
A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery



Abstract
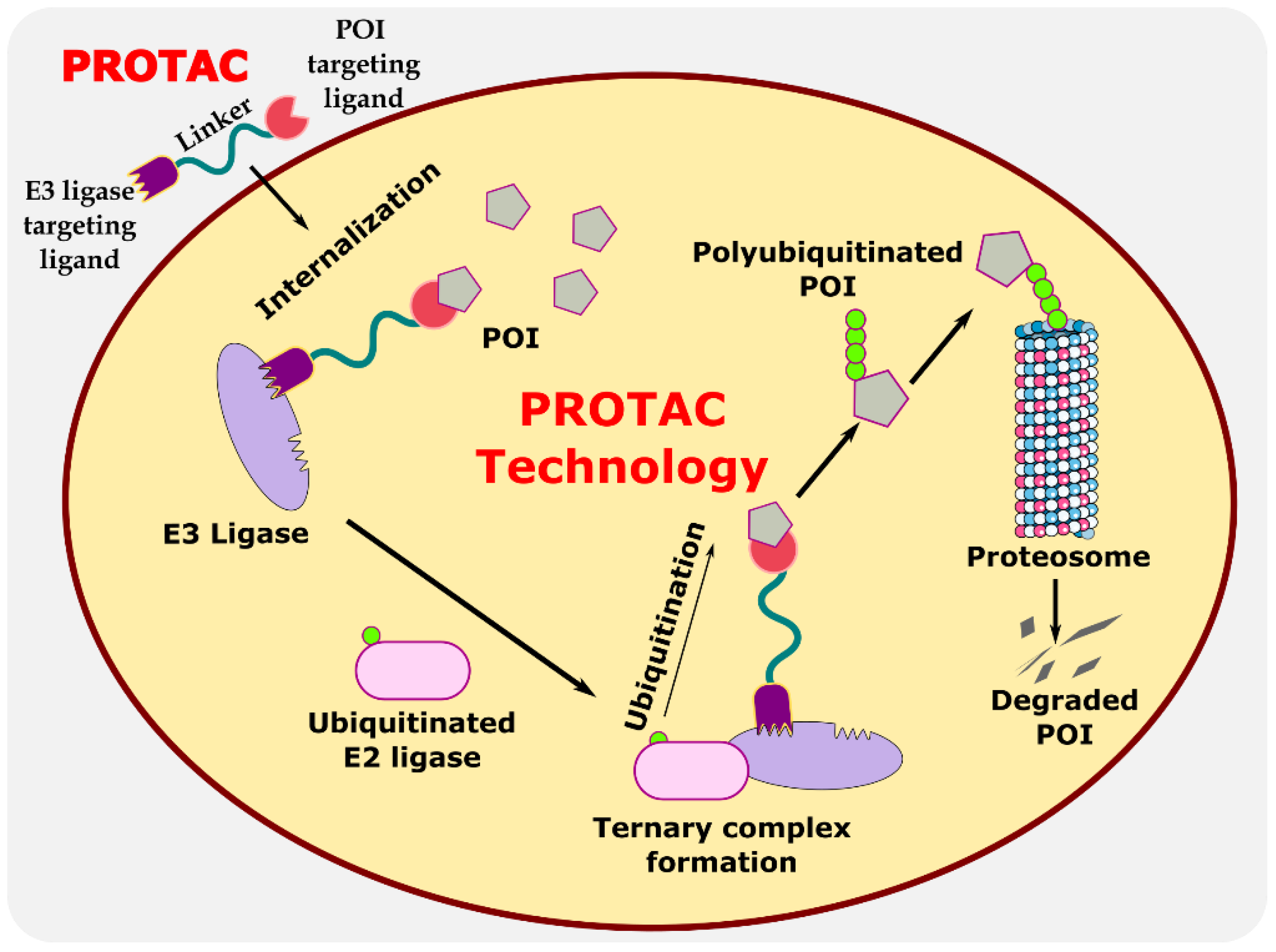
:1. Introduction
2. Eukaryotic System
2.1. Anti-Viral PROTACs
2.1.1. Degradation of Viral Protein
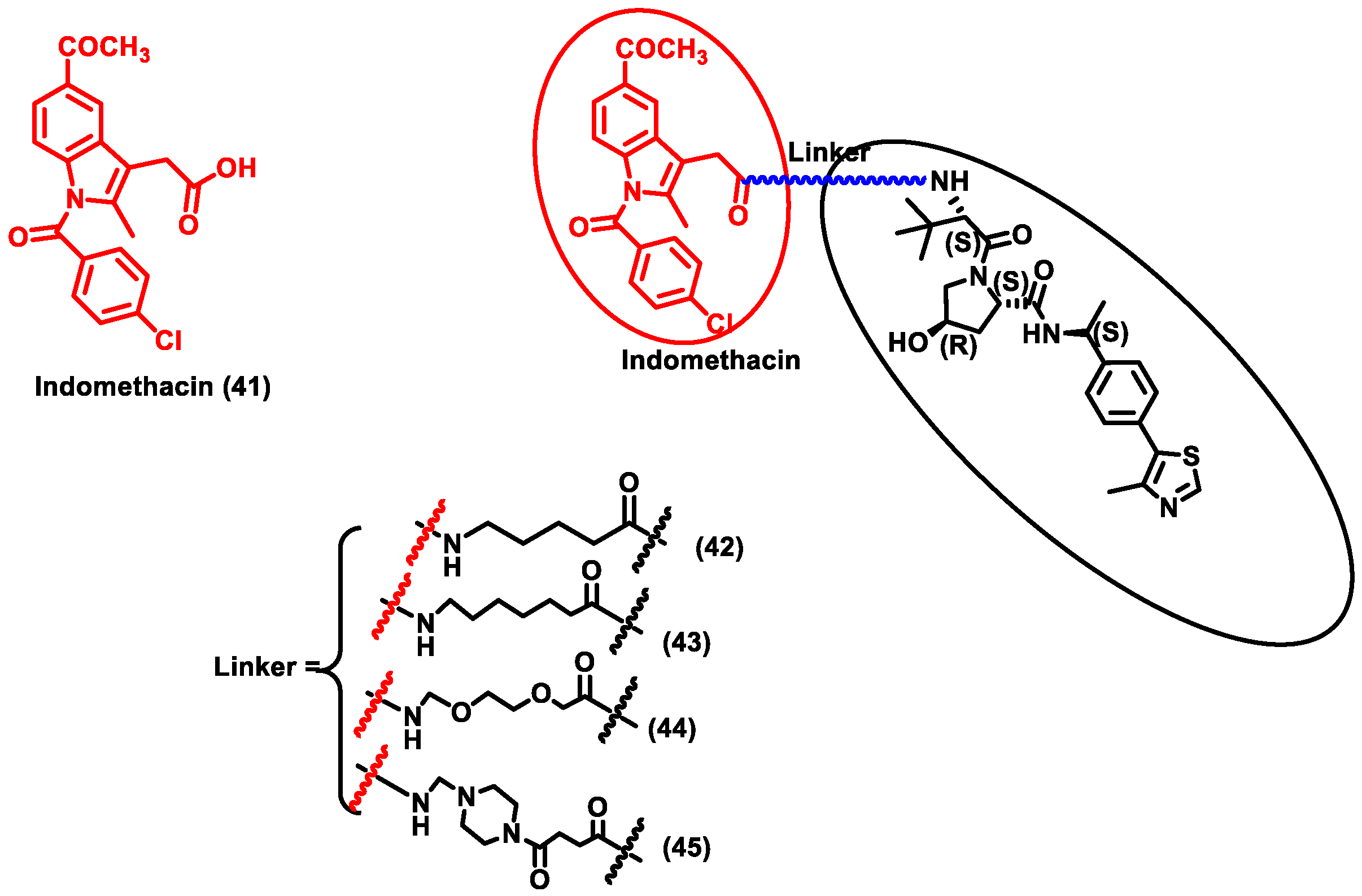
2.1.2. Degradation of Human Host Protein
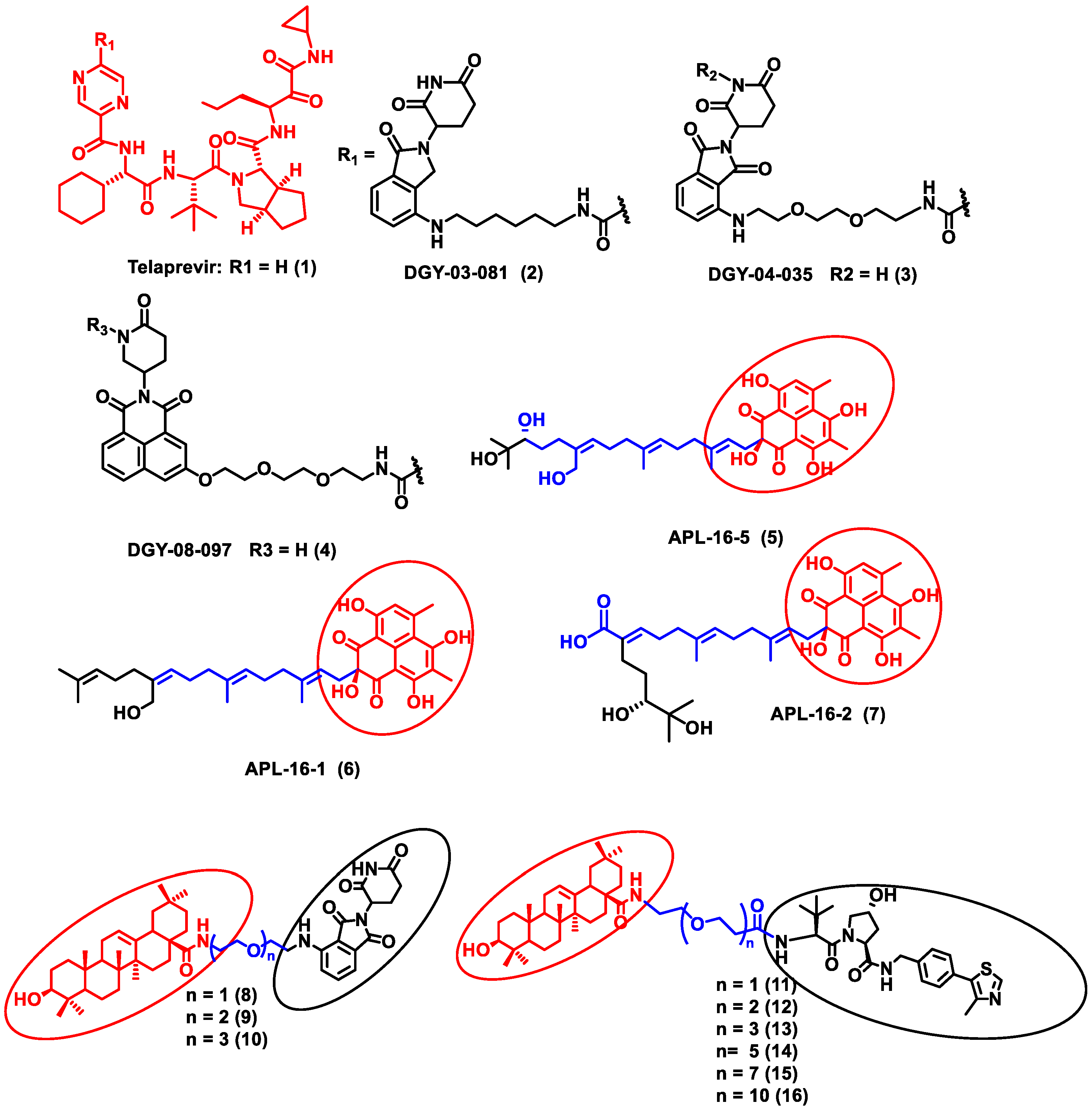
3. Prokaryotic System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Pathogen | Protein of Interest (POI) | POI Ligand | E3 Ligase Ligand | Research Outcome | Ref. |
|---|---|---|---|---|---|---|
| 1 | Bacillus subtilis | ClpCP protease | Phosphorylated arginine residue | Biotin |
| [62] |
| 2 | Mycobacterium tuberculosis | ClpC1P1P2 protease | Cyclomarin A (CymA) | Biotin JQ1(R) and JQ1(S) |
| [64] |
| 3 | Mycobacterium tuberculosis | ClpC1-NTD protease | Desoxycyclomarin derivative | - |
| [65] |
4. Patent Analysis and Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AH | Attenuator hairpin |
| BacPROTAC | Bacterial PROTAC |
| CDK | Cyclin-dependent kinase |
| CLpCP | Caseinolytic protease proteolytic |
| CRBN | Cereblon |
| CymA | Cyclomarin A |
| DNA | Deoxyribonucleic acid |
| eDGFR | Escherichia coli dihydrofolate reductase |
| HA | Hemagglutinin |
| HBV | Hepatitis B virus |
| HCC | Hepato cellular carcinoma |
| HCMV | Human cytomegalovirus |
| HIF | hypoxia-inducible factor |
| HyT | hydrophobic tagging |
| IFN-β | Interferon beta |
| IL-6 | Interleukin-6 |
| IMN | Indomethacin |
| LID | Ligand-induced degradation |
| LYTAC | Lysosome-targeting chimeras |
| MDCK cells | Madin–Darby canine kidney cells |
| MERS-CoV | Middle East respiratory syndrome |
| mRNA | Messenger RNA |
| NA | Neuraminidase |
| NATAC | Nucleic acid targeting chimeras |
| ODD | Oxygen-dependent degradation |
| ORF3 | Open reading frame 3 |
| PDT | Photo dynamic therapy |
| PGE-2 | Prostaglandin E2 |
| PKR | Protein kinase R |
| POI | Protein of interest |
| PROTAC | Proteolysis-targeting chimeras |
| PTG | Pentacyclic triterpenoid group |
| RIBOTAC | Ribonuclease targeting chimeras |
| RNA | Ribonucleic acid |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| TMP | Trimethoprim |
| UPS | Ubiquitin–proteasome-system |
| VHL | Von Hippel-Lindau |
References
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumar, A.K.; Kuppuswamy, V.; Chinnakkannu, P. Administration of Grape Seed Extract Alleviates Age-Associated Decline in Ubiquitin-Proteasome System and Cardiomyocyte Apoptosis in Rats. Adv. Biol. Chem. 2013, 3, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-Induced Genetic Degradation as a Tool for Target Validation. Drug Discov. Today Technol. 2019, 31, 91–98. [Google Scholar] [CrossRef]
- Choi, S.R.; Wang, H.M.; Shin, M.H.; Lim, H.-S. Hydrophobic Tagging-Mediated Degradation of Transcription Coactivator SRC-1. Int. J. Mol. Sci. 2021, 22, 6407. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorganic Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Sig. Transduct. Target 2019, 4, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Mayor-Ruiz, C.; Winter, G.E. Identification and Characterization of Cancer Vulnerabilities via Targeted Protein Degradation. Drug Discov. Today Technol. 2019, 31, 81–90. [Google Scholar] [CrossRef]
- Protacdb Server. Available online: http://cadd.zju.edu.cn/protacdb/ (accessed on 11 November 2022).
- Alabi, S.B.; Crews, C.M. Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef]
- Protein Degradation with PROTAC Protein Degraders. Available online: https://www.arvinas.com/ (accessed on 2 November 2022).
- Arvinas and Pfizer Announce Global Collaboration to Develop and Commercialize PROTAC® Protein Degrader ARV-471 | Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/arvinas-and-pfizer-announce-global-collaboration-develop (accessed on 2 November 2022).
- Accutar Biotech. Available online: https://www.accutarbio.com/workflow/ (accessed on 2 November 2022).
- Understanding Protein Degradation–Bristol Myers Squibb. Available online: https://www.bms.com/media/media-library/scientific-media-resources/understanding-protein-degradation-and-resources.html (accessed on 2 November 2022).
- Dialectic. Available online: https://www.dtsciences.com/ (accessed on 2 November 2022).
- Foghorntx Therapeutics Pipeline. Available online: https://foghorntx.com/pipeline/ (accessed on 2 November 2022).
- Therapeutic Pipeline. Available online: https://www.kymeratx.com/pipeline/ (accessed on 2 November 2022).
- Nurix Pipeline. Available online: https://www.nurixtx.com/pipeline/ (accessed on 2 November 2022).
- Targeted Protein Degradation–C4 Therapeutics. Available online: https://c4therapeutics.com/our-science/targeted-protein-degradation (accessed on 2 November 2022).
- Cullgen. Available online: https://www.cullgen.com (accessed on 3 November 2022).
- Mullard, A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef] [PubMed]
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C.; et al. Extended Pharmacodynamic Responses Observed upon PROTAC-Mediated Degradation of RIPK2. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, Y.; Wang, W.; Sun, D.; Zheng, M.; Zhou, Y.; Liang, J.; Zhu, M.; Li, H.; Chen, L. PROTAC Technology as a Novel Tool to Identify the Target of Lathyrane Diterpenoids. Biol. Med. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Proteolysis-Targeting Chimera (PROTAC) for Targeted Protein Degradation and Cancer Therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as Emerging Anticancer Therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Molecules for the Treatment of Autoimmune Disorders. ACS Med. Chem. Lett. 2019, 10, 276–277. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Hassan, M.I. Targeted Protein Degraders March towards the Clinic for Neurodegenerative Diseases. Ageing Res. Rev. 2022, 78, 101616. [Google Scholar] [CrossRef]
- Powell, M.; Blaskovich, M.A.T.; Hansford, K.A. Targeted Protein Degradation: The New Frontier of Antimicrobial Discovery? ACS Infect. Dis. 2021, 7, 2050–2067. [Google Scholar] [CrossRef]
- Desantis, J.; Goracci, L. Proteolysis Targeting Chimeras in Antiviral Research. Future Med. Chem. 2022, 14, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Trial of ARV-110 in Patients with Metastatic Castration Resistant Prostate Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03888612 (accessed on 8 November 2022).
- A Phase 1/2 Trial of ARV-471 Alone and in Combination with Palbociclib (IBRANCE®) in Patients with ER+/HER2- Locally Advanced or Metastatic Breast Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04072952 (accessed on 8 November 2022).
- Accutar Biotechnology Inc. A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Preliminary Anti-Tumor Activity of AC682 in Patients with Estrogen Receptor Positive/Human Epidermal Growth Factor Receptor 2 Negative (ER+/HER2-) Locally Advanced or Metastatic Breast Cancer. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- A Study of ARV-766 Given by Mouth in Men with Metastatic Castration-Resistant Prostate Cancer Who Have Progressed on Prior Approved Systemic Therapies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05067140 (accessed on 8 November 2022).
- Study to Evaluate the Safety and Tolerability of CC-94676 in Participants with Metastatic Castration-Resistant Prostate Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04428788 (accessed on 8 November 2022).
- A Study of DT2216 in Relapsed/Refractory Malignancies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04886622 (accessed on 8 November 2022).
- FHD-609 in Subjects with Advanced Synovial Sarcoma or Advanced SMARCB1-Loss Tumors-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04965753 (accessed on 9 November 2022).
- A Single and Multiple Ascending Dose Trial of KT-474 in Healthy Adult Volunteers and Patients with Atopic Dermatitis (AD) or Hidradenitis Suppurativa (HS)-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04772885 (accessed on 9 November 2022).
- Kymera Therapeutics, Inc. A Phase 1, Multicenter, Open-Label, Dose Escalation and Expansion Study to Evaluate the Safety, Tolerability, PK/PD, and Clinical Activity of Intravenously Administered KT-413 in Adult Patients with Relapsed or Refractory B-Cell NHL. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- Nurix Therapeutics, Inc. A Phase 1, Dose Escalation, Safety and Tolerability Study of NX-2127, a Bruton’s Tyrosine Kinase (BTK) Degrader, in Adults with Relapsed/Refractory B-Cell Malignancies. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- A Study of NX-5948 in Adults with Relapsed/Refractory B-Cell Malignancies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05131022 (accessed on 9 November 2022).
- A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05355753 (accessed on 9 November 2022).
- Effects of Systematic Proprioceptive-Tactile Stimulation with Use of the Protac MyFit®-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04173871 (accessed on 9 November 2022).
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar] [PubMed]
- Gopal, P.; Dick, T. Targeted Protein Degradation in Antibacterial Drug Discovery? Prog. Biophys. Mol. Biol. 2020, 152, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Montrose, K.; Krissansen, G.W. Design of a PROTAC That Antagonizes and Destroys the Cancer-Forming X-Protein of the Hepatitis B Virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740. [Google Scholar] [CrossRef]
- Lin, C. HCV NS3-4A Serine Protease. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.-L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; ISBN 978-1-904933-20-5. [Google Scholar]
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small Molecule Degraders of the Hepatitis C Virus Protease Reduce Susceptibility to Resistance Mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, J.; Pang, X.; Liu, Z.; Li, Q.; Yi, D.; Zhang, Y.; Fang, X.; Zhang, T.; Zhou, R.; et al. An Anti-Influenza A Virus Microbial Metabolite Acts by Degrading Viral Endonuclease PA. Nat. Commun. 2022, 13, 2079. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Ma, W.; Cheng, B.; Yi, Y.; Ma, X.; Xiao, S.; Zhang, L.; Zhou, D. Discovery of Pentacyclic Triterpenoid PROTACs as a Class of Effective Hemagglutinin Protein Degraders. J. Med. Chem. 2022, 65, 7154–7169. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, X.; Ma, X.; Zou, W.; Chen, Q.; Chen, F.; Deng, X.; Liang, J.; Dong, C.; Lan, K.; et al. Discovery of Oseltamivir-Based Novel PROTACs as Degraders Targeting Neuraminidase to Combat H1N1 Influenza Virus. Cell Insight 2022, 1, 100030. [Google Scholar] [CrossRef]
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-COV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RiboTAC) Degraders. ACS Cent. Sci. 2020, 6, 1713–1721. [Google Scholar] [CrossRef]
- Su, X.; Ma, W.; Feng, D.; Cheng, B.; Wang, Q.; Guo, Z.; Zhou, D.; Tang, X. Efficient Inhibition of SARS-CoV-2 Using Chimeric Antisense Oligonucleotides through RNase L Activation**. Angew. Chem. -Int. Ed. 2021, 60, 21662–21667. [Google Scholar] [CrossRef]
- Hahn, F.; Hamilton, S.T.; Wangen, C.; Wild, M.; Kicuntod, J.; Brückner, N.; Follett, J.E.L.; Herrmann, L.; Kheimar, A.; Kaufer, B.B.; et al. Development of a PROTAC-Based Targeting Strategy Provides a Mechanistically Unique Mode of Anti-Cytomegalovirus Activity. IJMS 2021, 22, 12858. [Google Scholar] [CrossRef] [PubMed]
- Desantis, J.; Mercorelli, B.; Celegato, M.; Croci, F.; Bazzacco, A.; Baroni, M.; Siragusa, L.; Cruciani, G.; Loregian, A.; Goracci, L. Indomethacin-Based PROTACs as Pan-Coronavirus Antiviral Agents. Eur. J. Med. Chem. 2021, 226, 113814. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, R.; Liu, S.; Disoma, C.; Du, A.; Li, S.; Chen, Z.; Dong, Z.; Zhang, Y.; Li, S.; et al. Host E3 Ligase HUWE1 Attenuates the Proapoptotic Activity of the MERS-CoV Accessory Protein ORF3 by Promoting Its Ubiquitin-Dependent Degradation. J. Biol. Chem. 2022, 298. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van Der Veken, P.; De Winter, H.; Augustyns, K. ClpP Protease, a Promising Antimicrobial Target. Int. J. Mol. Sci. 2019, 20, 2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentini, D.B.; Suskiewicz, M.J.; Heuck, A.; Kurzbauer, R.; Deszcz, L.; Mechtler, K.; Clausen, T. Arginine Phosphorylation Marks Proteins for Degradation by a Clp Protease. Nature 2016, 539, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Hajdusits, B.; Beveridge, R.; Heuck, A.; Vu, L.D.; Kurzbauer, R.; Hauer, K.; Thoeny, V.; Rumpel, K.; Mechtler, K.; et al. Structure of McsB, a Protein Kinase for Regulated Arginine Phosphorylation. Nat. Chem. Biol. 2019, 15, 510–518. [Google Scholar] [CrossRef]
- Morreale, F.E.; Kleine, S.; Leodolter, J.; Ovchinnikov, S.; Kley, J.; Kurzbauer, R.; Hoi, D.M.; Meinhart, A.; Hartl, M.; Haselbach, D.; et al. BacPROTACs Mediate Targeted Protein Degradation in Bacteria. Cell 2022, 185, 2338–2353.e18. [Google Scholar] [CrossRef] [PubMed]
- Weinhäupl, K.; Brennich, M.; Kazmaier, U.; Lelievre, J.; Ballell, L.; Goldberg, A.; Schanda, P.; Fraga, H. The Antibiotic Cyclomarin Blocks Arginine-Phosphate-Induced Millisecond Dynamics in the N-Terminal Domain of ClpC1 from Mycobacterium Tuberc. J. Biol. Chem. 2018, 293, 8379–8393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.S.; et al. The Natural Product Cyclomarin Kills Mycobacterium Tuberculosis by Targeting the ClpC1 Subunit of the Caseinolytic Protease. Angew. Chem. Int. Ed. Engl. 2011, 50, 5889–5891. [Google Scholar] [CrossRef]
- Junk, L.; Schmiedel, V.; Guha, S.; Greb, P.; Fischel, K.; Rumpel, K.; Kaur, P.; Krishnamurthy, R.; Narayanan, S.; Kofink, C.; et al. BacPROTAC-Induced Degradation of ClpC1 as a New Strategy against Drug-Resistant Mycobacteria. Chemrxiv 2022. [Google Scholar] [CrossRef]
- Gopal, P.; Sarathy, J.; Yee, M.; Ragunathan, P.; Shin, J.; Bhushan, S.; Zhu, J.; Akopian, T.; Kandror, O.; Lim, T.K.; et al. Pyrazinamide Triggers Degradation of Its Target Aspartate Decarboxylase. Nat. Commun. 2019, 11, 674416. [Google Scholar] [CrossRef] [Green Version]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor Mediated Protein Degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Lu, F.; Qi, F.; Wen, T.; Bai, M.; Wang, J. Compound for Degrading Deoxyribonucleic Acid (DNA) Polymerase, and Use Thereof. Available online: https://patentscope2.wipo.int/search/en/detail.jsf?docId=WO2022156764&_gid=202230 (accessed on 24 November 2022).
- Haibing, Z.; Wu, S.; Xu, Z. Oseltamivir PROTAC Compound, Preparation Method Thereof and Application Thereof in Anti-Influenza Virus Drugs. Eur. J. Med. Chem. 2022, 243, 114711. [Google Scholar]
- Gao, J.; Hou, B.; Zhu, Q.; Yang, L.; Jiang, X.; Zou, Z.; Li, X.; Xu, T.; Zheng, M.; Chen, Y.-H.; et al. Engineered Bioorthogonal POLY-PROTAC Nanoparticles for Tumour-Specific Protein Degradation and Precise Cancer Therapy. Nat. Commun. 2022, 13, 4318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Picaud, S.; Filippakopoulos, P.; D’Angiolella, V.; Bullock, A.N. Structural Basis for Recruitment of DAPK1 to the KLHL20 E3 Ligase. Structure 2019, 27, 1395–1404.e4. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.-T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted Degradation of Aberrant Tau in Frontotemporal Dementia Patient-Derived Neuronal Cell Models. eLife 2019, 8, e45457. [Google Scholar] [CrossRef] [PubMed]




| Sr. No. | Molecule | Route of Delivery (Dose) | Stage of the Trial | No. of Patients | Targeted Disease | Company | Follow Up Period | Clinical Trial No. | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | ARV-110 | Oral (Tablets) once or twice daily for 28 day cycles | Recruiting (Phase II) | 36 | Prostate cancer | Arvinas, USA | 28 days | NCT03888612 | [33] |
| 2 | ARV-471 | Oral | Recruiting (Phase II) | 36 | Breast cancer | Arvinas, Pfizer, USA | 28 days | NCT04072952 | [34] |
| 3 | AC682 | Oral | Recruiting (Phase I) | 42 | Breast cancer | Accutar Biotech, USA | 18 months | NCT05080842 | [35] |
| 4 | ARV-766 | Oral | Recruiting (Phase II) | 60 | Prostate cancer | Arvinas, USA | 6 weeks | NCT05067140 | [36] |
| 5 | CC-94676 | Oral | Recruiting (Phase I) | 40 | Prostate cancer | Celgene, USA | - | NCT04428788 | [37] |
| 6 | DT2216 | Intravenous administration | Recruiting (Phase I) | 24 | Liquid and solid tumors | Dialectic Therapeutics, USA | 28 days | NCT04886622 | [38] |
| 7 | FHD-609 | Intravenous administration | Recruiting (Phase I) | 70 | Synovial sarcoma | Foghorn Therapeutics, USA | 6 weeks | NCT04965753 | [39] |
| 8 | KT-474 | Oral | Completed (Phase I) | 124 | Autoimmune diseases (e.g., AD, HS, RA) | Kymera Therapeutics, USA | 28 days | NCT04772885 | [40] |
| 9 | KT-413 | Intravenous administration | Recruiting (Phase I) | 80 | Diffuse large B cell lymphoma (MYD88-mutant) | Kymera Therapeutics, USA | 18 months | NCT05233033 | [41] |
| 10 | KT-333 | Intravenous administration | Recruiting (Phase I) | 80 | Liquid and solid tumors | Kymera Therapeutics | 18 months | NCT05225584 | [41] |
| 11 | NX-2127 | Oral | Recruiting (Phase I) | 130 | B cell malignancies | Nurix Therapeutics, USA | 6 months | NCT04830137 | [42] |
| 12 | NX-5948 | Oral | Recruiting (Phase I) | 130 | B cell malignancies and autoimmune diseases | Nurix Therapeutics, USA | 100 days | NCT05131022 | [43] |
| 13 | CFT8634 | Oral | Recruiting (Phase II) | 110 | Synovial sarcoma | C4 Therapeutics | 90 days | NCT05355753 | [44] |
| 14 | Protac MyFit | Intravenous administration | Recruiting (Phase I) | 240 | Sensory impairment (SPD) | University of Southern Denmark | 21 days | NCT04173871 | [45] |
| S. No. | Pathogen | Protein of Interest (POI) | POI Ligand | E3 Ligase Ligand | Research Outcome | Ref. |
|---|---|---|---|---|---|---|
| 1 | Hepatitis B virus (HBV) | X-protein of the hepatitis B virus (HBV) | Oxygen-dependent degradation (ODD) domain of hypoxia-inducible factor (HIF-1a) | VHL ligand |
| [48] |
| 2 | Hepatitis C virus (HCV) | HCV NS3/4A protease | Telaprevir | CRBN ligand |
| [50] |
| 3 | Influenza A virus | Viral endonuclease PA | Asperphenalenone E |
| [51] | |
| 4 | Influenza A Virus | Influenza hemagglutinin | Pentacyclic triterpenoid | VHL and CRBN ligand |
| [52] |
| 5 | H1N1 influenza virus | Neuraminidase | Oseltamivir | VHL and CRBN ligand |
| [53] |
| 6 | SARS-CoV-2 virus | The RNA genome of the SARS-CoV-2 virus | RNA attenuator hairpin (AH) |
| [54] | |
| 7 | SARS-CoV-2 virus | Spike protein of SARS-CoV-2 | Antisense oligonucleotide |
| [55] |
| S. No. | Pathogen | Protein of Interest (POI) | POI Ligand | E3 Ligase Ligand | Research Outcome | Ref. |
|---|---|---|---|---|---|---|
| 1 | Human cytomegalovirus (HCMV) | Anti-HCMV AD169-GFP activity | Cyclin-dependent kinase (CDK) inhibitor SNSO32 | CRBN ligand |
| [56] |
| 2 | Syndrome coronavirus-2 (SARS-CoV-2). | Human prostaglandin E synthase type 2 (PGES-2) | Indomethacin | VHL ligand |
| [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatesan, J.; Murugan, D.; Rangasamy, L. A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery. Antibiotics 2022, 11, 1717. https://doi.org/10.3390/antibiotics11121717
Venkatesan J, Murugan D, Rangasamy L. A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery. Antibiotics. 2022; 11(12):1717. https://doi.org/10.3390/antibiotics11121717
Chicago/Turabian StyleVenkatesan, Janarthanan, Dhanashree Murugan, and Loganathan Rangasamy. 2022. "A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery" Antibiotics 11, no. 12: 1717. https://doi.org/10.3390/antibiotics11121717
APA StyleVenkatesan, J., Murugan, D., & Rangasamy, L. (2022). A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery. Antibiotics, 11(12), 1717. https://doi.org/10.3390/antibiotics11121717