
Phage–Antibiotic Therapy as a Promising Strategy to Combat Multidrug-Resistant Infections and to Enhance Antimicrobial Efficiency




Abstract
:1. Introduction
2. Phage–Antibiotic Synergy (PAS)
2.1. Antibiotic-Enhanced Phage Production
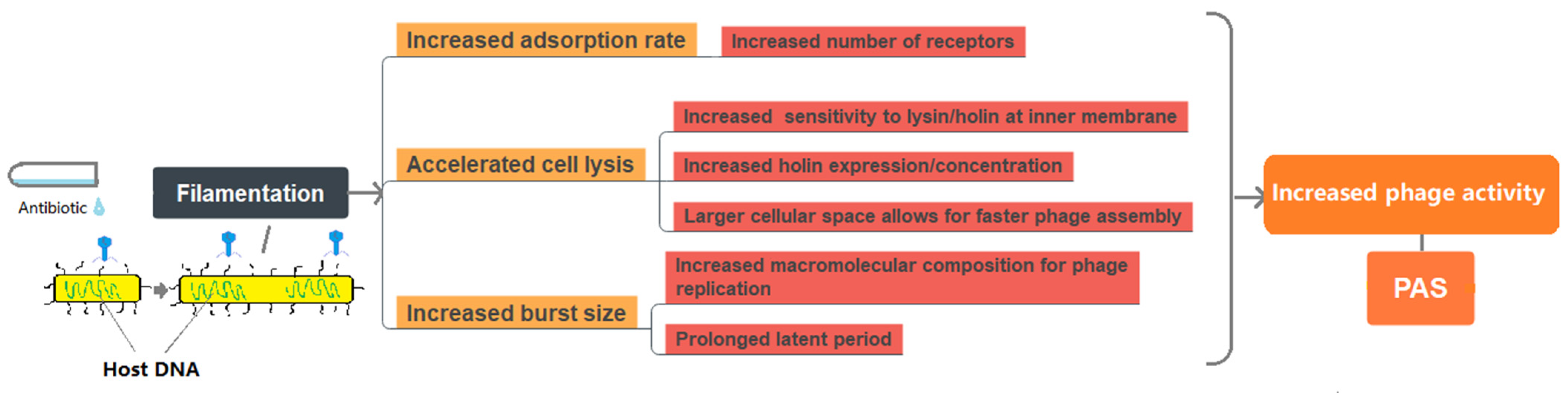
2.2. PAS Induced by Bacterial Filamentation
2.2.1. Improved Phage Adsorption Rate
2.2.2. Accelerated/Delayed Cell Lysis
2.2.3. Increased Single Burst Size
2.3. PAS with Temperate Phages
2.4. Limitations in Assessing PAS
3. The Development of Bacterial Resistance to Phages and Antibiotics
4. Applications of Phage–Antibiotic Therapy
4.1. Reduced Antibiotic Dose for Phage–Antibiotic Treatment
4.2. Effect of Administration Time and Sequence on Bacteria Inhibition
4.2.1. Aminoglycosides
4.2.2. Ciprofloxacin and Tetracycline
4.2.3. The Impact of Host Strains in Sequential Treatment and Host Environment on the Efficacy of PAS Treatment
4.3. In Vivo Efficiency of Phage and Antibiotic Therapy
4.4. Clinical Case Studies
5. Challenges
5.1. Polymicrobial System and Biofilm
5.2. Mutation Dynamics of Bacteria and Phages in the Presence of Anitbiotics
5.3. Phage–Antibiotic Pharmaceutics Development
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rossolini, G.M.; Arena, F.; Pecile, P.; Pollini, S. Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [Google Scholar] [CrossRef]
- De-Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 24 February 2022).
- Antibiotic Resistance Threats in the United States. 2019. Available online: https://stacks.cdc.gov/view/cdc/82532 (accessed on 3 April 2022).
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef]
- Housby, J.N.; Mann, N.H. Phage therapy. Drug Discov. Today 2009, 14, 536–540. [Google Scholar] [CrossRef]
- Eaton, M.D.; Bayne-Jones, S. Bacteriophage therapy—review of the principles and results of the use of bacteriophage in the treatment of infections. JAMA 1934, 103, 1769–1776. [Google Scholar] [CrossRef]
- Bao, J.; Wu, N.; Zeng, Y.; Chen, L.; Li, L.; Yang, L.; Zhang, Y.; Guo, M.; Li, L.; Li, J.; et al. Non-active antibiotic and bacteriophage synergism to successfully treat recurrent urinary tract infection caused by extensively drug-resistant Klebsiella pneumoniae. Emerg. Microbes Infect. 2020, 9, 771–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Abedon, S.T. Bacteriophage Host Range and Bacterial Resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.L.G.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [Green Version]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Leon, M.; Bastias, R. Virulence reduction in bacteriophage resistant bacteria. Front. Microbiol. 2015, 6, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akturk, E.; Oliveira, H.; Santos, S.B.; Costa, S.; Kuyumcu, S.; Melo, L.D.R.; Azeredo, J. Synergistic action of phage and antibiotics: Parameters to enhance the killing efficacy against mono and dual-species biofilms. Antibiotics 2019, 8, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopatina, A.; Tal, N.; Sorek, R. Abortive infection: Bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 2020, 7, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Rostøl, J.T.; Marraffini, L. (Ph)ighting phages: How bacteria resist their parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [Green Version]
- Engeman, E.; Freyberger, H.R.; Corey, B.W.; Ward, A.M.; He, Y.; Nikolich, M.P.; Filippov, A.A.; Tyner, S.D.; Jacobs, A.C. Synergistic killing and re-sensitization of Pseudomonas aeruginosa to antibiotics by phage-antibiotic combination treatment. Pharmaceuticals 2021, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Petsong, K.; Uddin, M.J.; Vongkamjan, K.; Ahn, J. Combined effect of bacteriophage and antibiotic on the inhibition of the development of antibiotic resistance in Salmonella typhimurium. Food Sci. Biotechnol. 2018, 27, 1239–1244. [Google Scholar] [CrossRef]
- Torres-Barcelo, C.; Hochberg, M.E. Evolutionary rationale for phages as complements of antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Tetart, F.; Trojet, S.N.; Prere, M.F.; Krisch, H.M. Phage-Antibiotic Synergy (PAS): Beta-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Chang, R.Y.K.; Britton, W.J.; Morales, S.; Kutter, E.; Chan, H.K. Synergy of nebulized phage PEV20 and ciprofloxacin combination against Pseudomonas aeruginosa. Int. J. Pharm. 2018, 551, 158–165. [Google Scholar] [CrossRef]
- Lin, Y.; Quan, D.; Chang, R.Y.K.; Chow, M.Y.T.; Wang, Y.; Li, M.; Morales, S.; Britton, W.J.; Kutter, E.; Li, J.; et al. Synergistic activity of phage PEV20-ciprofloxacin combination powder formulation-A proof-of-principle study in a P. aeruginosa lung infection model. Eur. J. Pharm. Biopharm. 2021, 158, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.M.; Mccutcheon, J.G.; Dennis, J.J. Aztreonam lysine increases the activity of phages E79 and phiKZ against Pseudomonas aeruginosa PA01. Microorganisms 2021, 9, 152. [Google Scholar] [CrossRef]
- Kim, M.; Jo, Y.; Hwang, Y.J.; Hong, H.W.; Hong, S.S.; Park, K.; Myung, H. Phage-Antibiotic Synergy via delayed lysis. Appl. Environ. Microbiol. 2018, 84, e02085-18. [Google Scholar] [CrossRef] [Green Version]
- Kirby, A.E. Synergistic action of gentamicin and bacteriophage in a continuous culture population of Staphylococcus aureus. PLoS ONE 2012, 7, e51017. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Harjai, K.; Chhibber, S. Methicillin-resistant Staphylococcus aureus phage plaque size enhancement using sublethal concentrations of antibiotics. Appl. Environ. Microbiol. 2012, 78, 8227–8233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex Phage-Antibiotic Synergy (PAS): Antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 2015, 81, 1132–1138. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T.; Yin, J. Bacteriophage plaques: Theory and analysis. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 1, pp. 161–174. [Google Scholar] [CrossRef]
- Koch, A.L. The growth of viral plaques during the enlargement phase. J. Theor. Biol. 1964, 6, 413–431. [Google Scholar] [CrossRef]
- Yin, J.; Mccaskill, J.S. Replication of viruses in a growing plaque: A reaction-diffusion model. Biophys. J. 1992, 61, 1540–1549. [Google Scholar] [CrossRef]
- Gallet, R.; Kannoly, S.; Wang, I.-N. Effects of bacteriophage traits on plaque formation. BMC Microbiol. 2011, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Fort, J.; Méndez, V. Time-delayed spread of viruses in growing plaques. Phys. Rev. Lett. 2002, 89, 178101. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Cejas, V.; Fort, J.; Mendez, V.; Campos, D. Approximate solution to the speed of spreading viruses. Phys. Rev. E 2004, 69, 031909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T.; Culler, R.R. Bacteriophage evolution given spatial constraint. J. Theor. Biol. 2007, 248, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; Habel, A.; Bläsi, U. Augmentation of the antimicrobial efficacy of antibiotics by filamented phages. Microb. Drug Resist. 2006, 12, 164–168. [Google Scholar] [CrossRef]
- Kong, K.-F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Otsuki, M.; Nishino, T. In vitro and in vivo activities of DQ-2556 and its mode of action. Antimicrob. Agents Chemother. 1992, 36, 2595–2601. [Google Scholar] [CrossRef] [Green Version]
- Curtis, N.A.; Orr, D.; Ross, G.W.; Boulton, M.G. Affinities of penicillins and cephalosporins for the penicillin-binding proteins of Escherichia coli K-12 and their antibacterial activity. Antimicrob. Agents Chemother. 1979, 16, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Horii, T.; Kobayashi, M.; Sato, K.; Ichiyama, S.; Ohta, M. An in-vitro study of carbapenem-induced morphological changes and endotoxin release in clinical isolates of gram-negative bacilli. J. Antimicrob. Chemother. 1998, 41, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Cushnie, T.P.; O’driscoll, N.H.; Lamb, A.J. Morphological and ultrastructural changes in bacterial cells as an indicator of antibacterial mechanism of action. Cell. Mol. Life Sci. 2016, 73, 4471–4492. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.S.J.; Shelton, A.; Greenwood, D. The response of Escherichia coli to ciprofloxacin and norfloxacin. J. Med. Microbiol. 1987, 23, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Pangborn, J.; Kilgore, W.W. Filamentous cells of Escherichia coli formed in the presence of mitomycin. J. Bacteriol. 1967, 93, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, C.; Amyes, S. The role of the SOS response in bacteria exposed to zidovudine or trimethoprim. J. Med. Microbiol. 1991, 34, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, A.M.; Heyer, A.; Kramer, C.; Grunberger, A.; Kohlheyer, D.; Frunzke, J. Analysis of SOS-induced spontaneous prophage induction in Corynebacterium glutamicum at the single-cell level. J. Bacteriol. 2014, 196, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Bergersen, F.J. Cytological changes induced in Bacterium coli by chloramphenicol. Microbiology 1953, 9, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilleland, L.B.; Gilleland, H.E.; Gibson, J.A.; Champlin, F.R. Adaptive resistance to aminoglycoside antibiotics in Pseudomonas aeruginosa. J. Med. Microbiol. 1989, 29, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Someya, A.; Tanaka, K.; Tanaka, N. Morphological changes of Escherichia coli induced by bicyclomycin. Antimicrob. Agents Chemother. 1979, 16, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Hadas, H.; Einav, M.; Fishov, I.; Zaritsky, A. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 1997, 143, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhuba, D.; Kolomiets, E.; Dey, E.S.; Novik, G. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145. [Google Scholar] [CrossRef]
- Cahill, J.; Young, R. Phage lysis: Multiple genes for multiple barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [CrossRef]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications, 1st ed.; CRC Press: Boca Raton, FL, USA, 2004; p. 528. [Google Scholar] [CrossRef]
- Ryan, E.M.; Alkawareek, M.Y.; Donnelly, R.F.; Gilmore, B.F. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 2012, 65, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easwaran, M.; De Zoysa, M.; Shin, H.J. Application of phage therapy: Synergistic effect of phage EcSw (PhiEcSw) and antibiotic combination towards antibiotic-resistant Escherichia coli. Transbound. Emerg. Dis. 2020, 67, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage latent-period evolution as a response to resource availability. Appl Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, W. Lysis inhibition in Escherichia coli infected with bacteriophage T4. J. Virol. 1967, 1, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T. Lysis of lysis-inhibited bacteriophage T4-infected cells. J. Bacteriol. 1992, 174, 8073–8080. [Google Scholar] [CrossRef] [Green Version]
- Al-Anany, A.M.; Fatima, R.; Hynes, A.P. Temperate phage-antibiotic synergy eradicates bacteria through depletion of lysogens. Cell Rep. 2021, 35, 109172. [Google Scholar] [CrossRef]
- Santos, S.B.; Carvalho, C.M.; Sillankorva, S.; Nicolau, A.; Ferreira, E.C.; Azeredo, J. The use of antibiotics to improve phage detection and enumeration by the double-layer agar technique. BMC Microbiol. 2009, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, D.; Kolter, R. Why are bacteria refractory to antimicrobials? Curr. Opin. Microbiol. 2002, 5, 472–477. [Google Scholar] [CrossRef]
- Chow, M.Y.T.; Chang, R.Y.K.; Li, M.; Wang, Y.; Lin, Y.; Morales, S.; Mclachlan, A.J.; Kutter, E.; Li, J.; Chan, H.K. Pharmacokinetics and time-kill study of inhaled antipseudomonal bacteriophage therapy in mice. Antimicrob. Agents Chemother. 2020, 65, e01470-20. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Chow, M.Y.T.; Wang, Y.; Liu, C.; Hong, Q.; Morales, S.; Mclachlan, A.J.; Kutter, E.; Li, J.; Chan, H.K. The effects of different doses of inhaled bacteriophage therapy for Pseudomonas aeruginosa pulmonary infections in mice. Clin. Microbiol. Infect. 2022; ahead of print. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Das, T.; Manos, J.; Kutter, E.; Morales, S.; Chan, H.K. Bacteriophage PEV20 and ciprofloxacin combination treatment enhances removal of Pseudomonas aeruginosa biofilm isolated from cystic fibrosis and wound patients. AAPS J. 2019, 21, 49. [Google Scholar] [CrossRef]
- Sturino, J.M.; Klaenhammer, T.R. Inhibition of bacteriophage replication in Streptococcus thermophilus by subunit poisoning of primase. Microbiology 2007, 153, 3295–3302. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Pereira, C.; Almeida, A. Sequential combined effect of phages and antibiotics on the inactivation of Escherichia coli. Microorganisms 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Blasco, L.; Ambroa, A.; Lopez, M.; Fernandez-Garcia, L.; Bleriot, I.; Trastoy, R.; Ramos-Vivas, J.; Coenye, T.; Fernandez-Cuenca, F.; Vila, J.; et al. Combined use of the Ab105-2phiDeltaCI lytic putant phage and different antibiotics in clinical isolates of multi-resistant Acinetobacter baumannii. Microorganisms 2019, 7, 556. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Concepcion-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef]
- Torres-Barcelo, C.; Arias-Sanchez, F.I.; Vasse, M.; Ramsayer, J.; Kaltz, O.; Hochberg, M.E. A window of opportunity to control the bacterial pathogen Pseudomonas aeruginosa combining antibiotics and phages. PLoS ONE 2014, 9, e106628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, P.; Yu, P.; Alvarez, P.J.J. Aminoglycosides antagonize bacteriophage proliferation, attenuating phage suppression of bacterial growth, biofilm formation, and antibiotic resistance. Appl. Environ. Microbiol. 2021, 87, e0046821. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, A.; Tanaka, N. Differential effects of aminoglycosides on cistron-specific initiation of protein synthesis. Biochem. Biophys. Res. Commun. 1972, 49, 951–957. [Google Scholar] [CrossRef]
- Jiang, Z.; Wei, J.; Liang, Y.; Peng, N.; Li, Y. Aminoglycoside antibiotics inhibit mycobacteriophage infection. Antibiotics 2020, 9, 714. [Google Scholar] [CrossRef]
- Constantinou, A.; Voelkel-Meiman, K.; Sternglanz, R.; Mccorquodale, M.M.; Mccorquodale, D.J. Involvement of host DNA gyrase in growth of bacteriophage T5. J. Virol. 1986, 57, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, G.; Ahn, J. Assessment of phage-mediated inhibition of Salmonella Typhimurium treated with sublethal concentrations of ceftriaxone and ciprofloxacin. FEMS Microbiol. Lett. 2020, 367, fnaa159. [Google Scholar] [CrossRef]
- Kumaran, D.; Taha, M.; Yi, Q.; Ramirez-Arcos, S.; Diallo, J.S.; Carli, A.; Abdelbary, H. Does treatment order matter? Investigating the ability of bacteriophage to augment antibiotic activity against Staphylococcus aureus biofilms. Front. Microbiol. 2018, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tkhilaishvili, T.; Trampuz, A. Adjunctive use of phage Sb-1 in antibiotics enhances inhibitory biofilm growth activity versus rifampin-resistant Staphylococcus aureus strains. Antibiotics 2020, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tkhilaishvili, T.; Bernal Andres, B.; Trampuz, A.; Gonzalez Moreno, M. Bacteriophage-antibiotic combinations against ciprofloxacin/ceftriaxone-resistant Escherichia coli in vitro and in an experimental Galleria mellonella model. Int. J. Antimicrob. Agents 2020, 56, 106200. [Google Scholar] [CrossRef]
- Liu, C.G.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-Antibiotic Synergy is driven by a unique combination of antibacterial mechanism of action and stoichiometry. MBio 2020, 11, e01462-20. [Google Scholar] [CrossRef]
- Thornton, L.A.; Burchell, R.K.; Burton, S.E.; Lopez-Villalobos, N.; Pereira, D.; Macewan, I.; Fang, C.; Hatmodjo, A.C.; Nelson, M.A.; Grinberg, A.; et al. The effect of urine concentration and pH on the growth of Escherichia coli in canine urine in vitro. J. Vet.-Intern. Med. 2018, 32, 752–756. [Google Scholar] [CrossRef]
- Pacios, O.; Fernández-García, L.; Bleriot, I.; Blasco, L.; González-Bardanca, M.; López, M.; Fernández-Cuenca, F.; Oteo, J.; Pascual, Á.; Martínez-Martínez, L.; et al. Enhanced antibacterial activity of repurposed mitomycin C and imipenem in combination with the lytic phage vB_KpnM-VAC13 against clinical isolates of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2021, 65, e0090021. [Google Scholar] [CrossRef]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.A. Synergistic interaction between phage therapy and antibiotics clears Pseudomonas Aeruginosa infection in endocarditis and reduces virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Harjai, K.; Chhibber, S. In vivo assessment of phage and linezolid based implant coatings for treatment of methicillin resistant S. aureus (MRSA) mediated orthopaedic device related infections. PLoS ONE 2016, 11, e0157626. [Google Scholar] [CrossRef] [Green Version]
- Chhibber, S.; Kaur, T.; Sandeep, K. Co-therapy using lytic bacteriophage and linezolid: Effective treatment in eliminating methicillin resistant Staphylococcus aureus (MRSA) from diabetic foot infections. PLoS ONE 2013, 8, e56022. [Google Scholar] [CrossRef]
- Tan, X.; Chen, H.; Zhang, M.; Zhao, Y.; Jiang, Y.; Liu, X.; Huang, W.; Ma, Y. Clinical experience of personalized phage therapy against carbapenem-resistant Acinetobacter baumannii lung infection in a patient with chronic obstructive pulmonary disease. Front. Cell. Infect. Microbiol. 2021, 11, 631585. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47.e34. [Google Scholar] [CrossRef] [PubMed]
- Gainey, A.B.; Burch, A.-K.; Brownstein, M.J.; Brown, D.E.; Fackler, J.; Horne, B.A.; Biswas, B.; Bivens, B.N.; Malagon, F.; Daniels, R. Combining bacteriophages with cefiderocol and meropenem/vaborbactam to treat a pan-drug resistant Achromobacter species infection in a pediatric cystic fibrosis patient. Pediatr. Pulmonol. 2020, 55, 2990–2994. [Google Scholar] [CrossRef]
- Moulton-Brown, C.E.; Friman, V.P. Rapid evolution of generalized resistance mechanisms can constrain the efficacy of phage-antibiotic treatments. Evol. Appl. 2018, 11, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Buckling, A.; Rainey, P.B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. Biol. Sci. 2002, 269, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, J.; Becks, L.; Jalasvuori, M.; Hiltunen, T. Sublethal streptomycin concentrations and lytic bacteriophage together promote resistance evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, A.M.L.; Fairhead, H.I.M. A commentary on the development of engineered phage as therapeutics. Drug Discov. Today 2021, 26, 2095–2098. [Google Scholar] [CrossRef]
- Kebriaei, R.; Lev, K.L.; Stamper, K.C.; Lehman, S.M.; Morales, S.; Rybak, M.J. Bacteriophage AB-SA01 cocktail in combination with antibiotics against MRSA-VISA strain in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 2020, 65, e01863-20. [Google Scholar] [CrossRef] [PubMed]
- Luscher, A.; Simonin, J.; Falconnet, L.; Valot, B.; Hocquet, D.; Chanson, M.; Resch, G.; Kohler, T.; Van Delden, C. Combined bacteriophage and antibiotic treatment prevents Pseudomonas aeruginosa infection of wild type and cftr-epithelial cells. Front. Microbiol. 2020, 11, 1947. [Google Scholar] [CrossRef]
- Kaur, P.; Gondil, V.S.; Chhibber, S. A novel wound dressing consisting of PVA-SA hybrid hydrogel membrane for topical delivery of bacteriophages and antibiotics. Int. J. Pharm. 2019, 572, 118779. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chang, R.Y.K.; Britton, W.J.; Morales, S.; Kutter, E.; Li, J.; Chan, H.K. Inhalable combination powder formulations of phage and ciprofloxacin for P. aeruginosa respiratory infections. Eur. J. Pharm. Biopharm. 2019, 142, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Gorski, A.; Borysowski, J.; Miedzybrodzki, R. Phage therapy: Towards a successful clinical trial. Antibiotics 2020, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Mutti, M.; Corsini, L. Robust approaches for the production of active ingredient and drug product for human phage therapy. Front. Microbiol. 2019, 10, 2289. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Animal Model | Bacterial Species | Drugs Used | Phage | Highlights |
|---|---|---|---|---|
| Moth larvae (Galleria mellonella) [85] | Klebsiella pneumoniae | Mitomycin C, imipenem (1/4 × MIC, 1/2 × MIC) | vB_KpnM-VAC13 (107 or 109 PFU/mL) | Survival rate of larvae significantly increased to 50% and 75% when phage co-treated with mitomycin C and imipenem, respectively, in treating resistant strain and persisted strain, compared to either monotherapy, except for phage/imipenem on resistant strain. |
| Moth larvae (Galleria mellonella) [71] | Acinetobacter baumannii | Imipenem, meropenem (1/4 × MIC, 1/8 × MIC) | Ab105-2φ∆CI (108 PFU/mL) | Combination therapy and meropenem alone had same survival rate; both survival rates were higher than phage monotreatment (p < 0.05); imipenem combined with phage showed high survival rate compared to monotherapy (p < 0.05). |
| Mouse: lung infection [25] | Pseudomonas aeruginosa | Ciprofloxacin (0.33 mg/mg) | PEV20 (106 PFU/mg) | PEV20 combined with ciprofloxacin significantly decreased bacterial concentration by 5.9 log, where either monotherapy showed no obvious bacterial reduction. |
| Moth larvae (Galleria mellonella) [82] | Escherichia coli | Fosfomycin (200 mg/kg) | fWL-3 (107 PFU) | Simultaneous treatment with phage and fosfomycin had higher survival rate than sequential treatment in both EC1 and ATCC 25922 strains. Phage and fosfomycin showed 75% of survival rate in ATCC 25922 strain. |
| Rat [86] | Pseudomonas aeruginosa | Ciprofloxacin (0.19 μg/mL) | Phage cocktail PP1131 Bolus injection (1 mL of 1010 PFU/mL in 1 min) Continuous infusion (0.1 mL/h of 1010 PFU/mL over 24 h) | Phage/ciprofloxacin exerted highest synergistic effects with 6 log bacterial reduction and achieved 64% reduction in bacterial infection. No phage-resistant mutants in vivo. |
| Mouse: prosthetic joint infection [87] | Methicillin-resistant Staphylococcus aureus | Linezolid (5% w/w) | MR-4 (109 PFU/mL) | The combined treatment with phage and linezolid maximised the mice locomotor activity, reduced oedema at the affected limb, and significantly reduced the bacterial burden (~4.5 log) as compared with the untreated control. |
| Mouse: diabetic foot infection [88] | Methicillin-resistant Staphylococcus aureus | Linezolid (25 mg/kg) | MR-10 (108 PFU/mL) | The combination of phage and linezolid demonstrated a high antimicrobial effect in reducing the bacterial load (5 log) and lesion level. Healing was accelerated at Day 7 after the co-treatment compared to the untreated control (Day 12). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Hong, Q.; Chang, R.Y.K.; Kwok, P.C.L.; Chan, H.-K. Phage–Antibiotic Therapy as a Promising Strategy to Combat Multidrug-Resistant Infections and to Enhance Antimicrobial Efficiency. Antibiotics 2022, 11, 570. https://doi.org/10.3390/antibiotics11050570
Liu C, Hong Q, Chang RYK, Kwok PCL, Chan H-K. Phage–Antibiotic Therapy as a Promising Strategy to Combat Multidrug-Resistant Infections and to Enhance Antimicrobial Efficiency. Antibiotics. 2022; 11(5):570. https://doi.org/10.3390/antibiotics11050570
Chicago/Turabian StyleLiu, Chengxi, Qixuan Hong, Rachel Yoon Kyung Chang, Philip Chi Lip Kwok, and Hak-Kim Chan. 2022. "Phage–Antibiotic Therapy as a Promising Strategy to Combat Multidrug-Resistant Infections and to Enhance Antimicrobial Efficiency" Antibiotics 11, no. 5: 570. https://doi.org/10.3390/antibiotics11050570
APA StyleLiu, C., Hong, Q., Chang, R. Y. K., Kwok, P. C. L., & Chan, H.-K. (2022). Phage–Antibiotic Therapy as a Promising Strategy to Combat Multidrug-Resistant Infections and to Enhance Antimicrobial Efficiency. Antibiotics, 11(5), 570. https://doi.org/10.3390/antibiotics11050570