
Covalent DNA Binding Is Essential for Gram-Negative Antibacterial Activity of Broad Spectrum Pyrrolobenzodiazepines


Abstract
:1. Introduction
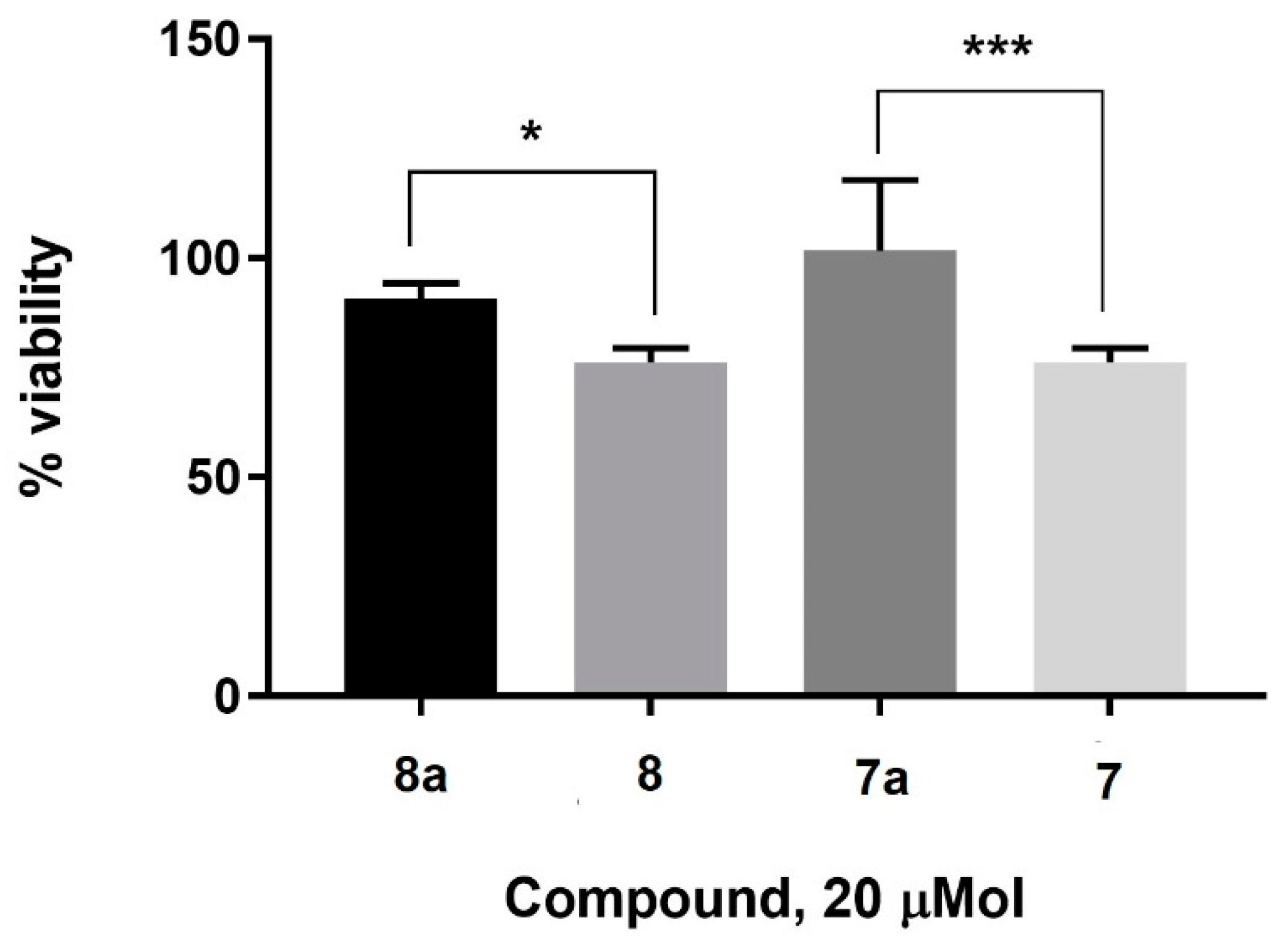
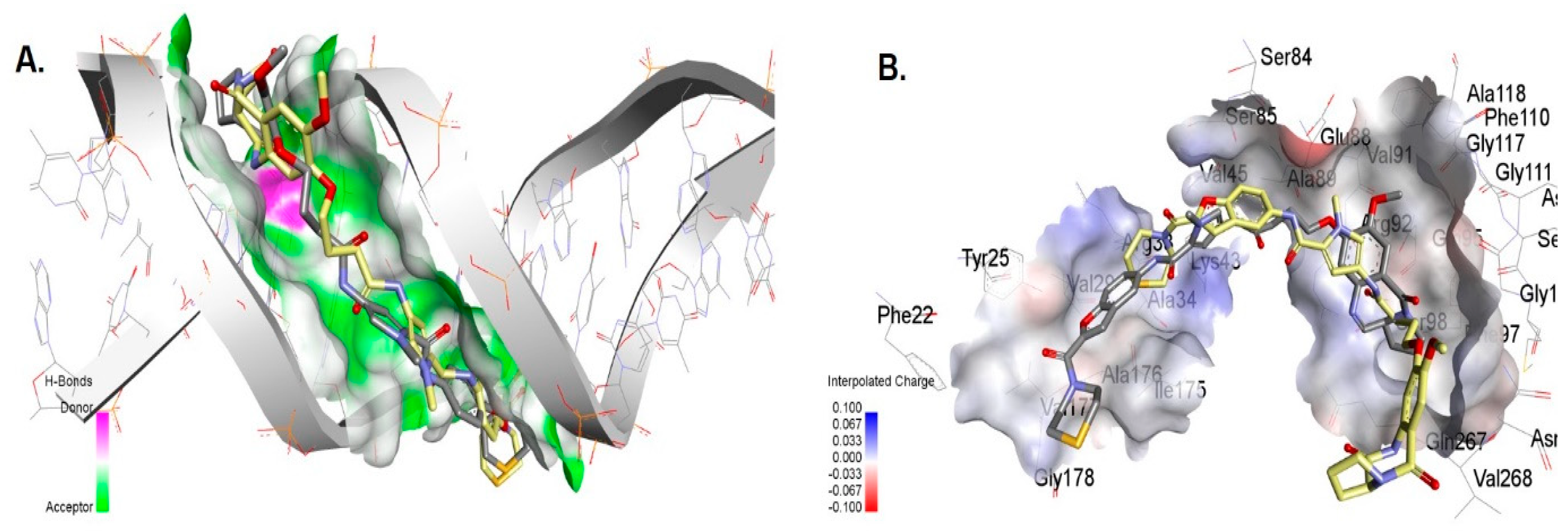
2. Results and Discussion
3. Materials and Methods
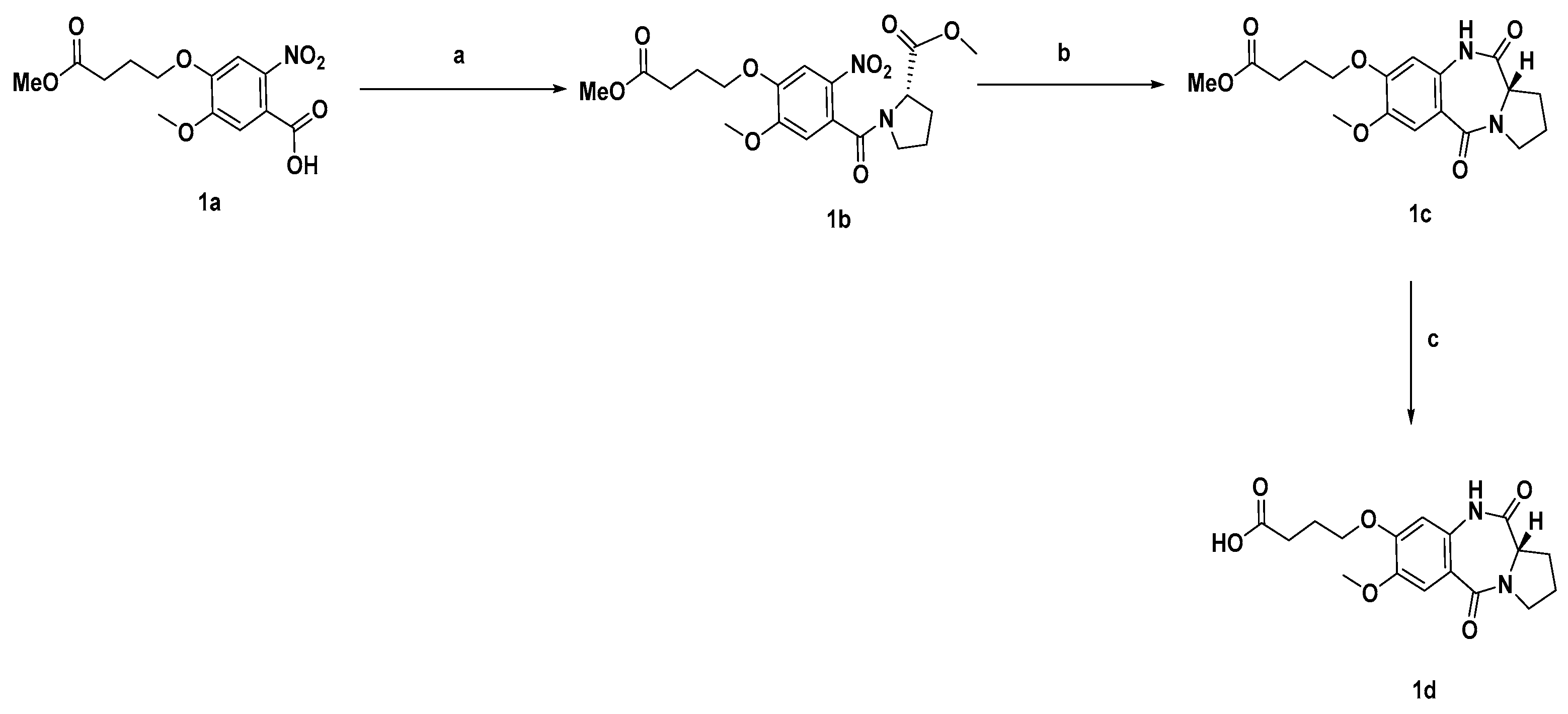
3.1. General Chemistry
3.2. Synthetic Procedures and Characterisation of Compounds
3.3. Cell Culture and MTT Assay
3.4. Bacterial Strains
3.5. Susceptibility Testing
3.6. Molecular Docking of Compounds 7 and 7a with DNA
3.7. Molecular Docking of the Compounds to Gyrase A
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dadgostar, P. Antimicrobial resistance: Implications and costs. Infect. Drug Resist. 2019, 12, 3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougan, G.; Dowson, C.; Overington, J.; Participants, N.G.A.D.S. Meeting the discovery challenge of drug-resistant infections: Progress and focusing resources. Drug Discov. Today 2019, 24, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Provenzani, A.; Hospodar, A.; Meyer, A.; Leonardi Vinci, D.; Hwang, E.; Butrus, C.; Polidori, P. Multidrug-resistant gram-negative organisms: A review of recently approved antibiotics and novel pipeline agents. Int. J. Clin. Pharm. 2020, 42, 1016–1025. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2019, 18, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piddock, L.J.; Paccaud, J.-P.; O’Brien, S.; Childs, M.; Malpani, R.; Balasegaram, M. A Nonprofit Drug Development Model Is Part of the Antimicrobial Resistance (AMR) Solution. Clin. Infect. Dis. 2022, 74, 1866–1871. [Google Scholar] [CrossRef]
- Coates, A.R.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picconi, P.; Hind, C.K.; Nahar, K.S.; Jamshidi, S.; Di Maggio, L.; Saeed, N.; Evans, B.; Solomons, J.; Wand, M.E.; Sutton, J.M.; et al. New Broad-Spectrum Antibiotics Containing a Pyrrolobenzodiazepine Ring with Activity against Multidrug-Resistant Gram-Negative Bacteria. J. Med. Chem. 2020, 63, 6941–6958. [Google Scholar] [CrossRef]
- Antonow, D.; Thurston, D.E. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. [Google Scholar] [CrossRef]
- Gerratana, B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leimgrub, W.; Stefanov, V.; Schenker, F.; Karr, A.; Berger, J. Isolation and Characterization of Anthramycin a New Antitumor Antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Jackson, P.J.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C. GC-targeted C8-linked pyrrolobenzodiazepine–biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, D.B.; Lewis, T.; Nahar, K.S.; Jamshidi, S.; Fegan, C.; Pepper, C.; Thurston, D.E.; Rahman, K.M. Effects of Systematic Shortening of Noncovalent C8 Side Chain on the Cytotoxicity and NF-κB Inhibitory Capacity of Pyrrolobenzodiazepines (PBDs). J. Med. Chem. 2019, 62, 2127–2139. [Google Scholar] [CrossRef]
- Rahman, K.M.; Vassoler, H.; James, C.H.; Thurston, D.E. DNA Sequence Preference and Adduct Orientation of Pyrrolo 2,1-c 1,4 benzodiazepine Antitumor Agents. ACS Med. Chem. Lett. 2010, 1, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.M.; Thompson, A.S.; James, C.H.; Narayanaswamy, M.; Thurston, D.E. The Pyrrolobenzodiazepine Dimer SJG-136 Forms Sequence-Dependent Intrastrand DNA Cross-Links and Monoalkylated Adducts in Addition to Interstrand Cross-Links. J. Am. Chem. Soc. 2009, 131, 13756–13766. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramesh, G.; Laxman, N.; Ramulu, P.; Srinivas, O.; Neelima, K.; Kondapi, A.K.; Sreenu, V.; Nagarajaram, H. Design, synthesis, and evaluation of new noncross-linking pyrrolobenzodiazepine dimers with efficient DNA binding ability and potent antitumor activity. J. Med. Chem. 2002, 45, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Quan, J.; Song, H.; Li, D.; Ma, E.; Wang, Y.; Ma, C. Novel pyrrolo [2,1-c][1,4] benzodiazepine-3,11-dione (PBD) derivatives as selective HDAC6 inhibitors to suppress tumor metastasis and invasion in vitro and in vivo. Bioorg. Chem. 2021, 114, 105081. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, L.; Araújo, A.C.; Airoldi, C.; Bini, D. Pyrrolo [2,1-c][1,4] benzodiazepine as a scaffold for the design and synthesis of anti-tumour drugs. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2009, 9, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberghien, A.C.; Levy, J.-N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and Synthesis of Tesirine, a Clinical Antibody-Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Lee, A. Loncastuximab tesirine: First approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Gregson, S.J.; Masterson, L.A.; Wei, B.; Pillow, T.H.; Spencer, S.D.; Kang, G.-D.; Yu, S.-F.; Raab, H.; Lau, J.; Li, G. Pyrrolobenzodiazepine dimer antibody–drug conjugates: Synthesis and evaluation of noncleavable drug-linkers. J. Med. Chem. 2017, 60, 9490–9507. [Google Scholar] [CrossRef] [PubMed]
- Donnell, A.F.; Zhang, Y.; Stang, E.M.; Wei, D.D.; Tebben, A.J.; Perez, H.L.; Schroeder, G.M.; Pan, C.; Rao, C.; Borzilleri, R.M. Macrocyclic pyrrolobenzodiazepine dimers as antibody-drug conjugate payloads. Bioorg. Med. Chem. Lett. 2017, 27, 5267–5271. [Google Scholar] [CrossRef]
- Andriollo, P.; Hind, C.K.; Picconi, P.; Nahar, K.S.; Jamshidi, S.; Varsha, A.; Clifford, M.; Sutton, J.M.; Rahman, K.M. C8-linked pyrrolobenzodiazepine monomers with inverted building blocks show selective activity against multidrug resistant Gram-positive bacteria. ACS Infect. Dis. 2018, 4, 158–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.M.; Rosado, H.; Moreira, J.B.; Feuerbaum, E.-A.; Fox, K.R.; Stecher, E.; Howard, P.W.; Gregson, S.J.; James, C.H.; de la Fuente, M.; et al. Antistaphylococcal activity of DNA-interactive pyrrolobenzodiazepine (PBD) dimers and PBD-biaryl conjugates. J. Antimicrob. Chemother. 2012, 67, 1683–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjivassileva, T.; Thurston, D.E.; Taylor, P.W. Pyrrolobenzodiazepine dimers: Novel sequence-selective, DNA-interactive, cross-linking agents with activity against Gram-positive bacteria. J. Antimicrob. Chemother. 2005, 56, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, P.J.; Kay, S.; Pysz, I.; Thurston, D.E. Use of pyrrolobenzodiazepines and related covalent-binding DNA-interactive molecules as ADC payloads: Is mechanism related to systemic toxicity? Drug Discov. Today Technol. 2018, 30, 71–83. [Google Scholar] [CrossRef]
- Iacobino, A.; Giannoni, F.; Fattorini, L.; Brucoli, F. Activity of DNA-targeted C8-linked pyrrolobenzodiazepine–heterocyclic polyamide conjugates against aerobically and hypoxically grown Mycobacterium tuberculosis under acidic and neutral conditions. J. Antibiot. 2018, 71, 831–834. [Google Scholar] [CrossRef]
- Antonow, D.; Jenkins, T.C.; Howard, P.W.; Thurston, D.E. Synthesis of a novel C2-aryl pyrrolo [2,1-c][1,4] benzodiazepine-5,11-dione library: Effect of C2-aryl substitution on cytotoxicity and non-covalent DNA binding. Bioorg. Med. Chem. 2007, 15, 3041–3053. [Google Scholar] [CrossRef] [PubMed]
- Laws, M.; Hind, C.; Favaron, A.; Jamshidi, S.; Evans, B.; Clifford, M.; Sutton, J.M.; Rahman, K.M. N1-Benzofused Modification of Fluoroquinolones Reduces Activity Against Gram-Negative Bacteria. ACS Omega 2020, 5, 11923–11934. [Google Scholar] [CrossRef]
- Picconi, P.; Jeeves, R.; Moon, C.W.; Jamshidi, S.; Nahar, K.S.; Laws, M.; Bacon, J.; Rahman, K.M. Noncytotoxic Pyrrolobenzodiazepine–Ciprofloxacin Conjugate with Activity against Mycobacterium Tuberculosis. ACS Omega 2019, 4, 20873–20881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gram-Positive Strains MIC (µg/mL) | ||||||
|---|---|---|---|---|---|---|
| VRE | VSE | MRSA | MSSA | |||
| Compound | NCTC 12201 | NCTC 12204 | NCTC 775 | NCTC 13616 | NCTC 13277 | ATCC 9144 |
| 7a | 8–16 | 8 | 4–8 | >32 | >32 | 4 |
| 7 | 0.03 | 0.25 | 1 | 2 | 2 | 0.5 |
| 8a | 8 | 8 | 16 | 32 | >32 | 8 |
| 8 | 0.03 | 0.03 | 0.03 | 0.03 | 0.06 | 0.03 |
| Gram-Negative Strains MIC (µg/mL) | ||||||
|---|---|---|---|---|---|---|
| A. baumannii | K. pneumoniae | P. aeruginosa | ||||
| Compound | AYE | ATCC 17978 | M6 | NCTC 13368 | PA01 | NCTC 13437 |
| 7a | >32 | >32 | >32 | >32 | >32 | >32 |
| 7 | 2 | 2 | 1 | 32 | >32 | >32 |
| 8a | >32 | >32 | >32 | >32 | >32 | >32 |
| 8 | 1 | 0.5 | 1 | 2 | >32 | >32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picconi, P.; Hind, C.K.; Sutton, J.M.; Rahman, K.M. Covalent DNA Binding Is Essential for Gram-Negative Antibacterial Activity of Broad Spectrum Pyrrolobenzodiazepines. Antibiotics 2022, 11, 1770. https://doi.org/10.3390/antibiotics11121770
Picconi P, Hind CK, Sutton JM, Rahman KM. Covalent DNA Binding Is Essential for Gram-Negative Antibacterial Activity of Broad Spectrum Pyrrolobenzodiazepines. Antibiotics. 2022; 11(12):1770. https://doi.org/10.3390/antibiotics11121770
Chicago/Turabian StylePicconi, Pietro, Charlotte K. Hind, J. Mark Sutton, and Khondaker Miraz Rahman. 2022. "Covalent DNA Binding Is Essential for Gram-Negative Antibacterial Activity of Broad Spectrum Pyrrolobenzodiazepines" Antibiotics 11, no. 12: 1770. https://doi.org/10.3390/antibiotics11121770
APA StylePicconi, P., Hind, C. K., Sutton, J. M., & Rahman, K. M. (2022). Covalent DNA Binding Is Essential for Gram-Negative Antibacterial Activity of Broad Spectrum Pyrrolobenzodiazepines. Antibiotics, 11(12), 1770. https://doi.org/10.3390/antibiotics11121770