LOX | 15-LOX | 5-LOX | 15-LOX | 5-LOX | 15-LOX | 5-LOX | 15-LOX | |
| 12 | −9.3 | −5.1 | −8.1 | −4.4 | −7.7 | −4.6 | 0.9 ± 0.1 | 17.4 ± 0.2 | 1.6 ± 0.3 | 25.3 ± 0.4 |
| 13 | −8.9 | −5.5 | −8.0 | −4.7 | −7.5 | −4.5 | 0.7 ± 0.2 | 14.3 ± 0.4 | 1.3 ± 0.1 | 23.6 ± 0.3 |
| 19 | −9.1 | −7.7 | −7.9 | −7.1 | −7.6 | −7.2 | 1.4 ± 0.2 | 4.2 ± 0.1 | 2.1 ± 0.4 | 6.7 ± 0.3 |
| NDGA ## | −7.2 | −6.9 | −7.0 | −6.5 | −6.8 | −6.5 | 6.9 ± 0.1 | 6.1 ± 0.2 | 8.8 ± 0.3 | 9.5 ± 0.5 |
| AA ## | −7.6 | −7.0 | −6.2 | −6.4 | −7.1 | −6.3 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzarea, S.I.; Elmaidomy, A.H.; Saber, H.; Musa, A.; Al-Sanea, M.M.; Mostafa, E.M.; Hendawy, O.M.; Youssif, K.A.; Alanazi, A.S.; Alharbi, M.; et al. Potential Anticancer Lipoxygenase Inhibitors from the Red Sea-Derived Brown Algae Sargassum cinereum: An In-Silico-Supported In-Vitro Study. Antibiotics 2021, 10, 416. https://doi.org/10.3390/antibiotics10040416
Alzarea SI, Elmaidomy AH, Saber H, Musa A, Al-Sanea MM, Mostafa EM, Hendawy OM, Youssif KA, Alanazi AS, Alharbi M, et al. Potential Anticancer Lipoxygenase Inhibitors from the Red Sea-Derived Brown Algae Sargassum cinereum: An In-Silico-Supported In-Vitro Study. Antibiotics. 2021; 10(4):416. https://doi.org/10.3390/antibiotics10040416
Chicago/Turabian StyleAlzarea, Sami I., Abeer H. Elmaidomy, Hani Saber, Arafa Musa, Mohammad M. Al-Sanea, Ehab M. Mostafa, Omnia Magdy Hendawy, Khayrya A. Youssif, Abdullah S. Alanazi, Metab Alharbi, and et al. 2021. "Potential Anticancer Lipoxygenase Inhibitors from the Red Sea-Derived Brown Algae Sargassum cinereum: An In-Silico-Supported In-Vitro Study" Antibiotics 10, no. 4: 416. https://doi.org/10.3390/antibiotics10040416
APA StyleAlzarea, S. I., Elmaidomy, A. H., Saber, H., Musa, A., Al-Sanea, M. M., Mostafa, E. M., Hendawy, O. M., Youssif, K. A., Alanazi, A. S., Alharbi, M., Sayed, A. M., & Abdelmohsen, U. R. (2021). Potential Anticancer Lipoxygenase Inhibitors from the Red Sea-Derived Brown Algae Sargassum cinereum: An In-Silico-Supported In-Vitro Study. Antibiotics, 10(4), 416. https://doi.org/10.3390/antibiotics10040416