
Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents

 , ,
, ,  , , ,
, , ,  , , , ,
, , , ,
Abstract
:1. Introduction
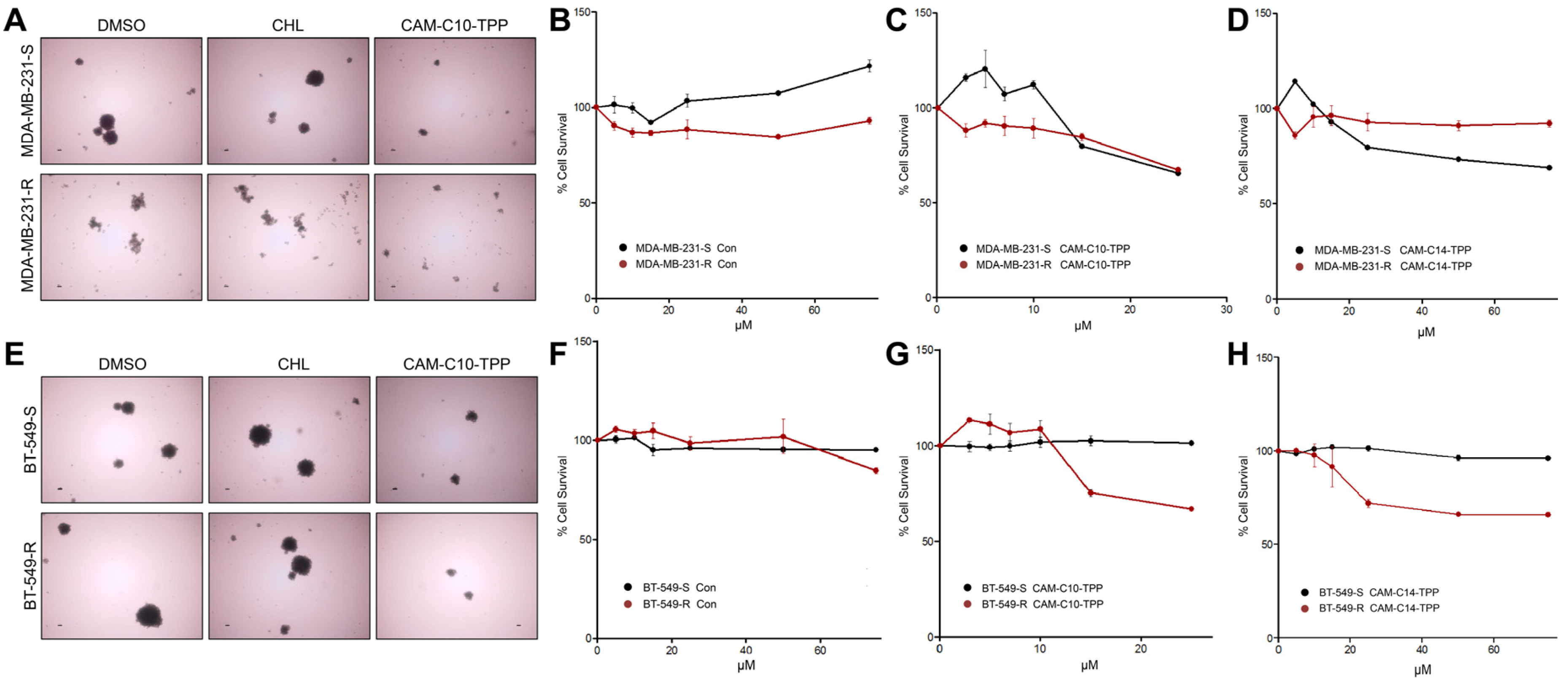
2. Results and Discussion
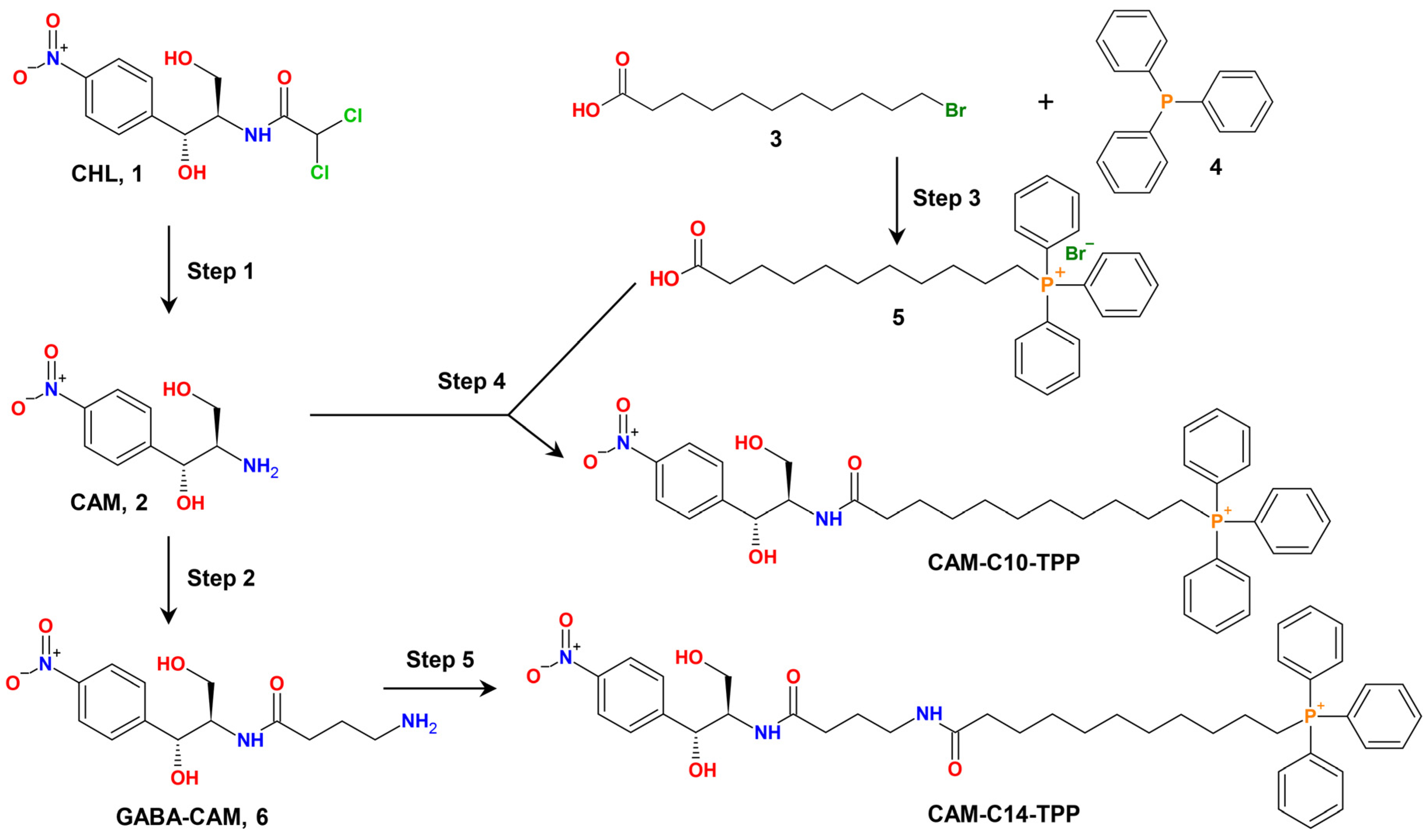
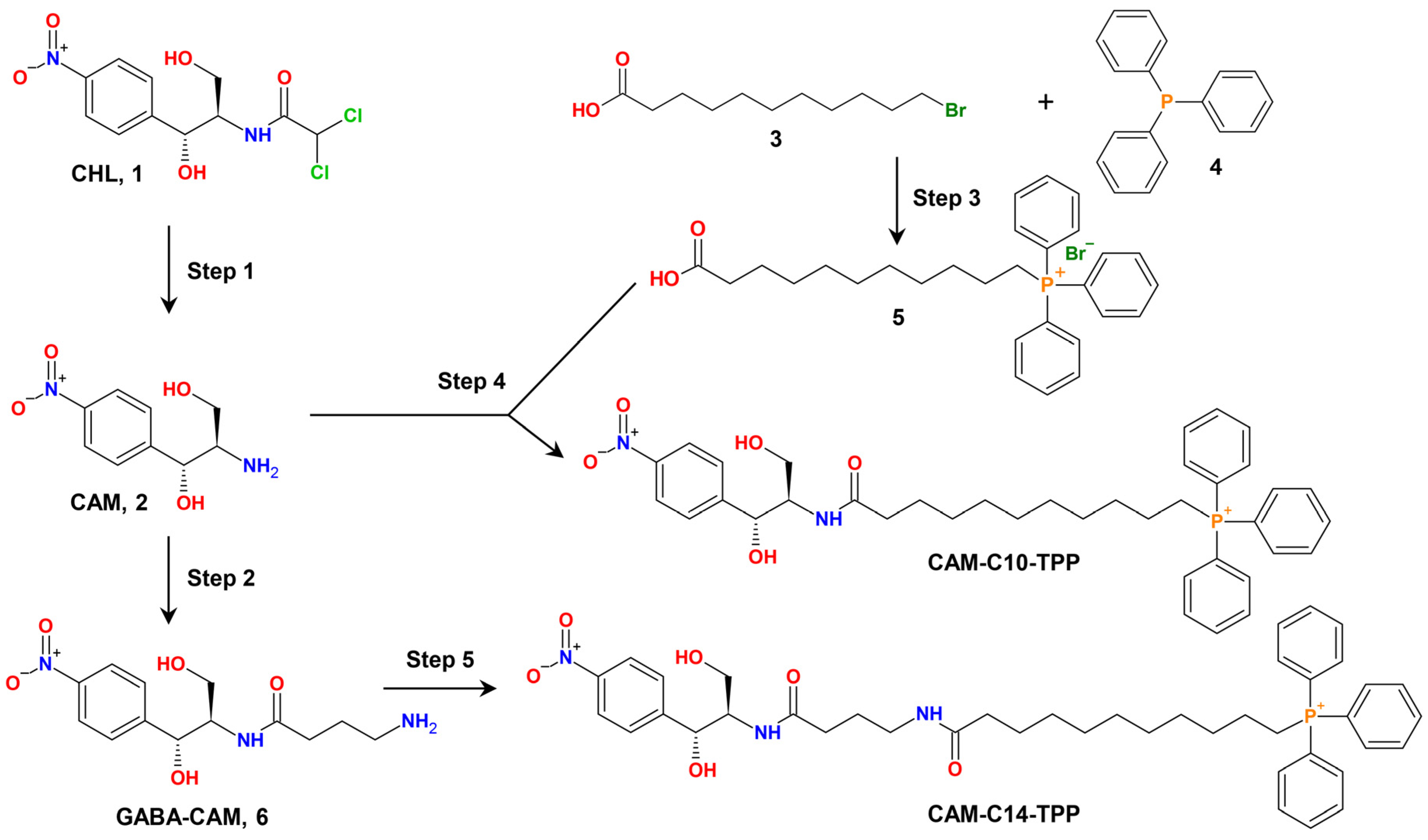
2.1. Synthesis of CAM-Cn-TPPs
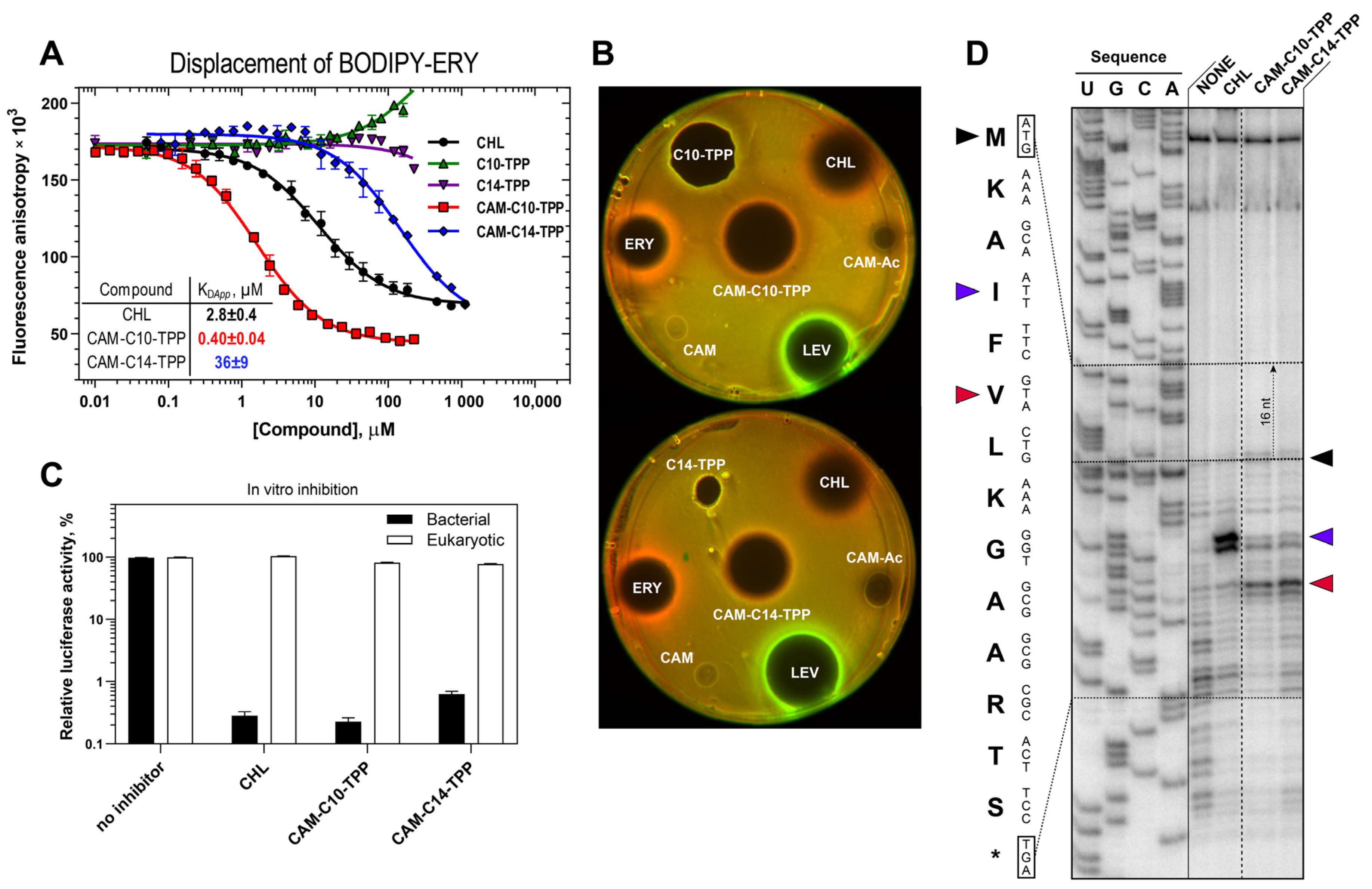
2.2. CAM-Cn-TPPs Bind to the Bacterial Ribosome with Different Affinities and Inhibit Protein Synthesis, Allowing the Formation of Short Peptides
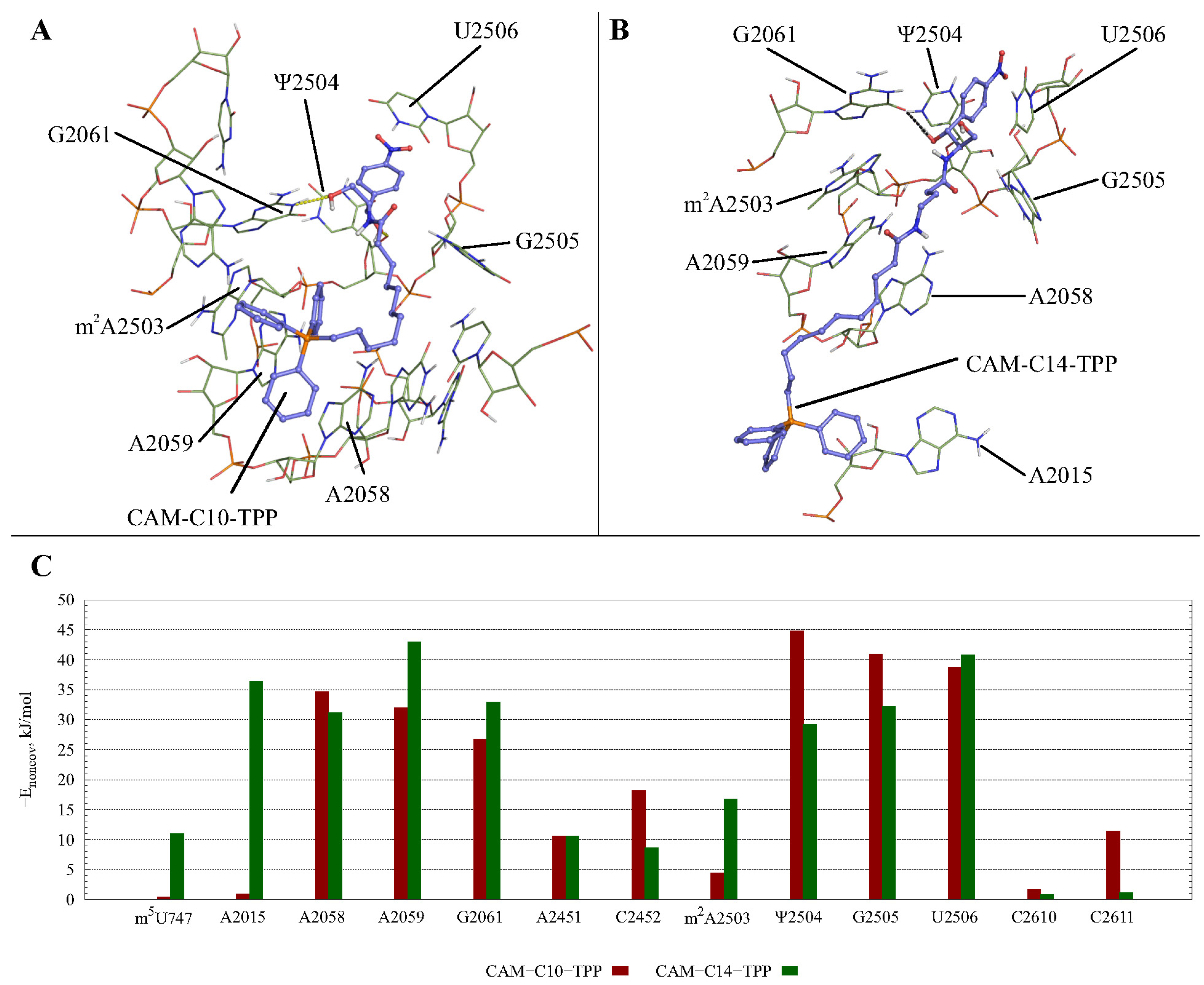
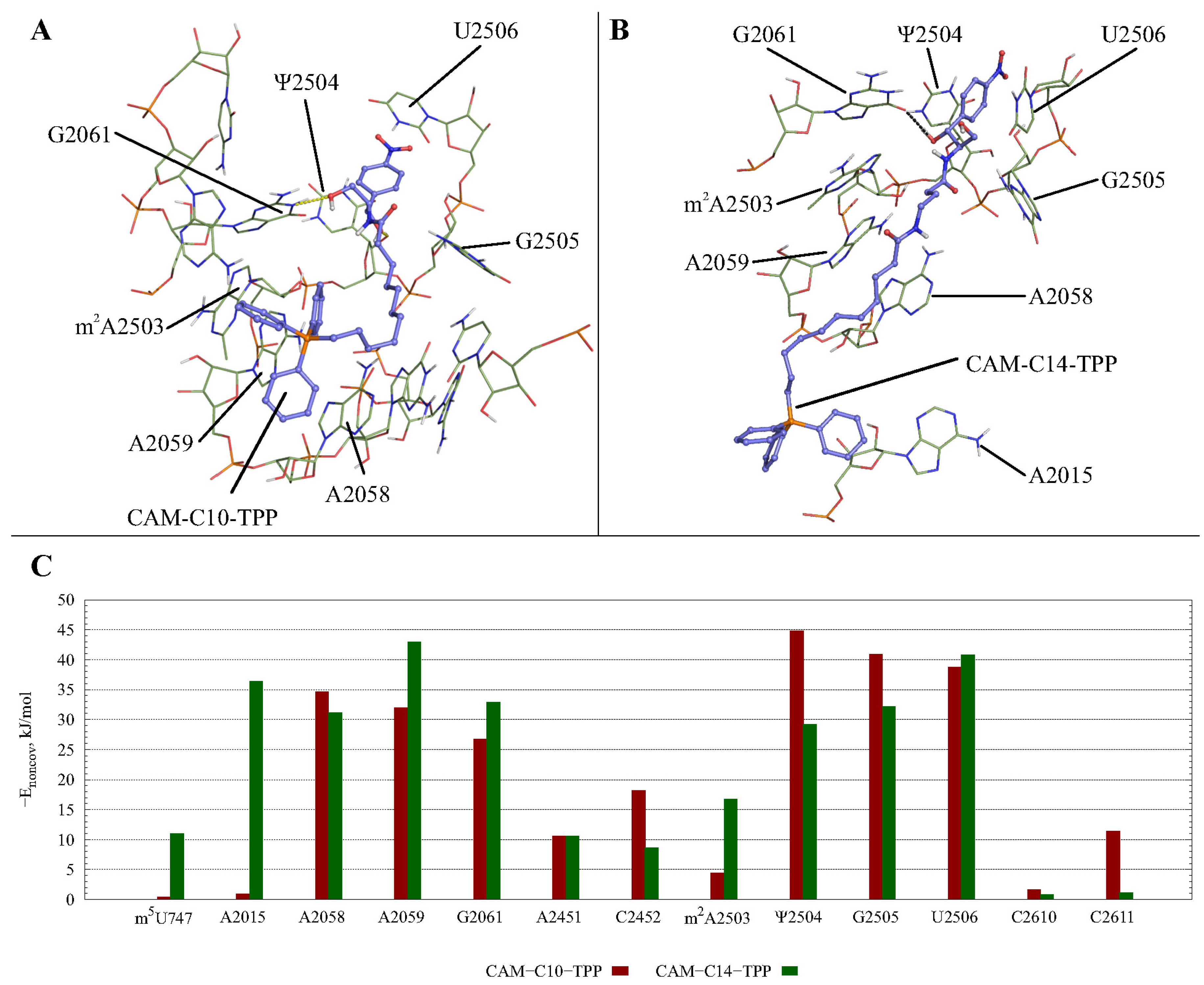
2.3. Possible CAM-Cn-TPPs Interaction Dynamics during Translation
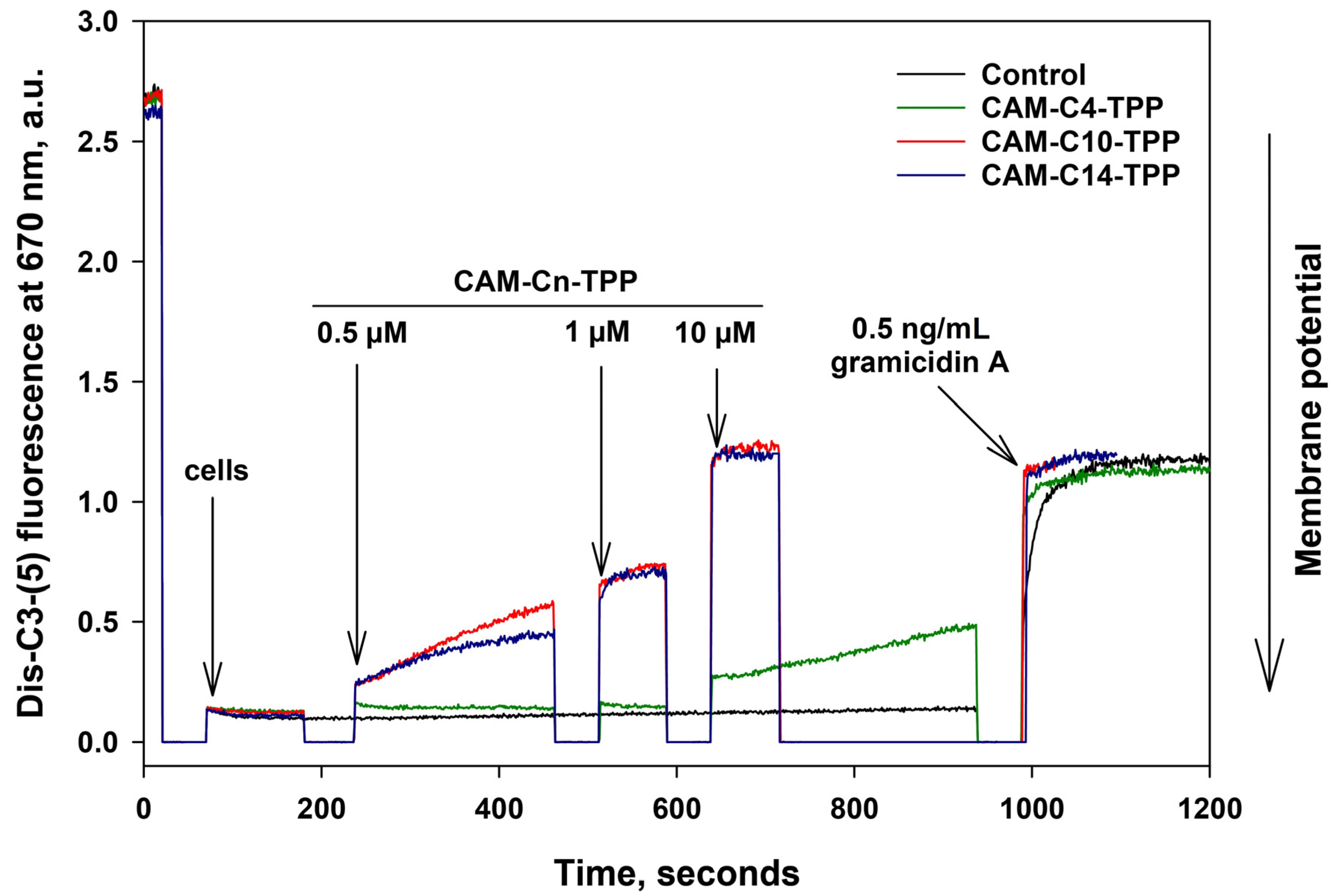
2.4. CAM-Cn-TPP Cause a Decrease in the Membrane Potential of B. subtilis
2.5. CAM-C10-TPP and CAM-C14-TPP Inhibit Bacterial Growth
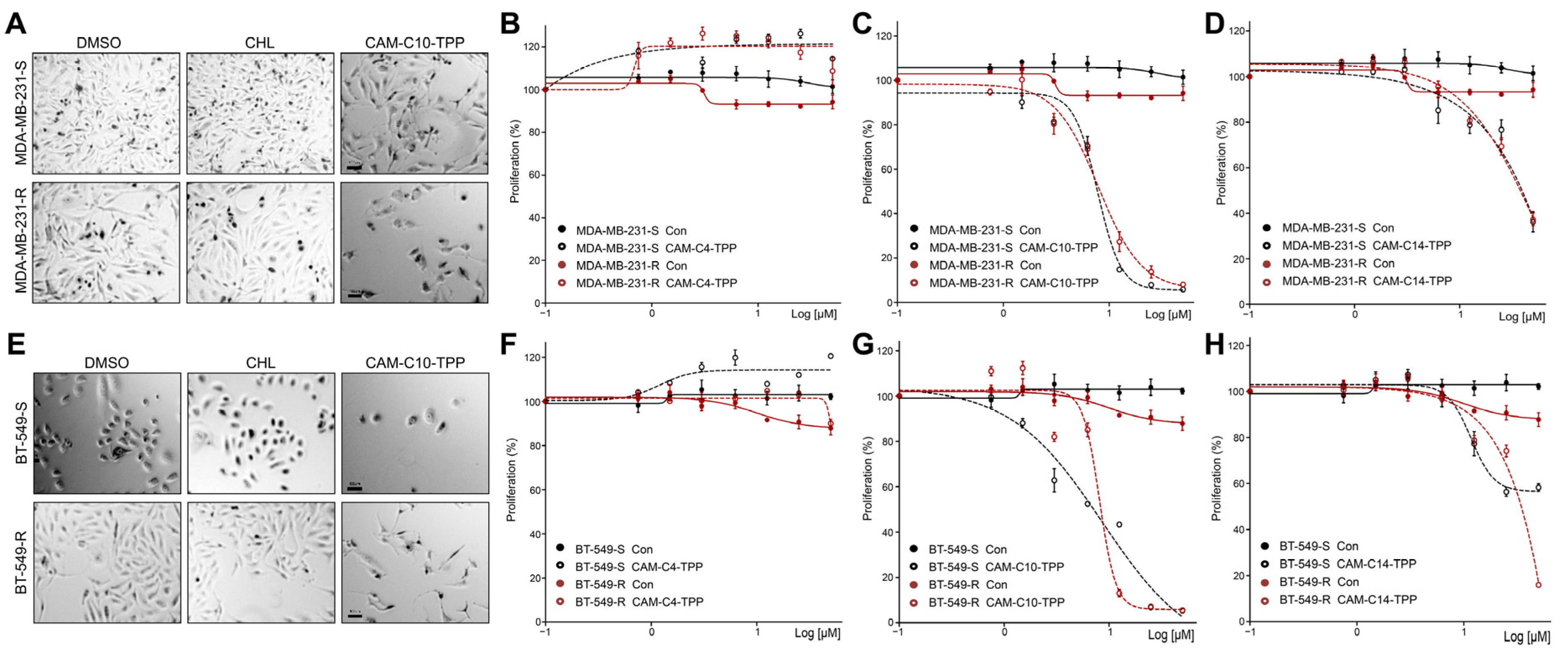
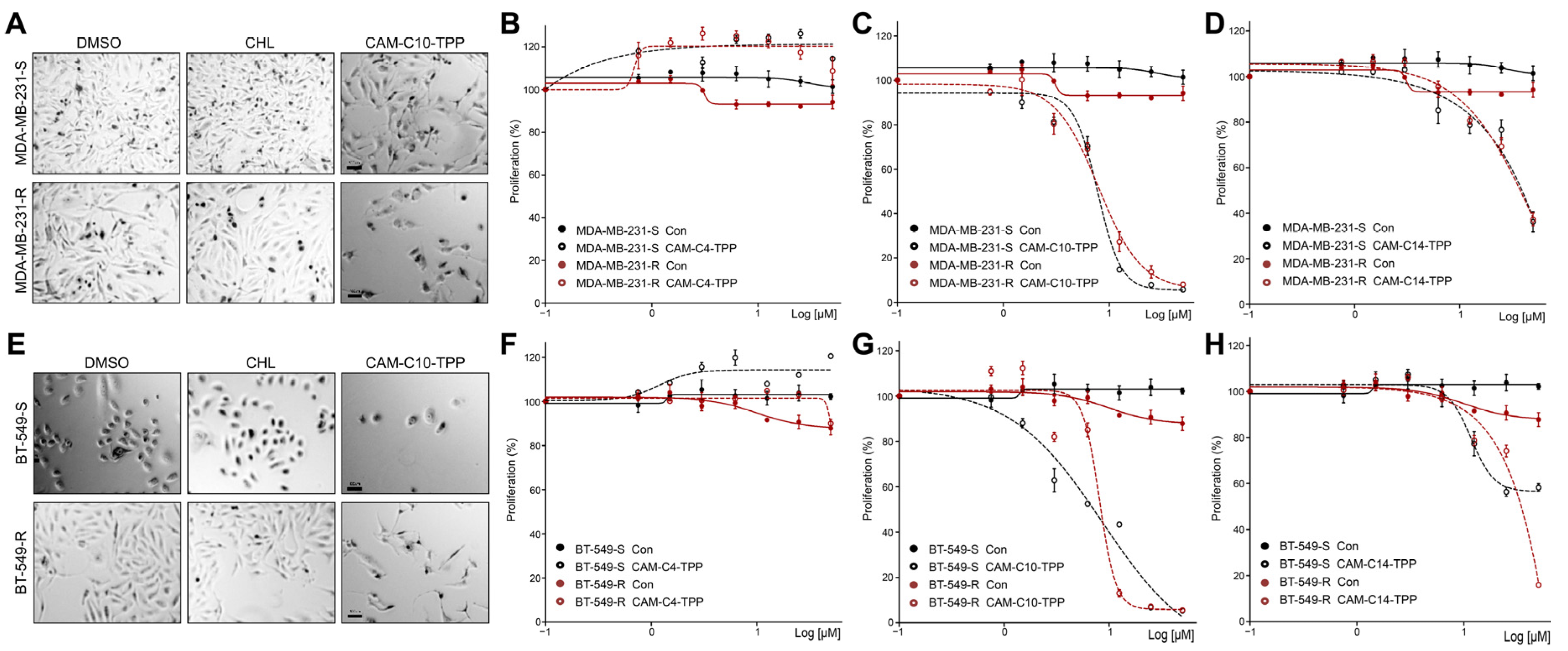
2.6. CAM-C10-TPP and CAM-C14-TPP Show a Reduced Toxicity Compared to Alkyl-TPP on Mammalian Cells
2.7. Anticancer Activities of CAM-C10-TPP and CAM-C14-TPP
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Synthetic Procedures
3.3. In Vitro Binding Assay
3.4. Detection of the Translation Inhibitors with a pDualrep2 Reporter
3.5. In Vitro Translation Inhibition Assay and Toeprinting Assays
3.6. Bacteria Inhibition Assays
3.6.1. Bacterial Strains
3.6.2. Plasmids
3.6.3. CAM-Cn-TPP-Dependent Bacterial Growth Suppression Screening of TolC-Requiring Transporters
3.6.4. MIC Determination
3.7. Measurement of the B. subtilis Membrane Potential
3.8. In Vitro Survival Assay (MTT Assay)
3.9. Cancer Cell Proliferation Assays
3.9.1. Cell Lines and Tumoursphere Formation
3.9.2. Cancer Cell Proliferation
3.10. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gabibov, A.G.; Dontsova, O.A.; Egorov, A.M. Overcoming antibiotic resistance in microorganisms: Molecular mechanisms. Biochemistry 2020, 85, 1289–1291. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2013, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. At the Crossroads of Bioenergetics and Antibiotic Discovery. Biochemistry 2020, 85, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Barbachyn, M.R. Recent advances in the discovery of hybrid antibacterial agents. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Elsevier Academic Press: San Diego, CA, USA, 2008; pp. 281–290. [Google Scholar] [CrossRef]
- Pokrovskaya, V.; Baasov, T. Dual-acting hybrid antibiotics: A promising strategy to combat bacterial resistance. Expert Opin. Drug Discov. 2010, 5, 883–902. [Google Scholar] [CrossRef]
- Tevyashova, A.N.; Olsufyeva, E.N.; Preobrazhenskaya, M.N. Design of dual action antibiotics as an approach to search for new promising drugs. Russ. Chem. Rev. 2015, 84, 61–97. [Google Scholar] [CrossRef]
- Parkes, A.L.; Yule, I.A. Hybrid antibiotics—Clinical progress and novel designs. Expert Opin. Drug Discov. 2016, 11, 665–680. [Google Scholar] [CrossRef]
- Tevyashova, A.N.; Bychkova, E.N.; Korolev, A.M.; Isakova, E.B.; Mirchink, P.E.; Osterman, I.A.; Erdei, R.; Szücs, Z.; Batta, G. Synthesis and evaluation of biological activity for dual-acting antibiotics on the basis of azithromycin and glycopeptides. Bioorg. Med. Chem. Lett. 2019, 29, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.B.; Ambrus, J.I.; Samosorn, S. Dual action-based approaches to antibacterial agents. Curr. Med. Chem. 2007, 14, 1459–1477. [Google Scholar] [CrossRef]
- Lewis, K. Persister Cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.; Gupta, V. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med Res. 2019, 149, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Efimova, S.S.; Tevyashova, A.N.; Olsufyeva, E.N.; Bykov, E.E.; Ostroumova, O.S. Pore-forming activity of new conjugate antibiotics based on amphotericin B. PLoS ONE 2017, 12, e0188573. [Google Scholar] [CrossRef] [Green Version]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of Coupling of Oxidative Phosphorylation and the Membrane Potential of Mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef]
- Feniouk, B.A.; Skulachev, V.P. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017, 10, 41–48. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A.J. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc. Natl. Acad. Sci. USA 2009, 107, 663–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khailova, L.S.; Nazarov, P.A.; Sumbatyan, N.V.; Korshunova, G.A.; Rokitskaya, T.I.; Dedukhova, V.I.; Antonenko, Y.N.; Skulachev, V.P. Uncoupling and toxic action of alkyltriphenylphosphonium cations on mitochondria and the bacterium Bacillus subtilis as a function of alkyl chain length. Biochemistry 2015, 80, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.A.; Osterman, I.A.; Tokarchuk, A.V.; Karakozova, M.V.; Korshunova, G.A.; Lyamzaev, K.G.; Skulachev, M.V.; Kotova, E.A.; Skulachev, V.P.; Antonenko, Y.N. Mitochondria-targeted antioxidants as highly effective antibiotics. Sci. Rep. 2017, 7, 1394. [Google Scholar] [CrossRef] [PubMed]
- Kolesińska, B.; Motylski, R.; Kamiński, Z.J.; Kwinkowski, M.; Kaca, W. Synthesis of P-triazinylphosphonium salts-hybrid molecules with potential antimicrobial activity. Acta Pol. Pharm. 2011, 68, 387–391. [Google Scholar] [PubMed]
- Kumari, S.; Jayakumar, S.; Gupta, G.D.; Bihani, S.C.; Sharma, D.; Kutala, V.K.; Sandur, S.K.; Kumar, V. Antibacterial activity of new structural class of semisynthetic molecule, triphenyl-phosphonium conjugated diarylheptanoid. Free Radical Biol. Med. 2019, 1, 140–145. [Google Scholar] [CrossRef]
- Kumari, S.; Jayakumar, S.; Bihani, S.C.; Shetake, N.; Naidu, R.; Kutala, V.K.; Sarma, H.D.; Gupta, G.D.; Sandur, S.K.; Kumar, V. Pharmacological characterization of a structurally new class of antibacterial compound, triphenyl-phosphonium conjugated diarylheptanoid: Antibacterial activity and molecular mechanism. J. Biosci. 2020, 45, 147. [Google Scholar] [CrossRef]
- Nazarov, P.A.; Kirsanov, R.S.; Denisov, S.S.; Khailova, L.S.; Karakozova, M.V.; Lyamzaev, K.G.; Korshunova, G.A.; Lukyanov, K.A.; Kotova, E.A.; Antonenko, Y.N. Fluorescein derivatives as antibacterial agents acting via membrane depolarization. Biomolecules 2020, 10, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaubasarova, I.R.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.I.; Silachev, D.N.; Danilina, T.I.; Plotnikov, E.Y.; Denisov, S.S.; Kirsanov, R.S.; Korshunova, G.A.; et al. Linking 7-nitrobenzo-2-oxa-1,3-diazole (NBD) to triphenylphosphonium yields mitochondria-targeted protonophore and antibacterial agent. Biochemistry 2020, 85, 1578–1590. [Google Scholar] [CrossRef]
- Churilov, M.N.; Denisenko, Y.V.; Batyushin, M.M.; Bren, A.B.; Chistyakov, V.A. Prospects of SkQ1 (10-(6′-plastoquinoyl) decyltriphenylphosphonium) application for prevention of oral cavity diseases. Rasayan J. Chem. 2018, 11, 1594–1603. [Google Scholar] [CrossRef]
- Kang, S.; Sunwoo, K.; Jung, Y.; Hur, J.K.; Park, K.H.; Kim, J.S.; Kim, D. Membrane-targeting triphenylphosphonium functionalized ciprofloxacin for methicillin-resistant Staphylococcus aureus (MRSA). Antibiotics 2020, 9, 758. [Google Scholar] [CrossRef]
- Pinto, T.; Banerjee, A.; Nazarov, P.A. Triphenyl phosphonium-based substances are alternatives to common antibiotics. Bull. Russ. State Med. Univ. 2018, 7, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Balczewski, P.; Skalik, J.; Nawrot, B.; Cieslak, M.; Kazmierczak-Baranska, J. Triphenylphosphonium Salts for Use in Cancer Therapy. European Patent Office 15,460,043.1, 13 August 2015. [Google Scholar]
- Sparey, T.; Ratcliffe, A.; Hallett, D.; Cochrane, E.; Lassalle, G.; Frodbise, A.; Stevenson, B. Triphenylphosphonium-Tethered Tetracycyclines for Use in Treating Cancer; World Intellectual Property Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Tsepaeva, O.V.; Nemtarev, A.V.; Salikhova, T.I.; Abdullin, T.I.; Grigor Eva, L.R.; Khozyainova, S.A.; Mironov, V.F. Synthesis, anticancer, and antibacterial activity of betulinic and betulonic acid C-28-triphenylphosphonium conjugates with variable alkyl linker length. Anticancer Agents Med Chem. 2020, 20, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Tsepaeva, O.V.; Salikhova, T.I.; Grigor’eva, L.R.; Ponomaryov, D.V.; Dang, T.; Ishkaeva, R.A.; Abdullin, T.I.; Nemtarev, A.V.; Mironov, V.F. Synthesis and in vitro evaluation of triphenylphosphonium derivatives of acetylsalicylic and salicylic acids: Structure-dependent interactions with cancer cells, bacteria, and mitochondria. Med. Chem. Res. 2021, 30, 925–939. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Dairkee, S.H.; Hackett, A.J. Differential retention of rhodamine 123 by breast carcinoma and normal human mammary tissue. Breast Cancer Res. Treat. 1991, 18, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta. Bioenergy 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Lleonart, M.E.; Abad, E.; Graifer, D.; Lyakhovich, A. Reactive oxygen species-mediated autophagy defines the fate of cancer stem cells. Antioxid. Redox Signal. 2018, 28, 1066–1079. [Google Scholar] [CrossRef]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting energy metabolism in cancer stem cells: Progress and challenges in leukemia and solid tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef]
- Han, X.; Su, R.; Huang, X.; Wang, Y.; Kuang, X.; Zhou, S.; Liu, H. Triphenylphosphonium-modified mitochondria-targeted paclitaxel nanocrystals for overcoming multidrug resistance. Asian J. Pharm. Sci. 2019, 14, 569–580. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Liu, H.N.; Guo, N.N.; Wang, T.T.; Guo, W.W.; Lin, M.T.; Huang-Fu, M.Y.; Vakili, M.R.; Xu, W.H.; Chen, J.J.; Wei, Q.C.; et al. Mitochondrial targeted doxorubicin-triphenylphosphonium delivered by hyaluronic acid modified and pH responsive nanocarriers to breast tumor: In vitro and in vivo studies. Mol. Pharm. 2018, 15, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Ozsvari, B.; Sotgia, F.; Lisanti, M.P. Exploiting mitochondrial targeting signal(s), TPP and bis-TPP, for eradicating cancer stem cells (CSCs). Aging 2018, 10, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Lleonart, M.E.; Grodzicki, R.; Graifer, D.M.; Lyakhovich, A. Mitochondrial dysfunction and potential anticancer therapy. Med. Res. Rev. 2017, 37, 1275–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging 2016, 8, 1593–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, A.; Vazquez, D. Cooperative and antagonistic interactions of peptidyl-tRNA and antibiotics with bacterial ribosomes. Eur. J. Biochem. 1977, 74, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol derivatives as antibacterial and anticancer agents: Historic problems and current solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and action of amino acid analogs of chloramphenicol upon the bacterial ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef]
- Mamos, P.; Krokidis, M.G.; Papadas, A.; Karahalios, P.; Starosta, A.L.; Wilson, D.N.; Kalpaxis, D.L.; Dinos, G.P. On the use of the antibiotic chloramphenicol to target polypeptide chain mimics to the ribosomal exit tunnel. Biochimie 2013, 95, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with polyamines enhances the antibacterial and anticancer activity of chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef]
- Giannopoulou, P.C.; Missiri, D.A.; Kournoutou, G.G.; Sazakli, E.; Papadopoulos, G.E.; Papaioannou, D.; Dinos, G.P.; Athanassopoulos, C.M.; Kalpaxis, D.L. New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity. Antibiotics 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and action of triphenylphosphonium analog of chloramphenicol upon the bacterial ribosome. Antibiotics 2021, 10, 390. [Google Scholar] [CrossRef]
- Yan, K.; Hunt, E.; Berge, J.; May, E.; Copeland, R.A.; Gontarek, R.R. Fluorescence polarization method to characterize macrolide-ribosome interactions. Antimicrob. Agents Chemother. 2005, 49, 3367–3372. [Google Scholar] [CrossRef] [Green Version]
- Tereshchenkov, A.G.; Shishkina, A.V.; Tashlitsky, V.N.; Korshunova, G.A.; Bogdanov, A.A.; Sumbatyan, N.V. Interaction of chloramphenicol tripeptide analogs with ribosomes. Biochemistry 2016, 81, 392–400. [Google Scholar] [CrossRef]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting out antibiotics’ mechanisms of action: A double fluorescent protein reporter for high-throughput screening of ribosome and DNA biosynthesis inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef] [Green Version]
- Rebstock, M.C.; Crooks, H.M.; Controulis, J.; Bartz, Q.R. Chloramphenicol (chloromycetin). IV. Chemical studies. J. Am. Chem. Soc. 1949, 71, 2458–2462. [Google Scholar] [CrossRef]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vázquez-Laslop, N.; Mankin, A.S. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartz, D.; McPheeters, D.S.; Traut, R.; Gold, L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988, 164, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Makarova, T.M. Investigation of Allosteric Phenomena in the Bacterial Ribosome by Molecular Dynamics Simulations Method. Ph.D. Thesis, Lomonosov Moscow State University, Moscow, Russia, 21 September 2020. [Google Scholar]
- Makarov, G.I.; Makarova, T.M. A noncanonical binding site of chloramphenicol revealed via molecular dynamics simulations. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2940–2947. [Google Scholar] [CrossRef]
- Nazarov, P.A.; Kotova, E.A.; Skulachev, V.P.; Antonenko, Y.N. Genetic variability of the AcrAB-TolC multidrug efflux pump underlies SkQ1 resistance in gram-negative bacteria. Acta Nat. 2019, 11, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyl-transferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Ross, M.F.; Prime, T.A.; Abakumova, I.; James, A.M.; Porteous, C.M.; Smith, R.A.; Murphy, M.P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 2008, 411, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Ralph, S.J.; Low, P.; Dong, L.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial targeted anti-cancer drugs as improved therapies and related patent documents. Recent Pat. Anticancer Drug Discov. 2006, 1, 327–346. [Google Scholar] [CrossRef]
- Abad, E.; García-Mayea, Y.; Mir, C.; Sebastian, D.; Zorzano, A.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A.; Lleonart, M.E. Common metabolic pathways implicated in resistance to chemotherapy point to a key mitochondrial role in breast cancer. Mol. Cell Proteom. 2019, 18, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Esner, M.; Graifer, D.; Lleonart, M.E.; Lyakhovich, A. Targeting cancer cells through antibiotics-induced mitochondrial dysfunction requires autophagy inhibition. Cancer Lett. 2017, 384, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Shishkina, A.V.; Makarov, G.I.; Tereshchenkov, A.G.; Korshunova, G.A.; Sumbatyan, N.V.; Golovin, A.V.; Svetlov, M.S.; Bogdanov, A.A. Conjugates of amino acids and peptides with 5-O-mycaminosyltylonolide and their interaction with the ribosomal exit tunnel. Bioconjug. Chem. 2013, 24, 1861–1869. [Google Scholar] [CrossRef]
- Wang, Z.X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Zakalyukina, Y.V.; Birykov, M.V.; Lukianov, D.A.; Shiriaev, D.I.; Komarova, E.S.; Skvortsov, D.A.; Kostyukevich, Y.; Tashlitsky, V.N.; Polshakov, V.I.; Nikolaev, E.; et al. Nybomycin-producing Streptomyces isolated from carpenter ant Camponotus vagus. Biochimie 2019, 160, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Szal, T.; Klepacki, D.; Shaw, K.J.; Vázquez-Laslop, N.; Mankin, A.S. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic. Acids Res. 2013, 41, e144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; Zhang, X.; Zhong, L.; Lu, Z. A modified electro-transformation method for Bacillus subtilis and its application in the production of antimicrobial lipopeptides. Biotechnol. Lett. 2011, 33, 1047–1051. [Google Scholar] [CrossRef]
- Lu, Y.P.; Zhang, C.; Lv, F.X.; Bie, X.M.; Lu, Z.X. Study on the electro-transformation conditions of improving transformation efficiency for Bacillus subtilis. Lett. Appl. Microbiol. 2012, 55, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Denisov, S.S.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.; Tashlitsky, V.N.; Firsov, A.M.; Korshunova, G.A.; Kotova, E.A. Alkyl-substituted phenylamino derivatives of 7-nitrobenz-2-oxa-1,3-diazole as uncouplers of oxidative phosphorylation and antibacterial agents: Involvement of membrane proteins in the uncoupling action. Biochim. Biophys. Acta Biomembr. 2017, 1859, 377–387. [Google Scholar] [CrossRef]
- Abad, E.; Civit, L.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A. Enhanced DNA damage response through RAD50 in triple negative breast cancer resistant and cancer stem-like cells contributes to chemoresistance. FEBS J. 2020. [Google Scholar] [CrossRef]
- Makarov, G.I.; Makarova, T.M. A noncanonical binding site of linezolid revealed via molecular dynamics simulations. J. Comput. Aided Mol. Des. 2019, 34, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101–014102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Athavale, S.S.; Petrov, A.S.; Hsiao, C.; Watkins, D.; Prickett, C.D.; Gossett, J.J.; Lie, L.; Bowman, J.C.; O’Neill, E.; Bernier, C.R.; et al. RNA folding and catalysis mediated by iron (II). PLoS ONE 2012, 7, e38024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Makarov, G.I.; Sumbatyan, N.V.; Bogdanov, A.A. Structural insight into interaction between C20 phenylalanyl derivative of tylosin and ribosomal tunnel. Biochemistry 2017, 82, 925–932. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Staphylococcus aureus | Listeria monocytogenes | Bacillus subtilis | Mycobacterium smegmatis | |
|---|---|---|---|---|
| CHL | 60 | 25 | 12 | 6 |
| CAM-C10-TPP | 5 | 6 | 2 | 2 |
| CAM-C14-TPP | 12 | 17 | 12 | 8 |
| C10-TPP | 2 | 5 | 2 | <2 |
| C14-TPP | 1.6 | 2 | 8 | 4 |
| E. coli ∆tolC | E. coli ∆tolC-CAT | Bacillus subtilis | B. subtilis pHT01-cat | B. subtilis pHT01-cfr | |
|---|---|---|---|---|---|
| CHL | 2.8 | >360 | 12 | 180 | 90 |
| CAM-C10-TPP | 3 | 25 | 2 | 6 | 6 |
| CAM-C14-TPP | 12.5 | 12.5 | 12 | 12.5 | 6 |
| C10-TPP | 3 | 3 | 2 | 1.6 | 0.8 |
| C14-TPP | 1.6 | 6 | 8 | 1.6 | 0.8 |
| HEK293T | MCF7 | A549 | VA13 | |
|---|---|---|---|---|
| CHL | >50 | >50 | >50 | >50 |
| CAM-C10-TPP | 0.62 ± 0.04 | 1.0 ± 0.1 | 0.7 ± 0.1 | 2.8 ± 0.5 |
| CAM-C14-TPP | 3.6 ± 0.5 | 5.8 ± 0.8 | 2.9 ± 0.4 | 5.2 ± 0.9 |
| C10-TPP | 0.08 ± 0.01 | 0.21 ± 0.03 | 0.07 ± 0.01 | 0.27 ± 0.05 |
| C14-TPP | 0.03 ± 0.02 | 0.02 ± 0.01 | 0.025 ± 0.009 | 0.07 ± 0.05 |
| Doxorubicin | 0.007 ± 0.001 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.18 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics 2021, 10, 489. https://doi.org/10.3390/antibiotics10050489
Pavlova JA, Khairullina ZZ, Tereshchenkov AG, Nazarov PA, Lukianov DA, Volynkina IA, Skvortsov DA, Makarov GI, Abad E, Murayama SY, et al. Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics. 2021; 10(5):489. https://doi.org/10.3390/antibiotics10050489
Chicago/Turabian StylePavlova, Julia A., Zimfira Z. Khairullina, Andrey G. Tereshchenkov, Pavel A. Nazarov, Dmitrii A. Lukianov, Inna A. Volynkina, Dmitry A. Skvortsov, Gennady I. Makarov, Etna Abad, Somay Y. Murayama, and et al. 2021. "Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents" Antibiotics 10, no. 5: 489. https://doi.org/10.3390/antibiotics10050489
APA StylePavlova, J. A., Khairullina, Z. Z., Tereshchenkov, A. G., Nazarov, P. A., Lukianov, D. A., Volynkina, I. A., Skvortsov, D. A., Makarov, G. I., Abad, E., Murayama, S. Y., Kajiwara, S., Paleskava, A., Konevega, A. L., Antonenko, Y. N., Lyakhovich, A., Osterman, I. A., Bogdanov, A. A., & Sumbatyan, N. V. (2021). Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics, 10(5), 489. https://doi.org/10.3390/antibiotics10050489