
High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15



Abstract
1. Introduction
2. Results and Discussion
2.1. CASTp3.0 Protein Topography Probe Result
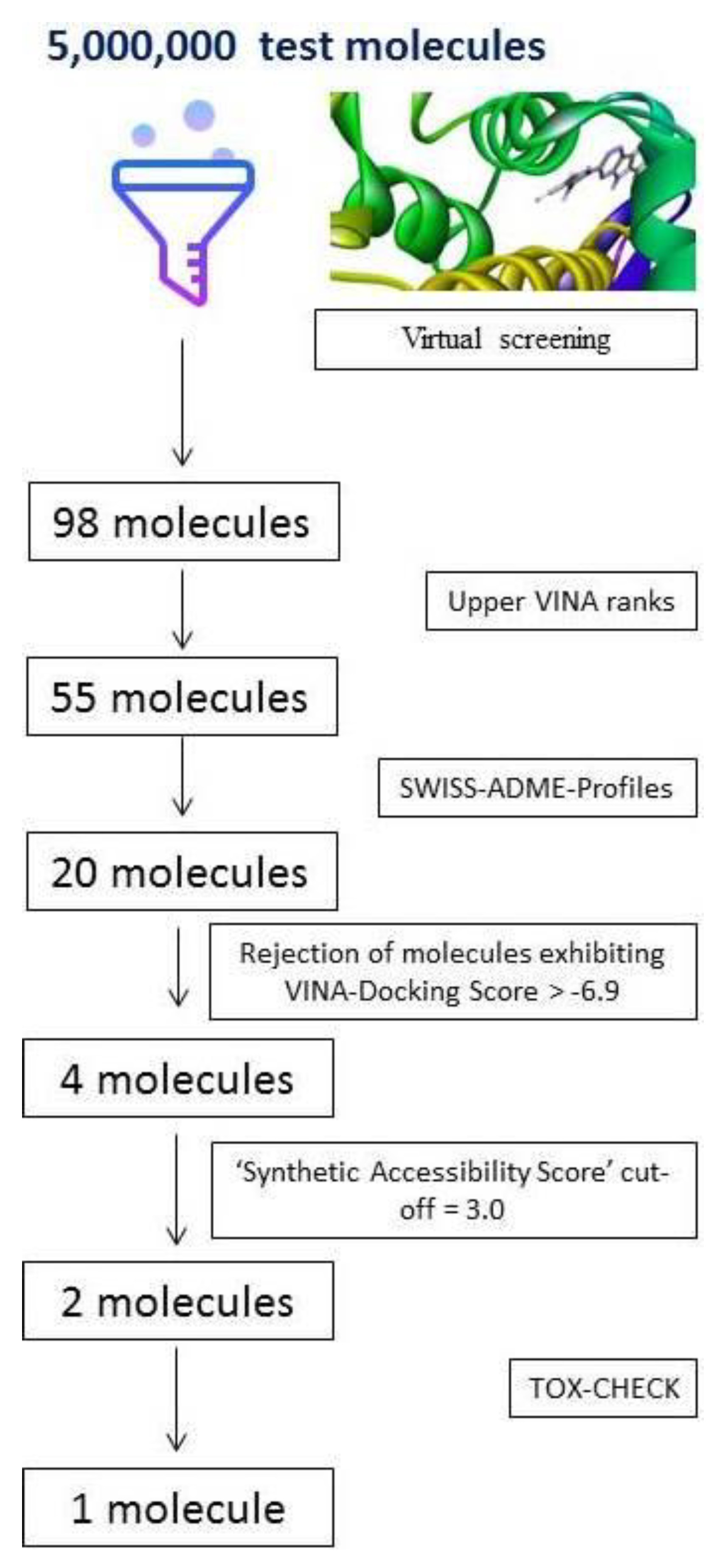
2.2. Outcome of the Screening Funnel
2.3. VINA Ranks and ADME-Analyses Results
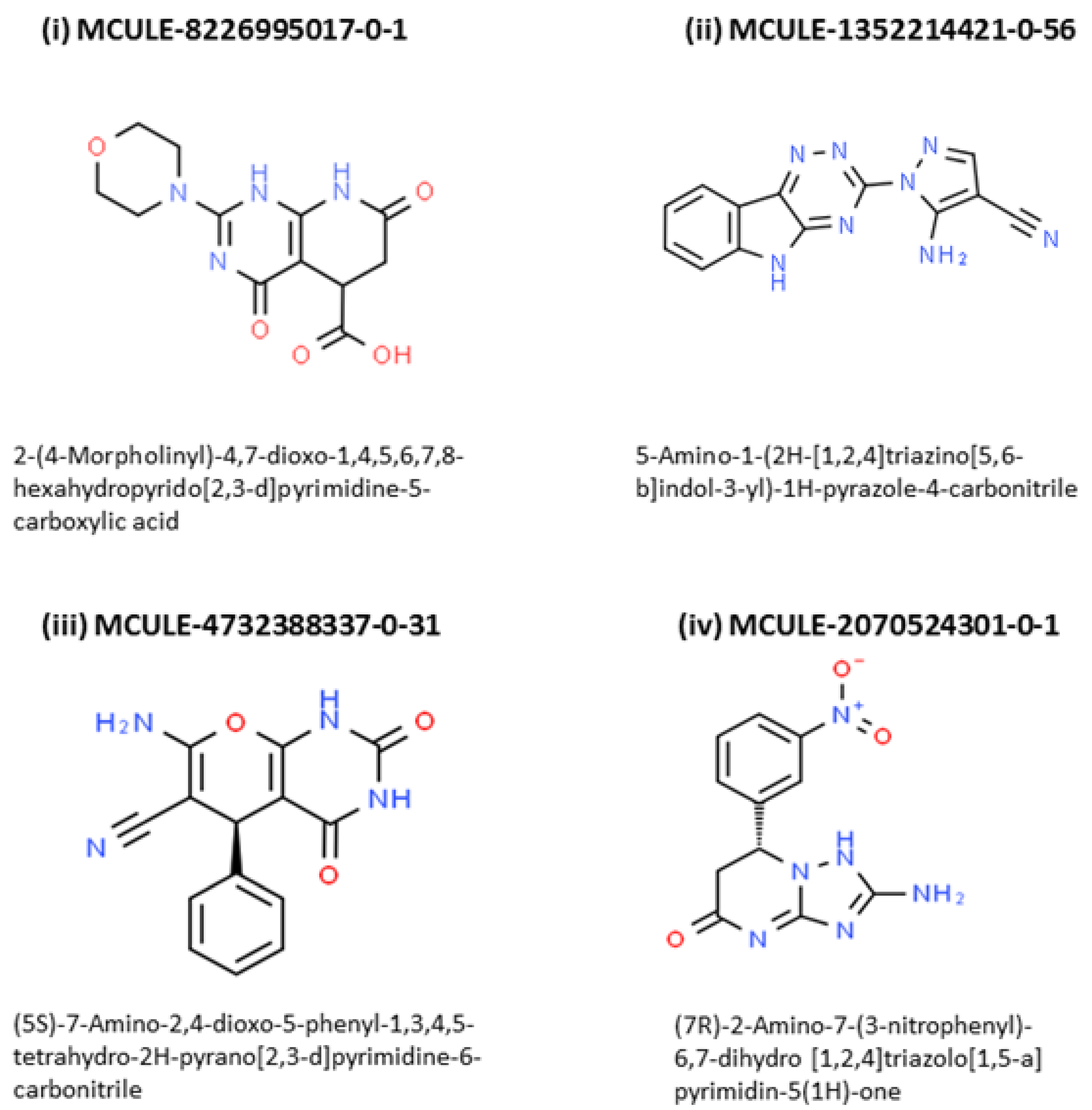
2.4. Outcome of Docking Score, ‘Zero RO5 Violation’, ‘Synthetic Accessibility Score’ and TOX-CHECK
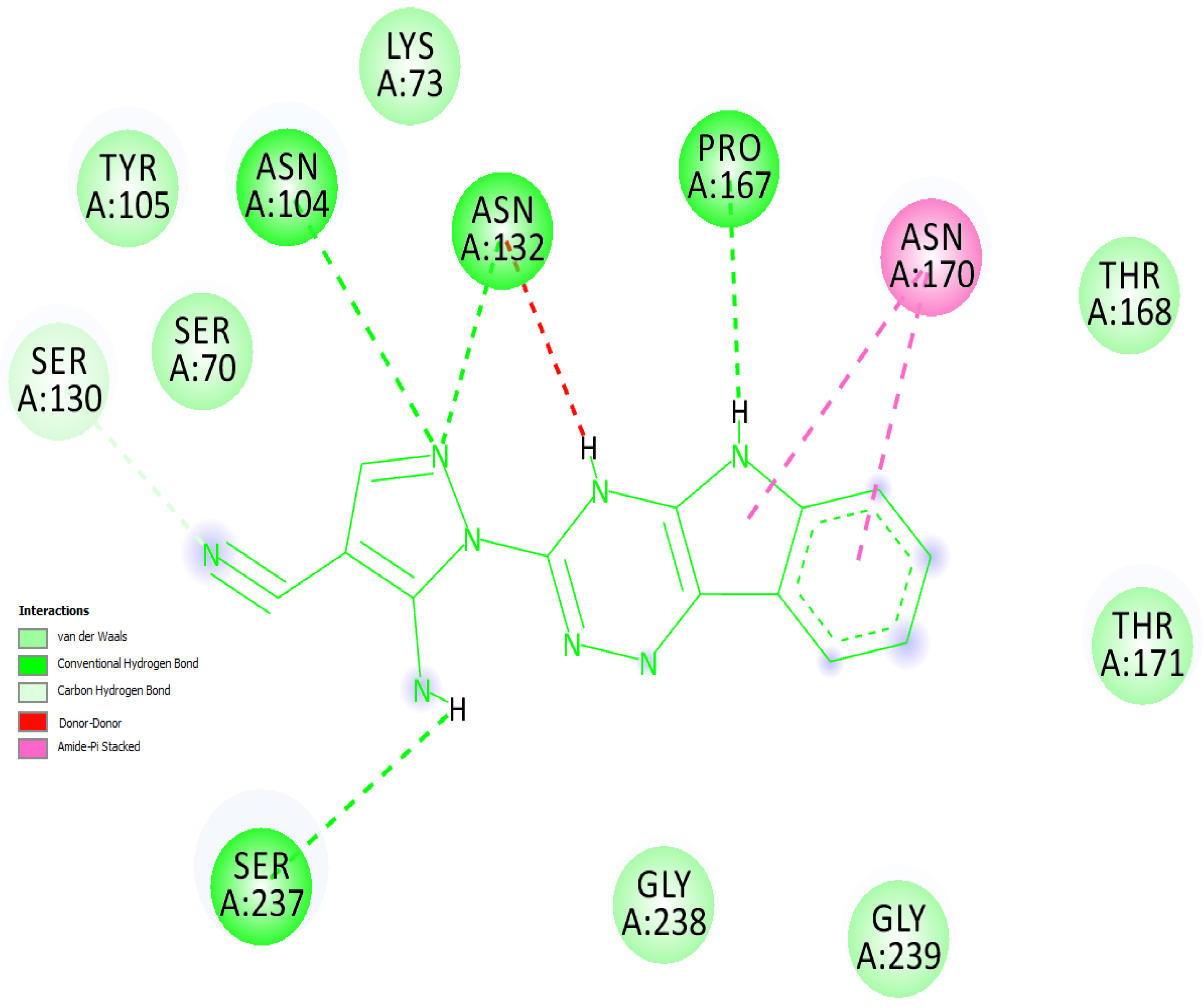
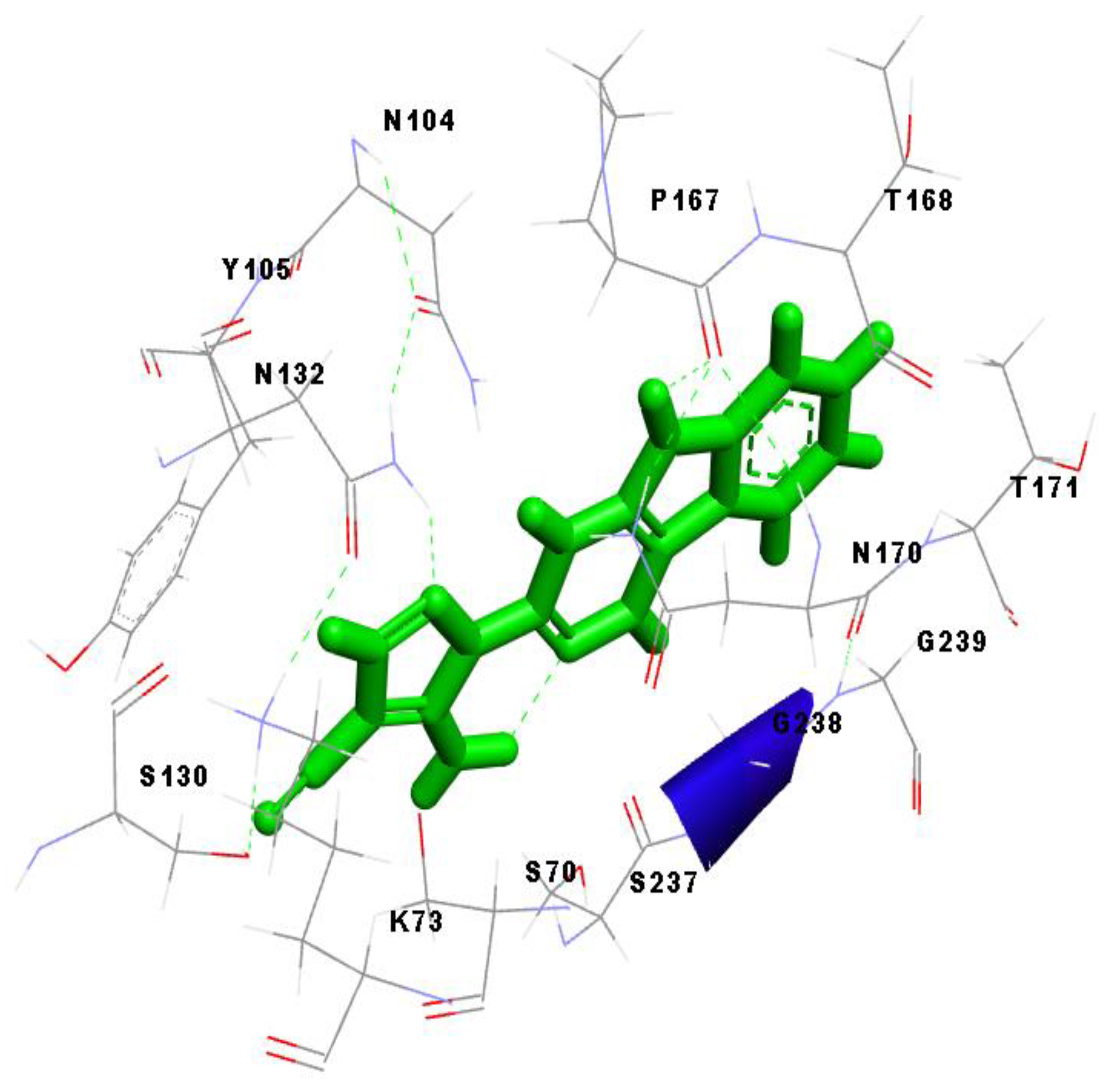
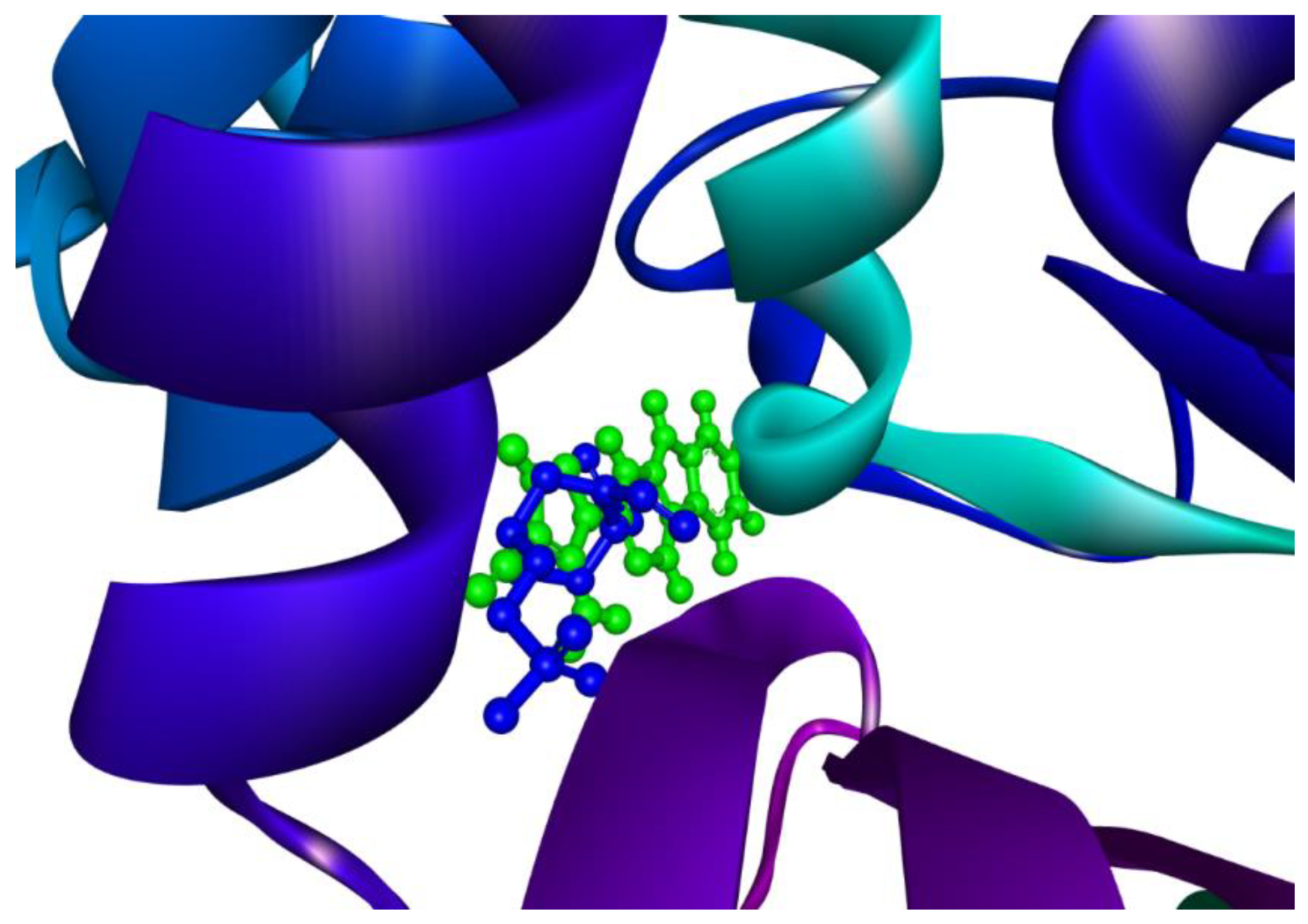
2.5. Molecular Overlay and Binding Interactions
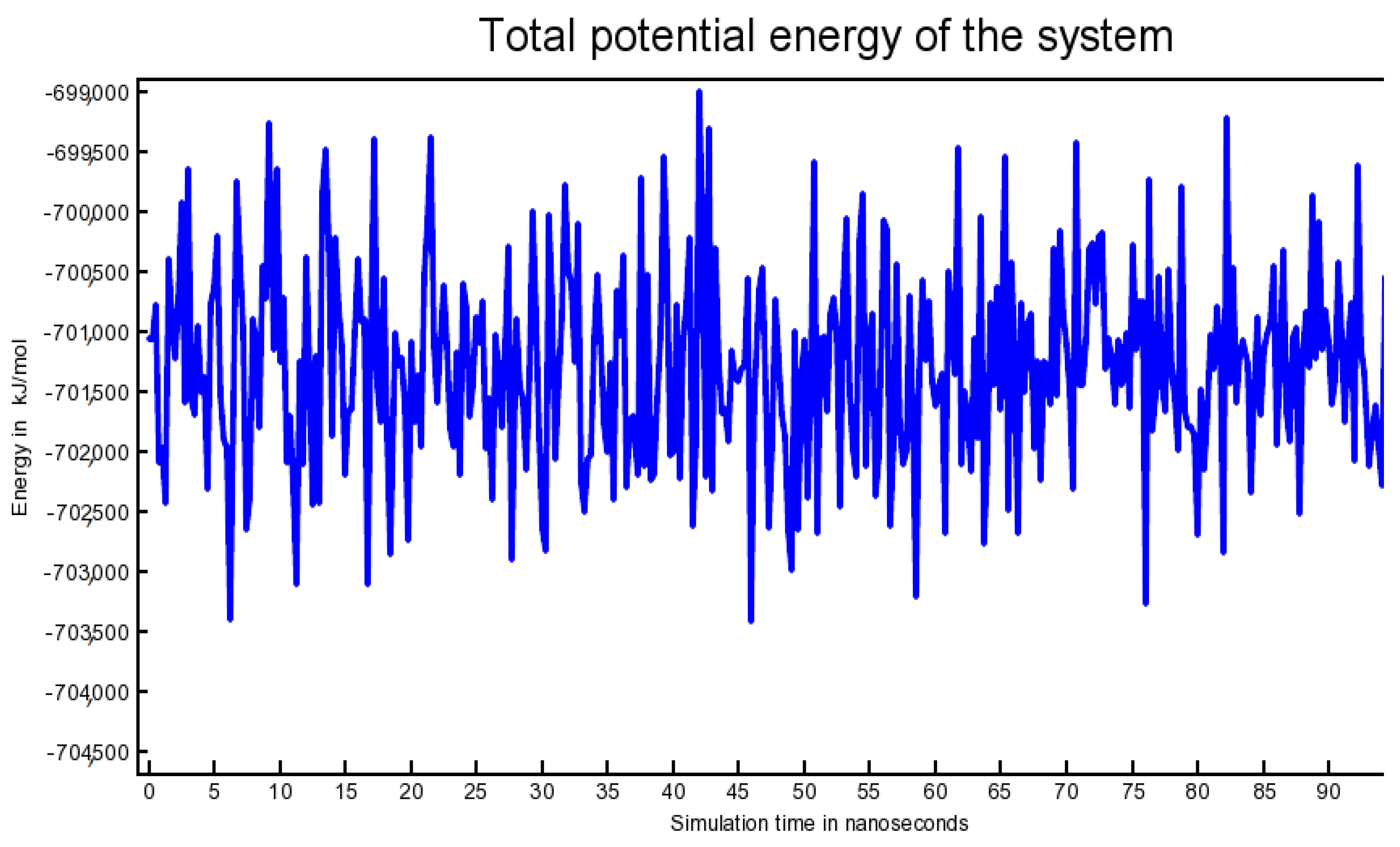
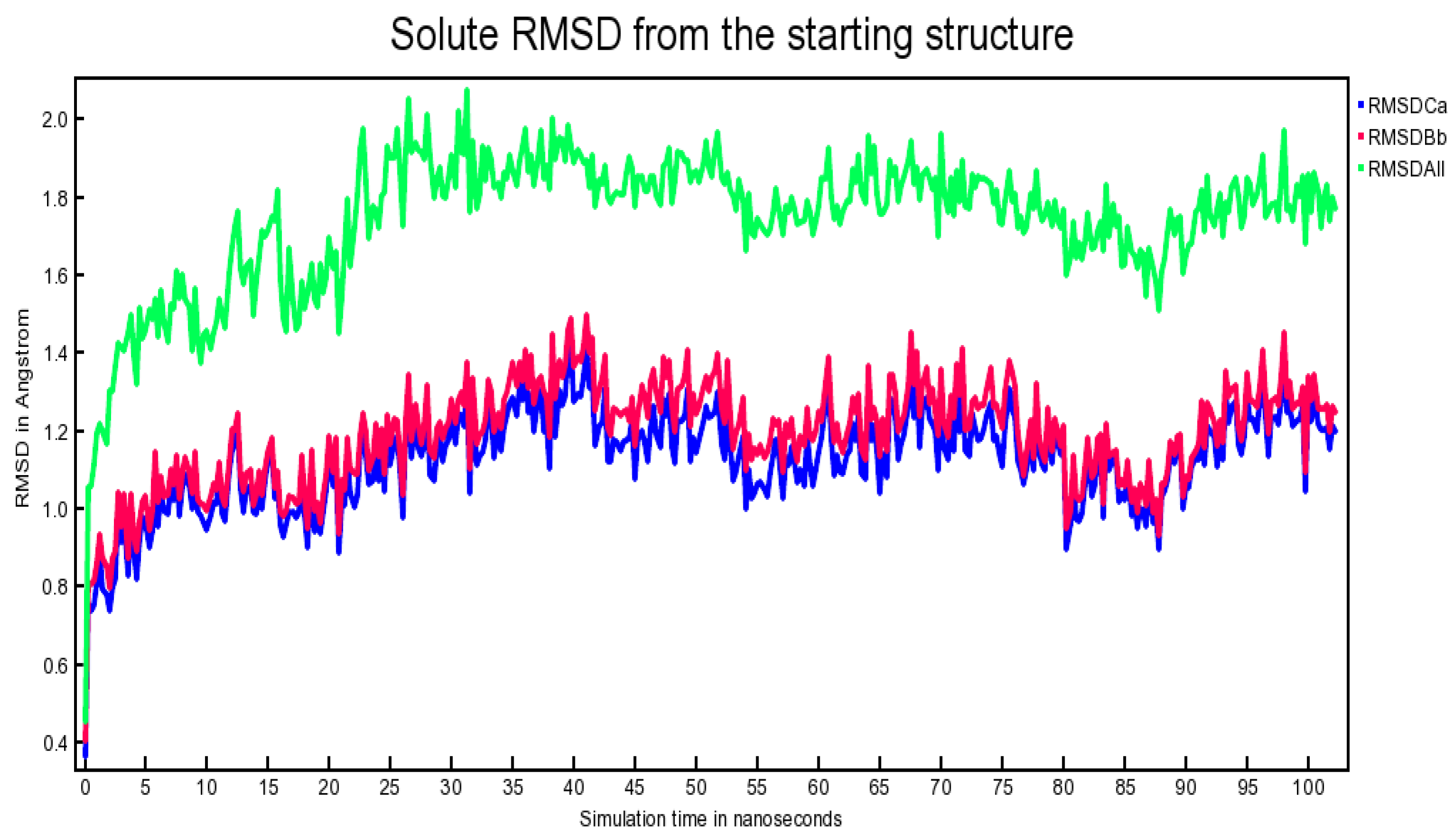
2.6. Outcome of Molecular Dynamics Simulation (102.25 ns)
3. Materials and Methods
3.1. Examination of the Binding Site by CASTp3.0
3.2. Structure Based Virtual Screening
Molecular Docking
3.3. VINA Ranking and ADME-Analyses
3.4. Docking Score, ‘Zero RO5 Violation’, ‘Synthetic Accessibility Score’ and TOX-CHECK
3.5. Binding Interactions of the Complex Involving the ‘Top CTX-M-15 Inhibitor’ & ‘Molecular Overlay’
3.6. Molecular Dynamics Simulation (102.25 Nanoseconds)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shakil, S.; Khan, R.; Zarrilli, R.; Khan, A.U. Aminoglycosides versus bacteria—A description of the action, resistance mecha-nism, and nosocomial battleground. J. Biomed. Sci. 2008, 15, 5–14. [Google Scholar] [CrossRef]
- Shakil, S.; Khan, A.U. Infected foot ulcers in male and female diabetic patients: A clinico-bioinformative study. Ann. Clin. Microbiol. Antimicrob. 2010, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Ajlan, A.M.; Shakil, S.; Jiman-Fatani, A.A.; Almasaudi, S.B.; Farman, M.; Baazeem, Z.M.; Baabdullah, R.; Alawi, M.; Al-Abdullah, N.; et al. Molecular characterization, antimicrobial resistance and clini-co-bioinformatics approaches to address the problem of extended-spectrum β-lactamase-producing Escherichia coli in west-ern Saudi Arabia. Sci. Rep. 2018, 8, 14847. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Ambler, R.P.; Coulson, A.F.W.; Frère, J.M.; Ghuysen, J.M.; Joris, B.; Forsman, M.; Levesque, R.C.; Tiraby, G.; Waley, S.G. A standard numbering scheme for the class A β-lactamases. Biochem. J. 1991, 276, 269–270. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2009, 54, 969–976. [Google Scholar] [CrossRef]
- Peirano, G.; Pitout, J.D.D. Extended-Spectrum β-Lactamase-Producing Enterobacteriaceae: Update on Molecular Epidemiology and Treatment Options. Drugs 2019, 79, 1529–1541. [Google Scholar] [CrossRef]
- Shakil, S.; Khan, A.U. Interaction of CTX-M-15 enzyme with cefotaxime: A molecular modelling and docking study. Bioinformation 2010, 4, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Fatima, J.; Shakil, S.; Rizvi, S.M.D.; Kamal, M.A. Antibiotic resistance and extended spectrum beta-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 2015, 22, 90–101. [Google Scholar] [CrossRef]
- Shaikh, S.; Rizvi, S.M.D.; Shakil, S.; Hussain, T.; Alshammari, T.M.; Ahmad, W.; Tabrez, S.; Al-Qahtani, M.H.; Abuzenadah, A.M. Syn-thesis and Characterization of Cefotaxime Conjugated Gold Nanoparticles and Their Use to Target Drug-Resistant CTX-M-Producing Bacterial Pathogens. J. Cell. Biochem. 2017, 118, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Bevan, E.R.; Jones, A.M.; Hawkey, P.M. Global epidemiology of CTX-M β-lactamases: Temporal and geographical shifts in genotype. J. Antimicrob. Chemother. 2017, 72, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, D.; Aung, M.S.; Carmona, Y.; González, M.K.; Pereda, N.; Hidalgo, M.; Rivero, M.; Zayas, A.; Del Campo, R.; Urushibara, N.; et al. High Prevalence of CTX-M Type Extended-Spectrum Beta-Lactamase Genes and Detection of NDM-1 Carbapenemase Gene in Extraintestinal Pathogenic Escherichia coli in Cuba. Pathogens 2020, 9, 65. [Google Scholar] [CrossRef]
- Buynak, J.D. β-Lactamase inhibitors: A review of the patent literature (2010–2013). Expert Opin. Ther. Pat. 2013, 23, 1469–1481. [Google Scholar] [CrossRef] [PubMed]
- McGeary, R.P.; Tan, D.T.C.; Selleck, C.; Monteiro Pedroso, M.; Sidjabat, H.E.; Schenk, G. Structure-activity relationship study and optimisation of 2-aminopyrrole-1-benzyl-4,5-diphenyl-1H-pyrrole-3-carbonitrile as a broad spectrum metallo-β-lactamase in-hibitor. Eur. J. Med. Chem. 2017, 137, 351–364. [Google Scholar] [CrossRef]
- Malani, M.H.; Dholakiya, B.Z. Synthesis, characterization and in vitro screening on bacterial, fungal and malarial strain of piprazinyl cyano biphenyl based compounds. Bioorganic Chem. 2013, 51, 16–23. [Google Scholar] [CrossRef]
- Al-Abdullah, E.S.; Al-Obaid, A.-R.M.; Al-Deeb, O.A.; Habib, E.E.; El-Emam, A.A. Synthesis of novel 6-phenyl-2,4-disubstituted pyrimidine-5-carbonitriles as potential antimicrobial agents. Eur. J. Med. Chem. 2011, 46, 4642–4647. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M. A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar] [PubMed]
- Shakil, S.; Baig, M.H.; Tabrez, S.; Rizvi, S.M.D.; Zaidi, S.K.; Ashraf, G.M.; Ansari, S.A.; Khan, A.A.P.; Al-Qahtani, M.H.; Abuzenadah, A.M.; et al. Molecular and enzoinformatics perspectives of targeting Polo-like kinase 1 in cancer therapy. Semin. Cancer Biol. 2019, 56, 47–55. [Google Scholar] [CrossRef]
- Shakil, S. Molecular interaction of anti-cancer ligands with human brain acetylcholinesterase. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Auto-mated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient opti-mization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Copeland, R.A. Conformational adaptation in drug–target interactions and residence time. Futur. Med. Chem. 2011, 3, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Edelsbrunner, H.; Mucke, E.P. Three-Dimensional Alpha-Shapes. ACM Trans. Graph. 1994, 13, 43–72. [Google Scholar] [CrossRef]
- Zhou, W.; Yan, H. Alpha shape and Delaunay triangulation in studies of protein-related interactions. Briefings Bioinform. 2014, 15, 54–64. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef]
- Potemkin, V.; Grishina, M.; Potemkin, A. Internet Resources for Drug Discovery and Design. Curr. Top. Med. Chem. 2019, 18, 1955–1975. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.-B. Minimization by Random Search Techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | MCULE- 8226995017-0-1 | MCULE- 1352214421-0-56 | MCULE- 4732388337-0-31 | MCULE- 2070524301-0-1 |
|---|---|---|---|---|
| IUPAC Name | 2-(4-Morpholinyl) -4,7-dioxo-1,4,5,6,7,8- hexahydropyrido [2,3-d]pyrimidine -5-carboxylic acid | 5-Amino-1-(2H- [1,2,4]triazino [5,6-b]indol-3-yl) -1H-pyrazole -4-carbonitrile | (5S)-7-Amino -2,4-dioxo-5-phenyl -1,3,4,5-tetrahydro -2H-pyrano[2,3-d] pyrimidine-6-carbonitrile | (7R)-2-Amino-7 -(3-nitrophenyl)-6,7 -dihydro[1,2,4] triazolo[1,5-a]pyrimidin-5(1H)-one |
| Chemical Formula | C12H14N4O5 | C13H8N8 | C14H10N4O3 | C11H10N6O3 |
| Molecular Weight | 294.26 | 276.26 | 282.25 | 274.24 |
| XLOGP3 | −2.49 | 1.31 | 0.62 | 0.33 |
| RO5 violations | 0 | 0 | 0 | 0 |
| H-bond acceptors | 6 | 5 | 4 | 5 |
| H-bond donors | 3 | 2 | 3 | 2 |
| Rotatable bonds | 2 | 1 | 1 | 2 |
| Toplogical PSA (Ų) | 124.62 | 122.09 | 124.76 | 131.65 |
| Molar Refractivity | 77.26 | 75.43 | 73.11 | 74.13 |
| GI Absorption | Low | High | High | High |
| BBB-permeation | No | No | No | No |
| Log S (Ali) | 0.42 | −3.47 | −2.81 | −2.66 |
| Lipinski-filter | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose-filter | No; 1 violation: WLOGP < −0.4 | Yes | Yes | Yes |
| Veber-filter | Yes | Yes | Yes | Yes |
| Egan-filter | Yes | Yes | Yes | No; 1 violation: TPSA > 131.6 |
| Muegge-filter | No; 1 violation: XLOGP3 < −2 | Yes | Yes | Yes |
| PAINS-filter | 0 alert | 0 alert | 0 alert | 0 alert |
| Brenk-filter | 0 alert | 0 alert | 0 alert | 1 alert: nitro group |
| Lead-likeness | Yes | Yes | Yes | Yes |
| Synthetic accessibility | 3.36 | 2.81 | 3.65 | 2.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakil, S.; Rizvi, S.M.D.; Greig, N.H. High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15. Antibiotics 2021, 10, 474. https://doi.org/10.3390/antibiotics10050474
Shakil S, Rizvi SMD, Greig NH. High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15. Antibiotics. 2021; 10(5):474. https://doi.org/10.3390/antibiotics10050474
Chicago/Turabian StyleShakil, Shazi, Syed M. Danish Rizvi, and Nigel H. Greig. 2021. "High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15" Antibiotics 10, no. 5: 474. https://doi.org/10.3390/antibiotics10050474
APA StyleShakil, S., Rizvi, S. M. D., & Greig, N. H. (2021). High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15. Antibiotics, 10(5), 474. https://doi.org/10.3390/antibiotics10050474