2.1. Data Collection and Pre-Processing
The models developed in this study were derived from the StarPep database [
32,
33]. This resource, as described by the authors, is a non-redundant compendium from 40 publicly available data sources, which encompasses annotations of more than 20 functions in approximately 45,000 AMPs, with nearly 8000 entries labelled as antibacterial peptides.
Before describing the construction of our training and test sets, we point out a shortcoming of several AMP-based predictors found in the literature [
16,
17,
18,
19,
20,
21,
22], whose models do not obey the first principle dictated by the Organisation for Economic Co-operation and Development (OECD) to build reliable Quantitative Structure–Activity Relationship (QSAR)/ML-based models [
34] (
https://doi.org/10.1787/9789264085442-en (accessed on 16 November 2022)). This principle is stated as “a defined endpoint”. Commonly, AMPs are annotated as such regardless of the target, mechanism, source, the method used to study the activity, to name some characteristics. The lack of such detailed information makes the discrimination between AMPs and non-AMPs a largely ambiguous endpoint for data analysis. In consequence, several criteria must be introduced to better define the modelled data and thus bring reliability to the predicted outcome. Notably, the most recent AMP predictors [
24,
25,
26,
27,
28,
29,
31] have designed their modeling approaches to break down the AMP annotation into three classes (typically antibacterial, antifungal, and antiviral peptides). This strategy is a suitable approach to fulfil the need for a defined endpoint.
Our work focused on the identification of ABPs. To this end, we extracted peptides from the StarPep database ranging between 5 and 50 residues, and whose composition contains only the 20 standard amino acids. To further refine the selection of ABPs, we only extracted those peptides annotated as active against at least one of the following targets: Acinetobacter baumannii, Bacillus subtilis, Enterococcus faecium, Escherichia coli, Klebsiella pneumonia, Listeria monocytogenes, Pseudomonas aeruginosa, Streptococcus agalactiae, and Staphylococcus aureus. In this way, we discarded entries that are annotated as ABPs without information of their targets, and those exclusively reported with activity against underrepresented targets in the entire database. The selected species cover a set of both Gram-positive and Gram-negative bacteria and are examples of relevant targets for therapeutic applications. The peptides labeled as non-ABP for our learning process are not annotated as antibacterial, against any target, in StarPep, but with a different function such as antifungal or anticancer, among others. This approach clearly carries the risk of mislabeling non-ABP in our dataset, due to insufficient annotation of the peptide in the original source. The pseudo-negative cases in the training data lead to a more stringent prediction of positive cases, and consequently lower false-positive rate and higher precision. The downside is the expected lower recall as the true positives can be also diminished. Nonetheless, the favourable precision is aligned with our stated goal of boosting the precision of the classifier instead of its recall or a combined metric such as accuracy or AUC.
Hence, we extracted a total of 22,707 peptides to design our training and testing schemes. This collection was partitioned into four datasets: training, development, validation, and test sets. The two first are intended for the learning process, while the others are meant for testing the models with hold-out data. The development (Dev) set was used to monitor the generalization of the models built during the optimization of the hyper-parameters in the learning algorithm. Usually, the terms development and validation set are applied indistinctively to a dataset used for the above-mentioned purpose. In this work, we made a distinction between these nomenclatures and reserved the term validation for a hold-out set, i.e., peptides that are not used in any step of the learning process. The difference between the validation and the strict test set is that we built the validation set in a way that its peptides share high similarity (≥90% identity) with at least one peptide in the training set (excluding identical matches). In turn, the test set was built in a way that its peptides share less than 90% identity among them, and with any peptide in the training data. Consequently, the test set comprises non-redundant peptides that are also not closely represented in our training. Challenging a peptide predictor in both scenarios, one that closely resembles the training conditions (without strict superposition), and another more distant setup, is important to assess the biasing effect on the generalization of the model due to the characteristics of the training data.
Finally, a production dataset was generated by combining the training and the development sets. The purpose of this set is to perform a final re-training of the model with an augmented dataset, while keeping the selection of descriptors and configuration of hyper-parameters as optimized with the training and development sets.
Figure 1 depicts the workflow followed to obtain the four datasets.
Together with the peptide sequences and their classification as ABP or non-ABP, we also extracted, from StarPep, the information about the Gram staining type of their known targets. Accordingly, we further categorized the ABPs into three activity classes: exclusively against Gram-positive targets (Gram+), exclusively against Gram-negative targets (Gram-), and broad-spectrum peptides. The four datasets resulting from the previous splitting were also used to train and assess the secondary classifier based on the Gram staining type of the targets. For this purpose, the non-ABP peptides were removed from such datasets.
Table 1 summarizes the number of peptides per type of Gram staining class in the four datasets.
2.2. Performance Measures
In this section, we summarize the formulations of the performance measures used to assess the different models described here. The measures are sensitivity (Sn), precision (Pr), accuracy (Acc), F1 score, and the Mathew Correlation Coefficient (MCC) [
35]. All of them are formulated in terms of the elements of a binary confusion matrix: true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN).
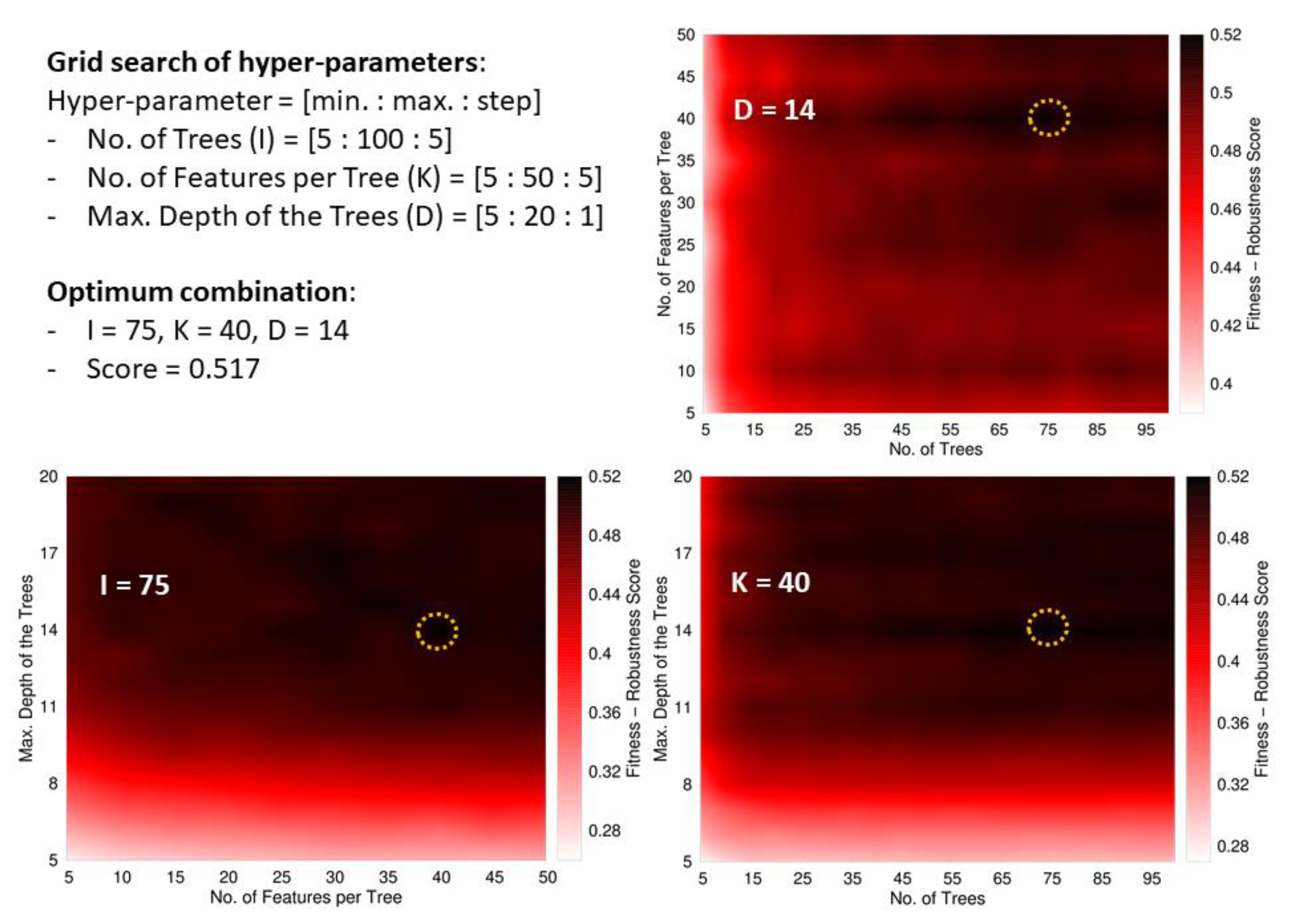
Besides, we define an ad-hoc measure named Fitness–Robustness Score (FRS) that is specifically used as a scoring function to tune the values of the hyper-parameters of the learning technique.
The FRS is a quality measure that provides a consolidated value for the performance of a particular model considering its goodness-of-fit, generalization, and robustness. The first term corresponds to the average performance in the following assessments: re-substitution (RT, fitting the training data), 10-fold cross-validation (RCV, within the training data), and generalization (RD, using the development set). The other two terms weigh the robustness of the model by measuring the deviations from the performance in training samples when the model is evaluated in hold-out data (cross-validation and development set). We formulated this ad-hoc measure as a function of another base quality measure, labelled as R, which should be evaluated in the different assessment schemes. For this study, we selected the MCC as the base measure to evaluate our fitness-robustness score. In the case of the multi-classifier trained to distinguish between the Gram staining classes, the average MCC value among the three classes was used as the base measure. The average was weighted according to the number of peptides in each class.
The FRS, when computed as a function of the MCC, has an optimum maximum value of one. We leveraged this score to identify optimum values for the hyper-parameters of the random forest [
36] algorithm used to develop our models.
2.3. Machine Learning Approach and Software
The classifiers developed in this work were random forest (RF) [
36] predictors, based on the implementation of this technique in the WEKA environment [
37]. RF belongs to the family of ensemble methods [
38] with base classifiers formed by decision trees. Recently, RF has been compared with deep learning approaches showing comparable performance for modeling AMP datasets [
39]. There, the authors conclude that no definitive evidence was found to support using deep-learning approaches for this problem, knowing the increased algorithmic complexity and computational cost of these methods.
Within RF, all the trees provide a prediction for every instance entering the forest, and the unified outcome is obtained as the majority vote among all the predictions. The hyper-parameters optimized during the learning process were the number of trees, the maximum number of descriptors used to build a tree (these descriptors are taken at the beginning of the training process from the global pool of attributes), and the maximum depth of the trees. In addition, the minimum number of instances in the final leaves of the trees was fixed to 10 in the case of the main classifier (ABPnon-ABP), and to five for the multi-classifier (Gram+/Gram−/broad spectrum).
The peptide descriptors fed to the learning algorithm were computed with the ProtDCal-Suite [
40] using the configuration files enclosed in the
Supplementary Material. The ProtDCal module [
41] is intended for the calculation of general-purpose and alignment-free descriptors of amino acid sequences and protein structures. These features are descriptive statistics (such as the variance, average, maximum, minimum, percentiles, etc.) of the distribution of amino acid properties (such as hydrophobicity, isoelectric point, molar weight, among others), in multiple groups of residues extracted from a given protein or peptide. The program possesses additional procedures that modify the intrinsic properties of a residue according to its vicinity in the sequence, thus adding connectivity information in the descriptors. The features derived from ProtDCal have been used by us and other authors to develop machine-learning-based predictors of posttranslational modifications [
42,
43], protein–protein interaction [
44], enzyme-like amino acid sequences [
45], residues critical for protein functions [
46], and antibacterial peptides [
47,
48], although with smaller databases. The project files enclosed in the
Supplementary Material contain the setup used to compute all the descriptors employed in this work.
2.4. Web Servers Available for ABPs Predictions
In this section, we briefly describe the most relevant state-of-the-art ABP predictors that are available via web server tools. ClassAMP was among the first methods that broke down the AMP family thus allowing the prediction of ABPs specifically [
25]. This tool was trained with peptides from the CAMP database [
18] and used RF and support vector machine (SVM) [
49] algorithms to identify antibacterial, antifungal, and antiviral peptides.
MLAMP, a multi-label classifier of AMPs was developed using a variant of Chou’s pseudo amino acid composition (PseACC) features [
50] to build an RF-based classifier that firstly distinguishes AMP from non-AMPs, and then subdivides the biological activity into antibacterial, anticancer, antifungal, antiviral, and anti-HIV [
26].
Similarly, the
iAMPpred predictor combines compositional, physicochemical, and structural features into Chou’s general PseACC as input variables for an SVM multi-classifier [
27]. This work reunited peptides from the databases CAMPR3 [
19], APD3 [
17], and AntiBP2 [
24]. The multi-classifier uses three categories in the outcome variable: antibacterial, antifungal, and antiviral peptides [
27].
The Antimicrobial Activity Predictor (AMAP) [
28], with a hierarchical multi-label classification scheme, was trained with AMPs annotated with 14 biological activities in the APD3 database and a designed subset of non-AMP. The models used amino acid composition features to feed SVM and XGboost tree [
51] algorithms.
The introduction of the AMP-Scanner webserver represented a significant improvement with respect to other predictors. AMP-Scanner vr.1 consists of two RF classifiers, trained with peptides selected from multiple sources [
18,
52,
53]. The first output of the classifier is the identification of ABPs. The second is a classifier trained to distinguish between peptides with Gram-positive or Gram-negative targets, using data of
S. aureus and
E. coli as reference targets. The authors refer that peptides predicted with scores within the range [0.4–0.6] for both classes should be considered as active against both types of targets (broad-spectrum peptides) [
29]. On the other hand, AMP-Scanner vr.2 is based on a Deep Neural Networks (DNN) classifier fed with ABP data only, obtained from the updated version of the ADP3 database [
19,
30].
Very recently, AMPDiscover [
31] was developed by mining AMP data from StarPep [
33]. AMPDiscover encompasses several binary (active/non-active) predictors of functions such as antibacterial, antiviral, antifungal, and antiparasitic peptides. The authors analyzed the performance of RF to model the antibacterial peptides data, which agrees with our choice of this learning scheme for our models.



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}