Abstract
Type B dihydrofolate reductases (DfrB) are intrinsically highly resistant to the widely used antibiotic trimethoprim, posing a threat to global public health. The ten known DfrB family members have been strongly associated with genetic material related to the application of antibiotics. Several dfrB genes were associated with multidrug resistance contexts and mobile genetic elements, integrated both in chromosomes and plasmids. However, little is known regarding their presence in other environments. Here, we investigated the presence of dfrB beyond the traditional areas of enquiry by conducting metagenomic database searches from environmental settings where antibiotics are not prevalent. Thirty putative DfrB homologues that share 62 to 95% identity with characterized DfrB were identified. Expression of ten representative homologues verified trimethoprim resistance in all and dihydrofolate reductase activity in most. Contrary to samples associated with the use of antibiotics, the newly identified dfrB were rarely associated with mobile genetic elements or antibiotic resistance genes. Instead, association with metabolic enzymes was observed, suggesting an evolutionary advantage unrelated to antibiotic resistance. Our results are consistent with the hypothesis that multiple dfrB exist in diverse environments from which dfrB were mobilized into the clinically relevant resistome. Our observations reinforce the need to closely monitor their progression.
1. Introduction
Trimethoprim (TMP) is a synthetic antibiotic that is intensively used worldwide as a result of its low cost and high effectiveness as a broad-spectrum treatment of bacterial infections [1]. TMP effectively inhibits bacterial dihydrofolate reductases (FolA) (e.g., Ki = 20 pM for Escherichia coli FolA), abrogating the metabolically essential reduction of dihydrofolate (DHF) into tetrahydrofolate (THF) [2]. Shortly after the clinical introduction of TMP in the late 1960′s, TMP-resistant dihydrofolate reductases were identified in clinical samples [1,3]. In addition to TMP-resistant homologues of FolA (known as DfrA) [4], the evolutionarily unrelated type B dihydrofolate reductase (DfrB) DfrB1 was identified. Originally named R67 DHFR, DfrB1 circumvents the inhibition of FolA by TMP through catalysis of the dihydrofolate reduction in the presence of the antibiotic [3,5].
At the outset of this work, there were ten known DfrB family members (DfrB1–11; there is no DfrB8, [6,7,8,9,10,11,12]). All procure high TMP resistance in E. coli (MIC > 600 µg/mL; Ki ~0.38 to 1.3 mM), and most were originally identified in clinical samples [6,7,8,9,10,11,12]. The turnover rates of DfrB enzymes for dihydrofolate reduction (kcat = 0.20–0.41 s−1) are at least 100-fold lower than bacterial FolA (e.g., kcat = 230 s−1 for E. coli FolA); nonetheless, a low level of DfrB expression suffices to confer TMP resistance [13,14].
The absence of structural homology or sequence similarity with the ubiquitous FolA family of enzymes indicates that the DfrB family has a distinct evolutionary origin, where the dihydrofolate reductase (Dfr) activity is a result of functional convergence [4,15]. DfrB are homotetrameric enzymes constituted of four identical, 78-residue SH3-like protomers (Figure 1) [16]. Their highly conserved SH3-like domain (Figure 1 and Figure S1) includes the ‘VQIY’ (V66, Q67, I68 and Y69) catalytic tetrad that forms the single active-site cavity of the homotetramer [17], W38 and W45 for assembly of the functional tetramer [18], and K32 to establish electrostatic interactions with the substrates [4]. Although structural evidence has been obtained only for DfrB1 [16], conservation of all functionally and structurally essential residues as well as their comparable Dfr activity are consistent with adoption of the same functional tetrameric assembly in all DfrB family members [13].
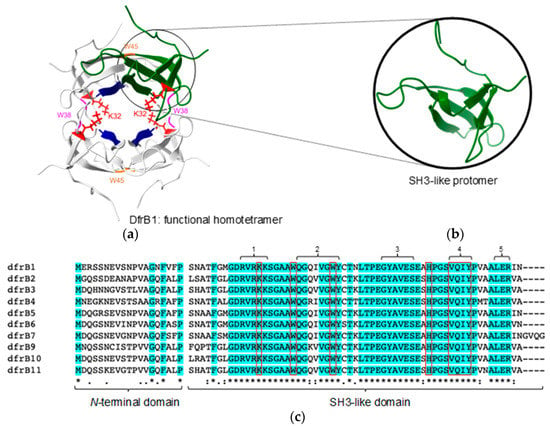
Figure 1.
Structure of DfrB1 and sequence alignment of the known DfrB family members (75–95% sequence identity). (a) The functional, homotetrameric DfrB1 (PDB 1VIE) is constituted of four identical SH3-like protomers (one shown in green) that form the single, central active-site tunnel. The VQIY catalytic tetrad (V66, Q67, I68, and Y69; dark blue) and key residues K32 (red), W38 (magenta) and W45 (orange), are identified. (b) The DfrB1 protomer adopts an SH3-like fold. (c) Multiple sequence alignment of DfrB1–DfrB11 (there is no DfrB8) shows amino acid conservation, using standard annotation beneath the alignment. Conserved residues are highlighted in cyan. The poorly conserved N-terminal domain and the highly conserved SH3-like domain are identified. Functionally and structurally important residues are framed in red.
We recently uncovered the mobility of dfrB genes found in pathogenic bacteria isolated from samples associated with human activity, such as clinical samples [11,12]. However, little is known about the presence or mobility of dfrB genes in environmental settings [19,20]. The small size of dfrB genes (~237 bp) and their unusual codon usage has impeded their discovery when using prokaryotic gene recognition tools, such as Prodigal, which discriminate against both these factors [12,21]. Recent bioinformatic developments facilitate identification of putative DfrB sequences. However, databases such as NCBI tend to be inherently biased towards clinical samples [22]. Metagenomic data can circumvent this limitation, as samples are collected from diverse environmental settings.
Our objective was to determine whether dfrB are identified predominantly in multidrug resistance contexts from samples associated with the use of antibiotics or whether they are also identified beyond those traditional areas of enquiry, without association to antibiotic resistance genes. To do so, we identified ten DfrB family members (DfrB12–DfrB21) from samples not associated with the use of antibiotics through a search of the JGI/IMG (Joint Genome Institute / Integrated Microbial Genomes) metagenomic database [23]. DfrB12–DfrB21 share 63% to 92% protein sequence identity with known DfrB family members (Figure S2). Expression in E. coli and characterization revealed that DfrB12–DfrB21 confer significant TMP resistance, and all but one display catalytic activities comparable to the known DfrB. Using similar search criteria, we identified ten further putative dfrB from the JGI/IMG database and ten more in NCBI to investigate their genomic context. Contrary to dfrB from samples associated with human activity, bioinformatic analyses revealed little association with mobility and multidrug resistance for environmental dfrB from samples not associated with the use of antibiotics. Identification of new dfrB in a variety of environments that are not directly associated with application of antibiotics confirms their widespread presence and suggests that the dfrB observed in the modern resistome may have originated from the mobilization of environmentally sourced dfrB.
2. Results and Discussion
2.1. Expansion of the DfrB Family
Following the recent identification and characterization of two new DfrB family members (DfrB10 and DfrB11) [12], our objective was to investigate whether further new DfrB homologues could be identified in environments that are less likely to be influenced by human activity. To this end, we queried the JGI/IMG metagenomic database and identified over 3000 dfrB gene homologues, from which ten sequences sharing 63 to 92% protein sequence identity were selected to be representative of sequence diversity (Figures S1 and S2). They were defined as DfrB12–DfrB21. High sequence identity of their SH3-like domain suggests that these DfrB12–DfrB21 should fold and tetramerize in a manner analogous to known DfrB enzymes, thus conferring high TMP resistance as a result of their Dfr activity. To investigate this, the minimal inhibitory concentration (MIC) of TMP for E. coli expressing DfrB12–DfrB21 was characterized, followed by determination of Dfr activity in E. coli lysate.
Remarkably, all homologues except DfrB12 provided TMP resistance in E. coli comparable to that of DfrB1, up to the highest soluble concentration of TMP (600 µg/mL) (Figure 2A). Furthermore, activity was clearly observed in clarified lysate of E. coli for all DfrB homologues except DfrB12, which conferred resistance up to 150 µg/mL of TMP (Figure 2B). This apparent discrepancy results from little Dfr activity being required to sustain microbial proliferation, such that MIC assays are more sensitive than activity assays in crude lysate [12].
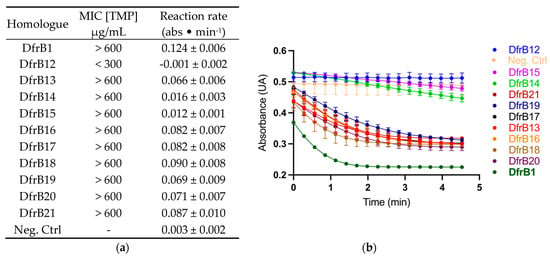
Figure 2.
Newly identified DfrB homologues confer TMP resistance and possess Dfr activity. (a) Minimal inhibitory concentrations were determined on solid media with [TMP] ranging between 0–600 µg/mL. The reported MIC was the lowest TMP concentration where no bacterial growth was observed. The initial rates of reaction were calculated from panel B (n = 3, mean ± SD). The rate of the most active variants is underestimated, since the initial rate was not captured. (b) Dfr activity was determined in E. coli lysate, monitoring substrate consumption as a function of time (n = 3, mean ± SD). The negative control (Neg. Ctrl) is E. coli expressing the cTEM-19m β-lactamase instead of a DfrB.
The lower TMP resistance and Dfr activity of DfrB12 relative to all other DfrB family members is most likely due to the Q67H substitution in the active-site VQIY tetrad (Figure S1). The Q67H mutation has been previously investigated: the mutation improves binding to both DHF and NADPH by 1–2 orders of magnitude compared to the native enzyme [24]. This favors the formation of the nonproductive DHF⋅DHF substrate or NADPH⋅NADPH cofactor complexes, resulting in an important decrease in activity. We note that this lower activity is sufficient to confer some TMP resistance. On the contrary, the 5- to 10-fold lower activity of DfrB14 and DfrB15 relative to DfrB1 is sufficient to confer the highest level of TMP resistance that we can measure. This work having been performed on crude lysate, we have not determined whether the reduced activity results from sequence variation outside of the conserved VQIY active site or other factors, such as reduced expression or stability.
These results confirm that DfrB12–DfrB21 constitute new DfrB family members. This demonstrates that identification of sequences sharing high sequence identity with dfrB1–dfrB11, including functionally and structurally important residues, is sufficient to identify new DfrB family members. This knowledge will facilitate robust identification of DfrB homologues in the future.
2.2. Genomic Context Analysis of DfrB12–21
The DNA sequences containing the newly identified dfrB genes originated from samples isolated from diverse environments not directly associated with the use of antibiotics (Table 1). Consistent with previous studies, dfrB genes were found in Proteobacteria [12]. As the identification of dfrB in diverse environments suggests their widespread presence, we investigated the mobility of the dfrB12–dfrB21 genes by determining whether mobile genetic elements (MGEs, e.g., plasmids, transposons, or integrons) were present in their vicinity. Other antibiotic resistance genes (ARG) were also sought, because a major public health concern is transmission of ARGs associated with MGEs in pathogenic bacteria [25]. To allow comparison to dfrB1–dfrB11, mostly isolated from samples associated with human activity, we characterized the genomic context of dfrB1–dfrB11 according to the same criteria (Figure S3).

Table 1.
Genomic context analysis of dfrB12–21.
First, sequences were classified as plasmidic or chromosomal using PlasForest and PlasFlow (Table S1) [26,27]. The resulting predictions obtained were often contradictory, such that it was difficult to conclude on their organization. The poor quality of predictions was expected, since the majority of analyzed contigs in that dataset were too short (<1 kbp) to allow for confident predictions [26].
The association of dfrB genes with integrons and transposon insertion sequences (IS) was investigated using IntegronFinder and ISFinder, respectively (Table 1) [28,29]. These tools rely on frequently updated databases as references and enable robust and precise identification of MGEs [28,29]. No contig contained transposon IS, but incomplete integrons (CALIN) were identified in the vicinity of dfrB12, dfrB19, and dfrB20 [30]. Both dfrB12 and dfrB19 were integrated within a CALIN element, indicative of potential mobility of those two genes. The dfrB20 gene was found outside of the CALIN identified in its genomic context, the longest obtained (nearly 10 kbp). Analysis of its content using BLASTP indicated 15 hypothetical or metabolism-associated proteins. Though not indicative of mobility of that dfrB, it demonstrates that genetic mobility occurred in the vicinity of the gene. Overall, this dataset contains evidence of genetic mobility in at most three among the dfrB12–dfrB21 genes, in contrast with our earlier findings based on samples closely linked to human activity [12].
The Resistance Gene Identifier (RGI) tool from the Comprehensive Antibiotic Resistance Database (CARD) was used to assess the association of dfrB12–dfrB21 with multidrug resistance (MDR, Table 1) [31]. In contrast to dfrB1–dfrB11, mostly identified in environments associated with the use of antibiotics and generally associated with MGE in a variety of MDR contexts (Table S3) [12], no ARGs were identified in this dfrB12–dfrB21 dataset.
A clear limitation of the current dataset is the short length of the contigs (Table 1). Most genetic contexts were of insufficient length to allow identification of additional genetic features with confidence, indicating that analyses on longer contexts should be conducted.
2.3. The Broader DfrB Sequence Space Includes DfrB of Concern
To gain further information on the genetic context of the dfrB gene family, we identified ten further putative dfrB from a BLASTP search conducted in NCBI (referred to as putative dfrB B1–B10) and ten more from the above-described metagenomic JGI/IMG database search (referred to as putative dfrB C1–C10). We selected sequences with analyzable genomic context (>1 kbp) identified from environments that are not directly associated with the use of antibiotics (e.g., river sediments, soil), although some may be associated with human activity (e.g., polluted river sediment, wastewater). One sequence from a clinical sample (B5) was included as a basis for comparison. Although these new putative DfrB homologues were not functionally characterized, high sequence identity with dfrB1–dfrB21 (63–92%) and conservation of all structurally and functionally important residues are consistent with their being functional DfrB homologues (Figure S3).
All sequences were predicted as chromosomal by PlasForest (Table 2), consistent with recent findings [12]. Complete integrons containing a dfrB gene (putative dfrB B1, B2, B5) and proximal transposases (putative dfrB B1, B5) were found only in contigs from samples collected in environments associated with human activity (Table 2). Strikingly, putative dfrB B1, B2, and B5 were also all associated with MDR (Table 2). Notably, previously known dfrB from clinical samples (dfrB1–5, 9–10; Table S3) are all associated with clinical integrons and are in MDR contexts [12]. This is consistent with human-associated settings procuring higher TMP selective pressure, thus inducing mobilization of dfrB and acting as reservoirs for ARGs [32,33,34]. Our findings suggest that these MGEs have contributed to propagating dfrB from diverse sources into clinically relevant settings.
Table 2.
Genomic context analyses of putative dfrB from BLASTP and JGI/IMG searches.
Additionally, putative dfrB B3 and B6 from water samples are from environments linked to human activity; they were found in MDR contexts but were not associated with MGEs (Table 2). This suggests vertical transmission or loss of mobility after acquisition of ARGs [35]. This was also the case for putative dfrB C3, which was isolated from soil in the Loxahatchee National Wildlife Refuge. The refuge accommodates a wide variety of recreational activities, although it is in a remote location; the relation of the sample to human activity is plausible but is not clear. Most ARGs found in the vicinity of putative dfrB B1–B3, B5-B6, and C3 are related to aminoglycoside resistance (aadA16, AAC(6′)-IIa, ParS, aadA, baeS), consistent with previous findings for dfrB of clinical origin [12]. Association with resistance to rifampin (arr2), chloramphenicol (catB3, cmlA6), beta-lactam (OXA-21), fosfomycin (fosX), polycationic antibiotics (parS), and macrolides (mtrA) was also noted. This demonstrates association of putative dfrB with MDR in environments linked to human activity beyond clinical contexts.
Conversely, indications of genetic mobility were found in the genomic context of putative dfrB B4 and B10 isolated from soil and putative dfrB C1 isolated from freshwater sediment, without association with MDR (Table 2). Strikingly, whereas analyses using CARD reveal no ARGs associated with those putative dfrB, BLASTP analyses of the integron content in the vicinity of putative dfrB C1 indicate the presence of ten proteins associated with metabolism and detoxification. This suggests the coevolution of putative dfrB C1 with metabolism- and defense-associated genes, rather than with antibiotic resistance genes. The remaining putative dfrB isolated from soil (C5, C6, C8–C10) and from water samples (B7, B8, C2, and C7) were not associated with MGEs or MDR (Table 2). These findings suggest that DfrB may confer an evolutionary advantage in environmental contexts that are not directly associated with the use of antibiotics.
All putative dfrB genes were isolated from Proteobacteria as for all known dfrB, most of which have been reported in clinical settings, often linked to ARGs and mobility (Table S3) [12]. Our findings highlight a new pattern: with few exceptions, the 12 putative dfrB genes identified in settings that are not associated with human activity were not associated with ARGs or mobility. Exceptions include the putative dfrB C4 linked with β-lactam resistance (OmpA) and an incomplete transposase (Table 2) and observation of dfrB7 in a clinical integron (Table S3), despite both having been isolated from environmental sources. These examples could be the result of environmental contamination with clinically relevant pathogens [36]. Inversely, putative dfrB B9 was found in a wastewater sample but is not associated with MDR nor MGEs.
As the isolation sources and genomic contexts of these putative dfrB are heterogeneous, more information is needed to conclude on the influence of environment on mobility and prevalence of dfrB. Nonetheless, the association of dfrB from environmental sources with MGEs and/or ARGs demonstrates that the broader DfrB sequence space includes DfrB of concern and justifies the need to closely monitor them.
2.4. DfrB Genes with Similar Level of Mobility Share Closer Evolutionary Relationships
The identification of dfrB genes in various settings led us to investigate whether closer phylogenetic relationships exist between dfrB isolated from similar environments because of a higher likelihood of horizontal gene transfer [37]. To this end, the phylogeny of dfrB1–dfrB21 and the 20 putative dfrB (B1–B10, C1–C10) was reconstructed using IQ-Tree [38].
These results highlight evolutionary proximity between sequences that have similar levels of mobility (Figure 3). For instance, most dfrB contained in integrons and associated with MDR (dfrB1–dfrB5, dfrB9, dfrB10, putative dfrB B1, dfrB B2, dfrB B5) share their closest ancestor with another integron-associated sequence.
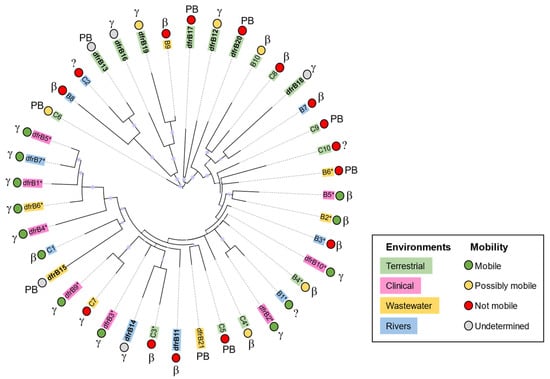
Figure 3.
Phylogenetic tree of dfrB1–dfrB21 and associated putative dfrB genes. Sequences are classified according to their source environment, predicted mobility, and taxonomy. Sequences were aligned using MAFFT; the tree was obtained using IQ-tree and visualized with iTOL. Bootstrap confidence levels are indicated by the size of the circle before each node. Information pertaining to source environment, taxonomy, and predicted mobility is reported in Table 2 and Table S3. The newly identified dfrB12–dfrB21 are in bold. Sequences associated with MDR are marked with an asterisk (*). Taxonomy: “β” for β-Proteobacteria, “γ” for γ-Proteobacteria, “PB” for Proteobacteria; “?” indicates undetermined, as taxonomic information was not available in some cases.
These results also indicate evolutionary proximity between sequences from similar environments in the absence of indicators of mobility. For instance, pairs of dfrB from terrestrial samples (dfrB16 and dfrB19; dfrB12 and dfrB17; putative dfrB C8 and B10) share their most proximal common ancestor, although none hold clear markers of genetic mobility (Figure 3). This could suggest high conservation of the dfrB sequence owing to similar selection pressures from a similar environment and/or loss of mobility of an ancestral dfrB. In contrast, dfrB from samples isolated in aquatic or wastewater that are not associated with mobility are evenly distributed throughout the tree, suggesting that various evolutionary paths define their relationships.
Because all dfrB analyzed were found in Proteobacteria (Tables S2 and S3), it is difficult to distinguish events that are due to taxonomy from those due to horizontal gene transfer in our reconstructed phylogeny. Interestingly, dfrB from the same genus (e.g., Rhodoferax sp., putative dfrB B3 and B9) are not associated with mobility and do not share a close common ancestor. This could reflect different evolutionary pressures from different environments, as putative dfrB B3 was isolated from groundwater, whereas putative dfrB B9 was isolated from activated sludge. This also indicates that dfrB genes can exist in bacterial strains that are not typically associated with clinical settings [39], suggesting that DfrB enzymes could confer an evolutionary advantage in environmental contexts.
3. Conclusions
The results reported here demonstrate, for the first time, the widespread presence of dfrB in a diversity of environments. Most dfrB genes from samples not related to the use of antibiotics were not associated with markers of mobility nor of antibiotic resistance. Their association with metabolically relevant proteins and diverse evolutionary paths suggests that dfrB confer an evolutionary advantage unrelated to antibiotic resistance. Our results are consistent with the hypothesis that such environmentally sourced dfrB have been mobilized into the clinically relevant resistome, where they are associated with markers of mobility and antibiotic resistance. This work highlights the need to closely investigate and monitor their dissemination within the framework of developing therapeutic interventions to counter TMP resistance.
4. Materials and Methods
4.1. Identification of Putative dfrB Genes
Metagenomes deposited in the JGI/IMG database (https://img.jgi.doe.gov/) were queried using a Pfam search for “DHFR_2” on 8 May 2020. This returned a list of 2702 metagenomes, which were filtered for the Pfam keyword “pfam06442” [40]. This resulted in 3116 putative dfrB genes. Non-redundant sequences shorter than 100 amino acids approximating a full-length DfrB (78 amino acids) and starting with a methionine were filtered with CD-HIT [41]. Ten representative sequences sharing at most 95% protein sequence identity with any of the dfrB1–dfrB11 genes were codon-optimized for E. coli and synthesized by Twist Bioscience (South San Francisco, CA, USA). These sequences had been subcloned into expression vector pET29 under control of the IPTG-inducible lac promoter.
Additional putative dfrB genes were identified by filtering the JGI/IMG metagenomic search results based on their nucleotide identity with dfrb1–dfrB21 genes (<95%) and the length of their genomic context (>1 kbp). Complete coding sequences (234 nt = 78 amino acids) were prioritized. To identify further putative dfrB genes, the dfrB1 sequence (Uniprot ID P00383) was used as a query for a BLASTP analysis using default parameters (10 January 2022). Results were filtered with CD-HIT to retain only sequences starting with a methionine and containing 78 amino acids while sharing 60–95% protein sequence identity with dfrB1–dfrB21.
4.2. Minimal Inhibitory Concentration (MIC)
MICs were determined in triplicates using the agar microdilution method. This was done as previously reported [12], with the following modifications. E. coli BL21(DE3) harboring one of the dfrB12–dfrB21 genes, dfrB1 (positive control), and TEM-1 β-lactamase variant cTEM-19m [42] (negative control) were propagated in 1 mL Luria-Bertani (LB) broth for 16–18 h at 37 °C with agitation at 230 rpm. LB–agar plates were prepared containing 0.25 mM IPTG (ThermoFisher). Plates were inoculated with 104 colony-forming units per mL (CFU/mL) and incubated for 16–18 h at 37 °C. The lowest TMP concentration inhibiting visible bacterial growth was considered the MIC.
4.3. Dihydrofolate Reductase Activity Assays in E. coli Lysate
DfrB12–DfrB21, DfrB1 (positive control), and cTEM-19m (negative control) were overexpressed in E. coli BL21(DE3). An overnight (16–18 h) culture in LB (50 µg/mL kanamycin) was used to inoculate 1 mL ZYP-5052 autoinduction media [43] (for 1 L of media: 928 mL of ZY (1% tryptone, 0.5% yeast extract), 50 mL 20 × P (50 mM Na2HPO4, 50 mM KH2PO4, 25 mM (NH4)2SO4), 20 mL 50 × 5052 (0.5% glycerol, 0.05% glucose, 0.2% a-lactose), 2 mL MgSO4 (2 mM), and 0.2 mL 1000 × trace elements (50 mM FeCl3, 20 mM CaCl2, 10 mM MnCl2, 10 mM ZnSO4, 2 mM CoCl2, 2 mM CuCl2, 2 mM NiCl2, 2 mM Na2MoO4, 2 mM Na2SeO3, and 2 mM H3BO3) with 50 µg/mL kanamycin to obtain an initial OD600nm of 0.1. The cultures were incubated at 37 °C, 230 rpm until the OD600nm reached 0.7–1. Incubation was continued at 22 °C, 230 rpm for 16–18 h to allow protein expression. Cells were pelleted at 20,800× g for 30 min at 21 °C, and the pellets were stored at –72 °C until use. The pellets were thawed at room temperature and resuspended in 400 µL of lysis buffer (0.1 M KH2PO4-K2HPO4 (pH 8), 10 mM MgSO4 (Anachemia), 1 mM dithiothreitol (Fisher), 0.5 mg/mL lysozyme (MP Biomedicals), 0.4 U DNAse (Thermo), 1.5 mM benzamidine (Fisher), and 0.25 mM phenylmethylsulfonyl fluoride (Bioshop)) and kept for 2 h at RT with vigorous shaking. The lysates were centrifuged at 20,800× g for 30 min at 21 °C. The clarified lysates were used in subsequent assays.
DHF and NADPH in 50 mM KH2PO4-K2HPO4 (pH 7) were quantified by spectrophotometry (Cary 100 Bio UV-Visible, Agilent) using eDHF282nm 28 400 M−1·cm−1 and eNADPH340nm 6200 M−1·cm−1. In 96-well UV-transparent plates (Corning), 10 µL of clarified lysate was added to 100 µM NADPH and 100 µM DHF in 50 mM KH2PO4-K2HPO4 (pH 7) for a final volume of 100 µL. Enzyme activity was determined by monitoring the depletion of DHF/NADPH at 340 nm with a plate reader (Beckman Coulter DTX880) over 5 min. The initial rate of the reaction was determined by linear regression of the initial rate (first 20% of substrate consumption or the first minute) of depletion of both substrates (∆e340nm 12 300 M−1·cm−1). Assays were carried out in triplicate.
4.4. Genomic Context Analysis
The contigs were classified as plasmidic or chromosomal using PlasForest [26] with the latest release of the NCBI database. Integrons were identified in contigs using IntegronFinder [28]. To perform this search, the local detection (--local-max) and search for promoter and attI sites (--promoter-attI) options were used. Transposon insertion sequences (IS) were identified in contigs using ISFinder BLASTN [29]. Antibiotic resistance genes were identified in contigs using the Resistance Gene Identifier (RGI) tool from the Comprehensive Antibiotic Resistance Database (CARD) [44].
4.5. Phylogenetic Tree
Amino acid sequences were aligned using MAFFT [45] with the default options. A phylogenetic tree was constructed using IQ-Tree [38] with the Ultrafast bootstrap analysis (1000 alignments, 1000 iterations, 0.99 minimum correlation coefficient). Branch support was determined using the SH-aLRT branch test (1000 replicates) and the Approximate Bayes test. The JTT+G4 substitution model was selected using the automatic model selection option. The resulting consensus tree was visualized using iTOL and rooted using the Midpoint root function [46].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics11121768/s1, Figure S1: Multiple sequence alignment of the newly identified dfrB12–dfrB21; Figure S2: Similarity and identity shared between dfrB1–dfrB21; Figure S3: Multiple sequence alignment of 20 newly identified dfrB homologues; Table S1: Prediction of chromosomal or plasmidic location; Table S2: Additional characteristics of the putative dfrB genes; Table S3: Genomic context analyses of dfrB6, dfrB7, dfrB9 and dfrB11 genes.
Author Contributions
S.C.-G. and K.L. conceptualization, methodology, data acquisition and analysis, writing—original draft preparation; C.L.-S.-D. conceptualization, methodology, data acquisition and analysis; P.T. data acquisition; A.B.-B. data acquisition; J.N.C. conceptualization, data acquisition and analysis; J.N.P. conceptualization, writing—review and editing, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NSERC discovery grants RGPIN-N-2018-04686 and the Canada Research Chair in Engineering of Applied Proteins CRC-2020-00171. C.L.-S.-D. and S.C.-G. are grateful to NSERC, FQRNT, and Université de Montréal for scholarships. K.L. was supported by a CREATE-APRENTICE scholarship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or in the supporting information. The accession numbers of analyzed sequences listed are accessible in publicly available databases.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huovinen, P. Trimethoprim resistance. Antimicrob. Agents Chemother. 1987, 31, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Tjong, E.; Dimri, M.; Mohiuddin, S.S. Biochemistry, Tetrahydrofolate. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fleming, M.P.; Datta, N.; Grüneberg, R.N. Trimethoprim resistance determined by R factors. Br. Med. J. 1972, 1, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Howell, E.E. Searching sequence space: Two different approaches to dihydrofolate reductase catalysis. Chembiochem 2005, 6, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, G.M.; Huovinen, P. Resistance to Trimethoprim-Sulfamethoxazole. Clin. Infect. Dis. 2001, 32, 1608–1614. [Google Scholar] [CrossRef]
- L’Abée-Lund, T.M.; Sørum, H. Class 1 integrons mediate antibiotic resistance in the fish pathogen Aeromonas salmonicida worldwide. Microb. Drug Resist. 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Kadlec, K.; Kehrenberg, C.; Schwarz, S. Molecular basis of resistance to trimethoprim, chloramphenicol and sulphonamides in Bordetella bronchiseptica. J. Antimicrob. Chemother. 2005, 56, 485–490. [Google Scholar] [CrossRef]
- Tennstedt, T.; Szczepanowski, R.; Braun, S.; Pühler, A.; Schlüter, A. Occurrence of integron-associated resistance gene cassettes located on antibiotic resistance plasmids isolated from a wastewater treatment plant. FEMS Microbiol. Ecol. 2003, 45, 239–252. [Google Scholar] [CrossRef]
- Barlow, R.S.; Pemberton, J.M.; Desmarchelier, P.M.; Gobius, K.S. Isolation and characterization of integron-containing bacteria without antibiotic selection. Antimicrob. Agents Chemother. 2004, 48, 838–842. [Google Scholar] [CrossRef]
- Sunde, M. Prevalence and characterization of class 1 and class 2 integrons in Escherichia coli isolated from meat and meat products of Norwegian origin. J. Antimicrob. Chemother. 2005, 56, 1019–1024. [Google Scholar] [CrossRef]
- Toulouse, J.L.; Edens, T.J.; Alejaldre, L.; Manges, A.R.; Pelletier, J.N. Integron-Associated DfrB4, a Previously Uncharacterized Member of the Trimethoprim-Resistant Dihydrofolate Reductase B Family, Is a Clinically Identified Emergent Source of Antibiotic Resistance. Antimicrob. Agents Chemother. 2017, 61, e02665-16. [Google Scholar] [CrossRef]
- Lemay-St-Denis, C.; Diwan, S.-S.; Pelletier, J.N. The Bacterial Genomic Context of Highly Trimethoprim-Resistant DfrB Dihydrofolate Reductases Highlights an Emerging Threat to Public Health. Antibiotics 2021, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Toulouse, J.L.; Shi, G.; Lemay-St-Denis, C.; Ebert, M.C.C.J.C.; Deon, D.; Gagnon, M.; Ruediger, E.; Saint-Jacques, K.; Forge, D.; Vanden Eynde, J.J.; et al. Dual-Target Inhibitors of the Folate Pathway Inhibit Intrinsically Trimethoprim-Resistant DfrB Dihydrofolate Reductases. ACS Med. Chem. Lett. 2020, 11, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Brisson, N.; Hohn, T. Nucleotide sequence of the dihydrofolate-reductase gene borne by the plasmid R67 and conferring methotrexate resistance. Gene 1984, 28, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Lemay-St-Denis, C.; Alejaldre, L.; Jemouai, Z.; Lafontaine, K.; St-Aubin, M.; Hitache, K.; Valikhani, D.; Weerasinghe, N.W.; Létourneau, M.; Thibodeaux, C.J.; et al. A conserved SH3-like fold in diverse putative proteins tetramerises into an oxidoreductase providing an antimicrobial resistance phenotype. Philos. Trans. R. Soc. B Biol. Sci. 2022. [Google Scholar] [CrossRef]
- Narayana, N.; Matthews, D.A.; Howell, E.E.; Nguyen-huu, X. A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nat. Struct. Biol. 1995, 2, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Krahn, J.M.; Jackson, M.R.; Derose, E.F.; Howell, E.E.; London, R.E. Crystal Structure of a Type II Dihydrofolate Reductase Catalytic Ternary Complex. Biochemistry 2007, 46, 14878–14888. [Google Scholar] [CrossRef]
- West, F.W.; Seo, H.-S.; Bradrick, T.D.; Howell, E.E. Effects of Single-Tryptophan Mutations on R67 Dihydrofolate Reductase. Biochemistry 2000, 39, 3678–3689. [Google Scholar] [CrossRef]
- Kneis, D.; Berendonk, T.U.; Forslund, S.K.; Hess, S. Antibiotic Resistance Genes in River Biofilms: A Metagenomic Approach toward the Identification of Sources and Candidate Hosts. Environ. Sci. Technol. 2022, 56, 14913–14922. [Google Scholar] [CrossRef]
- Szczepanowski, R.; Linke, B.; Krahn, I.; Gartemann, K.H.; Gützkow, T.; Eichler, W.; Pühler, A.; Schlüter, A. Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiology 2009, 155, 2306–2319. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Ebmeyer, S.; Kristiansson, E.; Larsson, D.G.J. A framework for identifying the recent origins of mobile antibiotic resistance genes. Commun. Biol. 2021, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Nordberg, H.; Shabalov, I.; Aerts, A.; Cantor, M.; Goodstein, D.; Kuo, A.; Minovitsky, S.; Nikitin, R.; Ohm, R.A.; et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012, 40, D26–D32. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bradrick, T.D.; Howell, E.E. A glutamine 67--> histidine mutation in homotetrameric R67 dihydrofolate reductase results in four mutations per single active site pore and causes substantial substrate and cofactor inhibition. Protein Eng. 1997, 10, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L.; Coque, T.M.; Baquero, F. Prioritizing risks of antibiotic resistance genes in all metagenomes. Nat. Rev. Microbiol. 2015, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Pradier, L.; Tissot, T.; Fiston-Lavier, A.S.; Bedhomme, S. PlasForest: A homology-based random forest classifier for plasmid detection in genomic datasets. BMC Bioinform. 2021, 22, 349. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E.P.C. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P.J. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol. 2018, 26, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Pruden, A.; Virta, M.; Zhang, T. Editorial: Antibiotic Resistance in Aquatic Systems. Front. Microbiol. 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Riesenfeld, C.S.; Goodman, R.M.; Handelsman, J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ. Microbiol. 2004, 6, 981–989. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug efflux pumps of gram-negative bacteria. J. Bacteriol. 1996, 178, 5853–5859. [Google Scholar] [CrossRef]
- Abella, J.; Fahy, A.; Duran, R.; Cagnon, C. Integron diversity in bacterial communities of freshwater sediments at different contamination levels. FEMS Microbiol. Ecol. 2015, 91, fiv140. [Google Scholar] [CrossRef] [PubMed]
- Ghaly, T.M.; Gillings, M.R.; Penesyan, A.; Qi, Q.; Rajabal, V.; Tetu, S.G. The Natural History of Integrons. Microorganisms 2021, 9, 2212. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Imhoff, J.F. The Phototrophic Beta-Proteobacteria. In The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 593–601. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Gobeil, S.M.C.; Gagné, D.; Doucet, N.; Pelletier, J.N. 15N, 13C and 1H backbone resonance assignments of an artificially engineered TEM-1/PSE-4 class A β-lactamase chimera and its deconvoluted mutant. Biomol. NMR Assign. 2016, 10, 93–99. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).