
Antimicrobial Resistance and Comparative Genomic Analysis of Elizabethkingia anophelis subsp. endophytica Isolated from Raw Milk

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Isolation and Phenotypic Characterization of the E. anophelis Subsp. endophytica ML-44 Strain
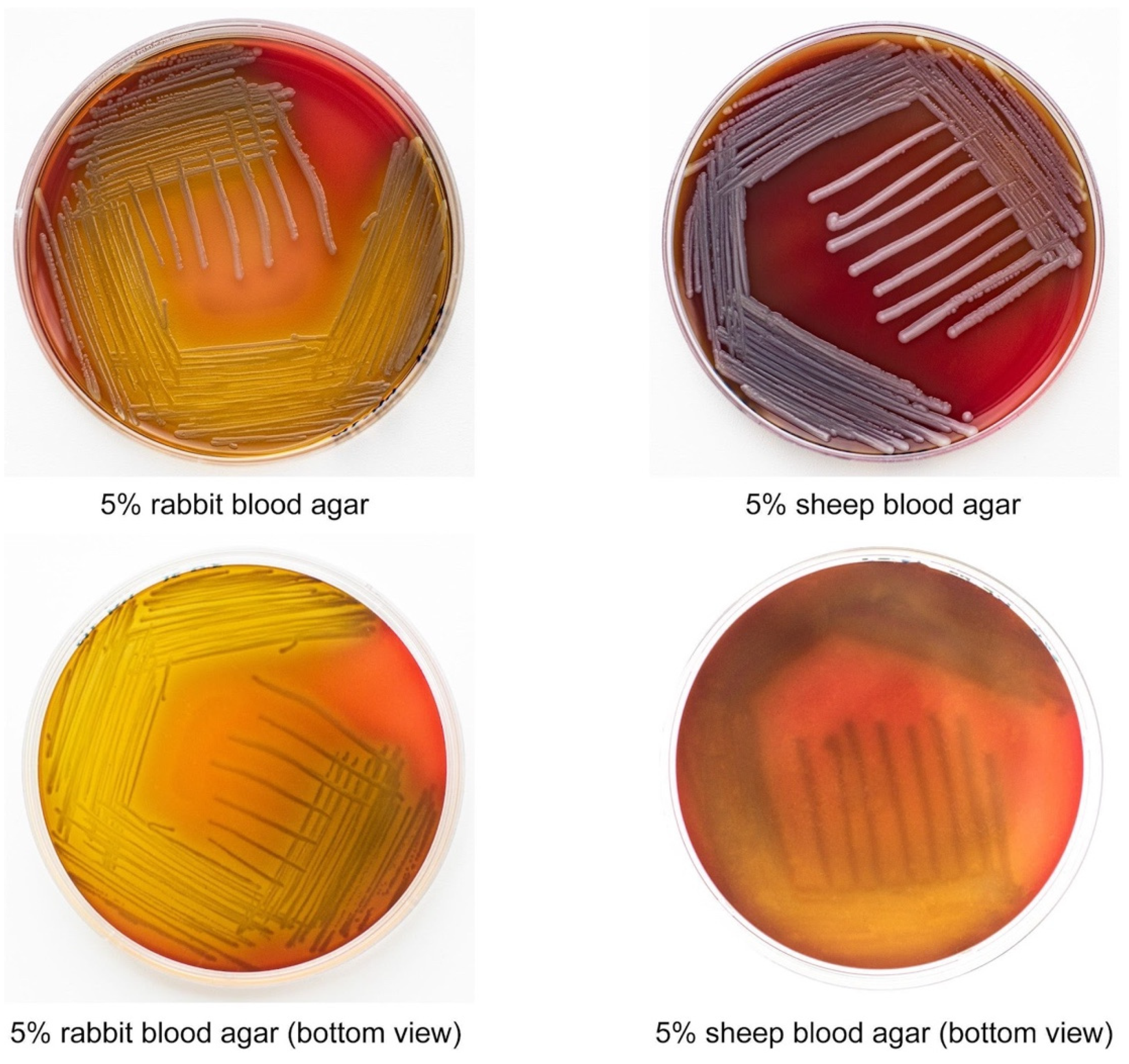
2.2. E. anophelis ML-44 Possesses an Alpha-Hemolytic Activity
2.3. E. anophelis ML-44 Showed a Multidrug-Resistant Phenotype
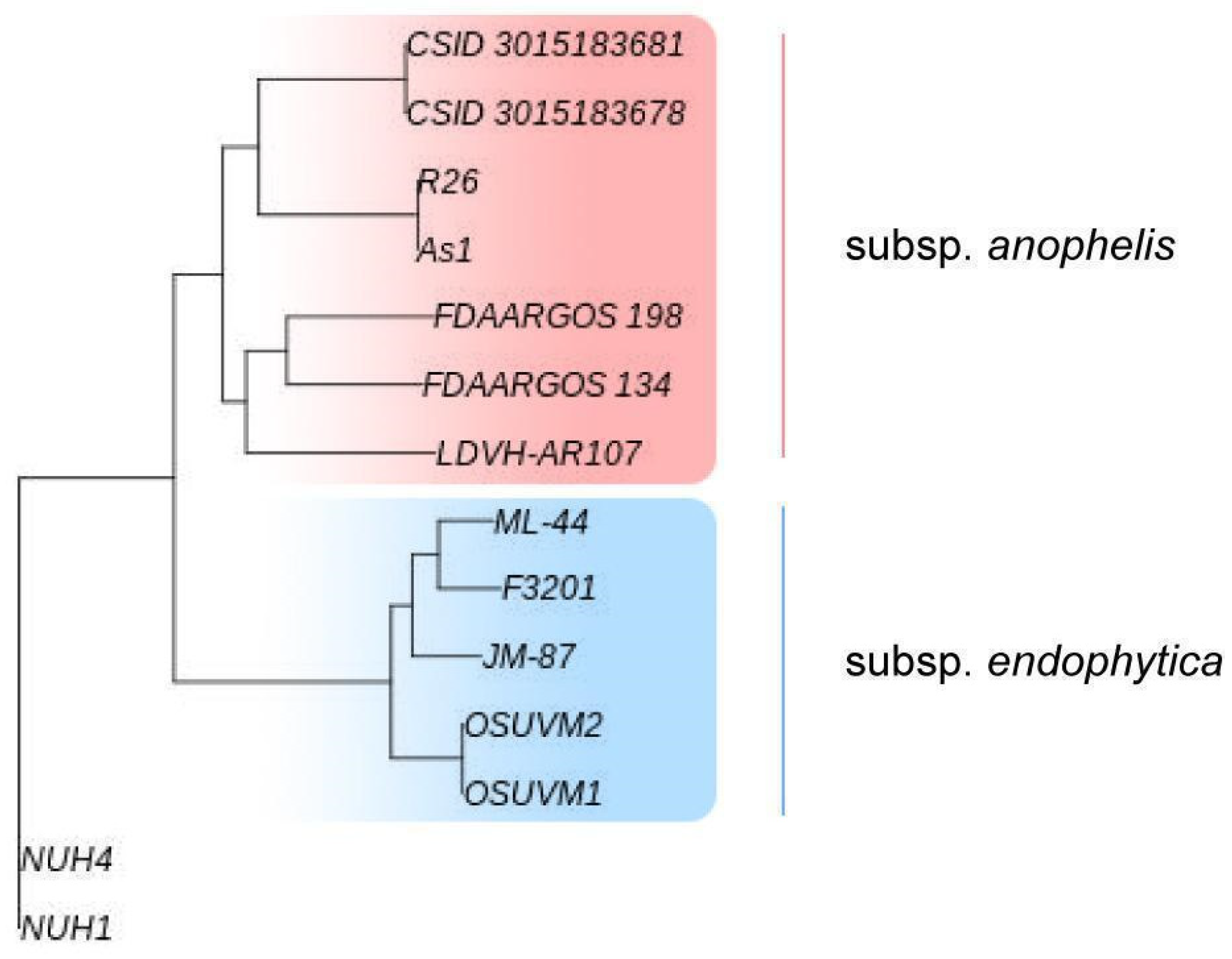
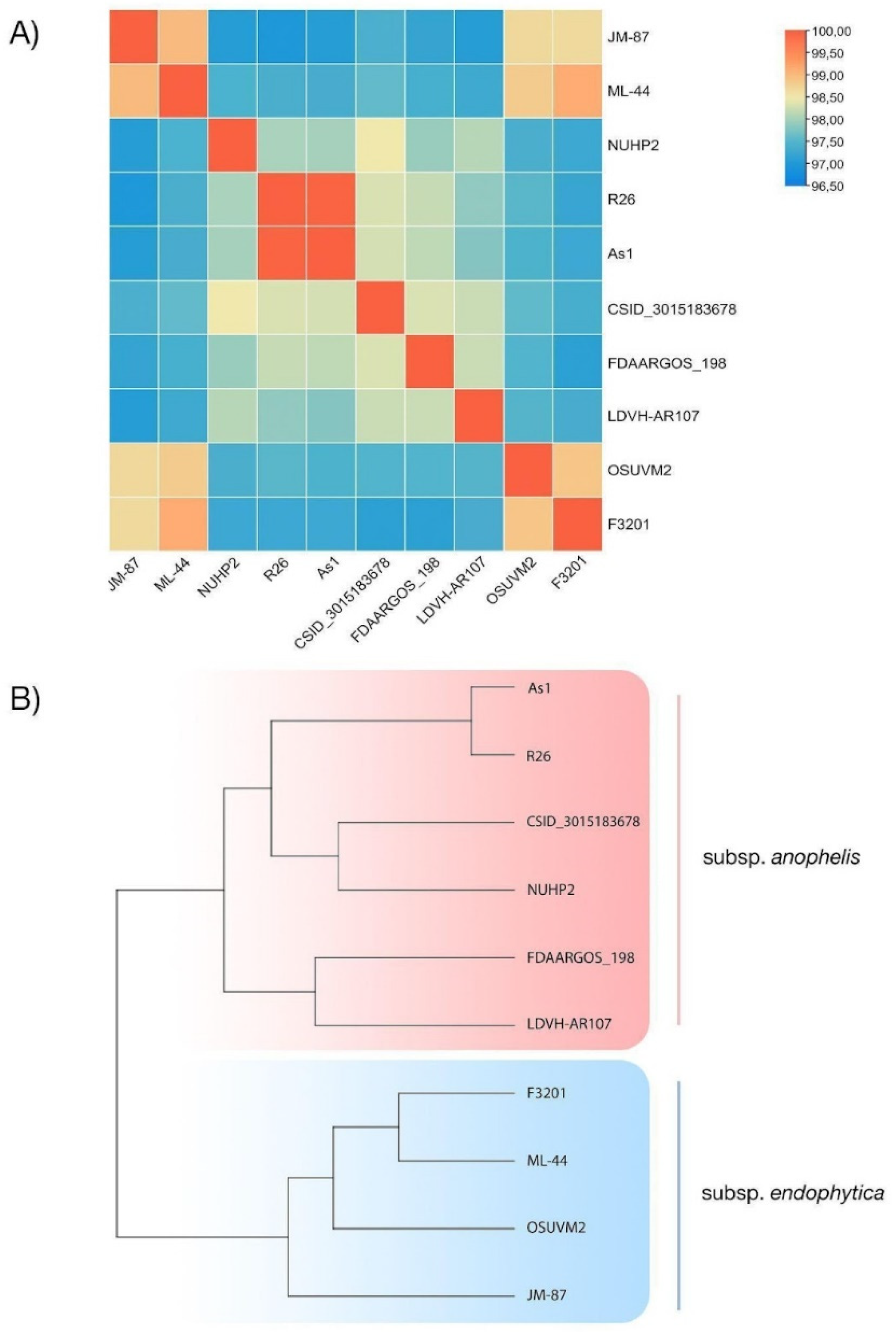
2.4. Genomic Traits of E. anophelis ML-44
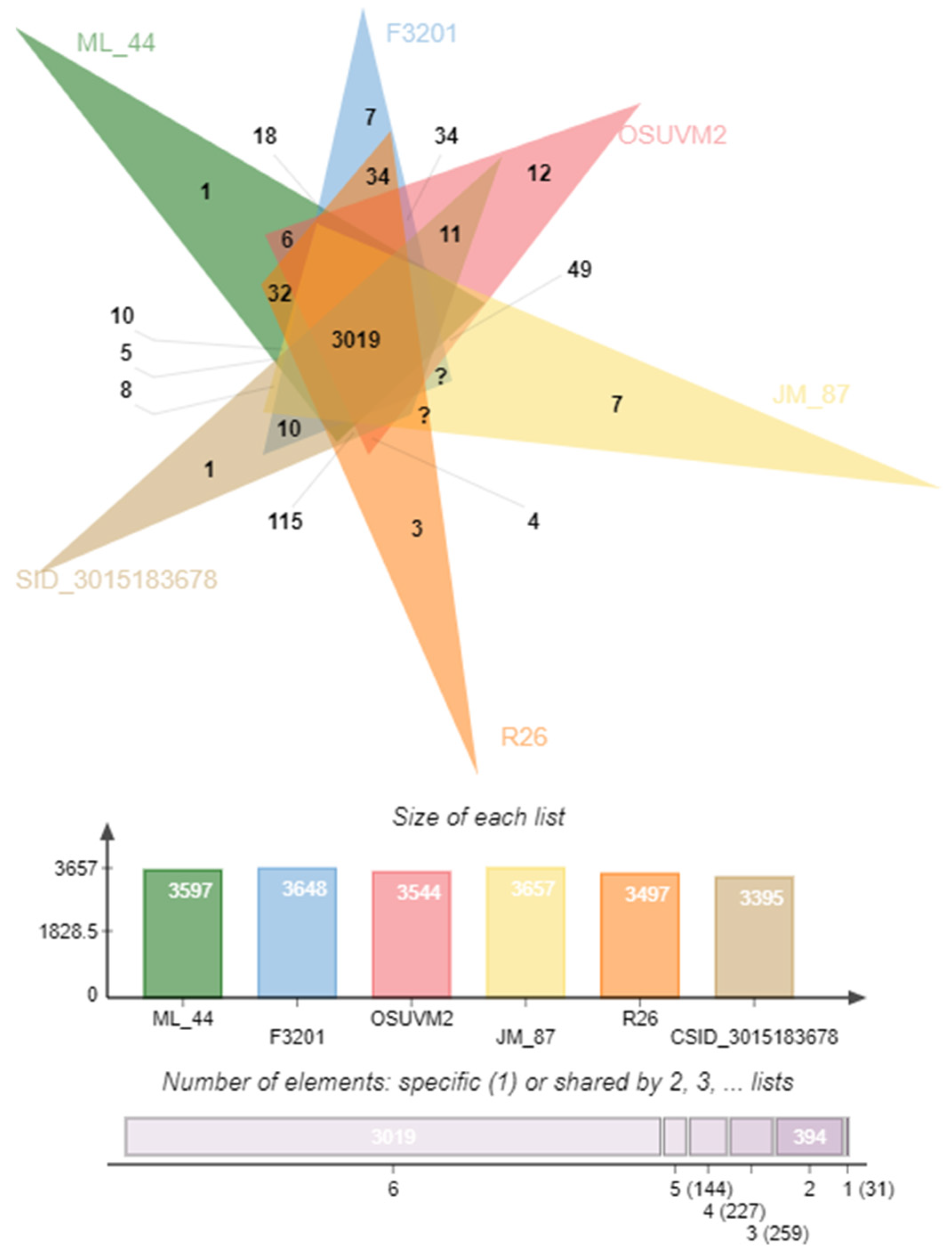
2.5. Comparison of Coding Sequences
2.6. Comparative Resistome Analysis
2.7. Virulome Analysis
2.8. Prophage and CRISPR Comparative Investigation
3. Discussion
4. Materials and Methods
4.1. E. anophelis Isolation and Identification
4.2. Physiological and Biochemical Characterization of E. anophelis ML-44
4.3. Antimicrobial Susceptibility Testing
4.4. Hemolysis Assays
4.5. Whole-Genome Sequencing, Assembly, and Annotation
4.6. Comparative Genomic Analysis of E. anophelis Strains
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kämpfer, P.; Matthews, H.; Glaeser, S.P.; Martin, K.; Lodders, N.; Faye, I. Elizabethkingia Anophelis Sp. Nov., Isolated from the Midgut of the Mosquito Anopheles Gambiae. Int. J. Syst. Evol. Microbiol. 2011, 61, 2670–2675. [Google Scholar] [CrossRef]
- Breurec, S.; Criscuolo, A.; Diancourt, L.; Rendueles, O.; Vandenbogaert, M.; Passet, V.; Caro, V.; Rocha, E.P.C.; Touchon, M.; Brisse, S. Genomic Epidemiology and Global Diversity of the Emerging Bacterial Pathogen Elizabethkingia Anophelis. Sci. Rep. 2016, 6, 30379. [Google Scholar] [CrossRef] [Green Version]
- Janda, J.M.; Lopez, D.L. Mini Review: New Pathogen Profiles: Elizabethkingia Anophelis. Diagn. Microbiol. Infect. Dis. 2017, 88, 201–205. [Google Scholar] [CrossRef]
- Frank, T.; Gody, J.C.; Nguyen, L.B.L.; Berthet, N.; Le Fleche-Mateos, A.; Bata, P.; Rafaï, C.; Kazanji, M.; Breurec, S. First Case of Elizabethkingia Anophelis Meningitis in the Central African Republic. Lancet 2013, 381, 1876. [Google Scholar] [CrossRef]
- Teo, J. First Case of E Anophelis Outbreak in an Intensive-Care Unit. Lancet 2013, 382, 855–856. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Chow, W.N.; Foo, C.H.; Curreem, S.O.T.; Lo, G.C.S.; Teng, J.L.L.; Chen, J.H.K.; Ng, R.H.Y.; Wu, A.K.L.; Cheung, I.Y.Y.; et al. Elizabethkingia Anophelis Bacteremia Is Associated with Clinically Significant Infections and High Mortality. Sci. Rep. 2016, 6, 26045. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.S.P.; Owens, D.S.; Jepson, A.; Turton, J.F.; Ashworth, S.; Donaldson, H.; Holmes, A.H. Waterborne Elizabethkingia Meningoseptica in Adult Critical Care—Volume 22, Number 1—January 2016—Emerging Infectious Diseases Journal—CDC. Emerg. Infect. Dis. 2016, 22, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.L.; Liu, K.M.; Chang, H.L.; Lin, J.S.; Kung, F.Y.; Ho, C.M.; Lin, K.H.; Chen, Y.T. A Dominant Strain of Elizabethkingia Anophelis Emerged from a Hospital Water System to Cause a Three-Year Outbreak in a Respiratory Care Center. J. Hosp. Infect. 2021, 108, 43–51. [Google Scholar] [CrossRef]
- Perrin, A.; Larsonneur, E.; Nicholson, A.C.; Edwards, D.J.; Gundlach, K.M.; Whitney, A.M.; Gulvik, C.A.; Bell, M.E.; Rendueles, O.; Cury, J.; et al. Evolutionary Dynamics and Genomic Features of the Elizabethkingia Anophelis 2015 to 2016 Wisconsin Outbreak Strain. Nat. Commun. 2017, 8, 15483. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.L.; Tarpgaard, I.H.; Fuglsang-Damgaard, D.; Thomsen, P.K.; Brisse, S.; Dalager-Pedersen, M. Rare Elizabethkingia Anophelis Meningitis Case in a Danish Male. JMM Case Rep. 2018, 5, e005163. [Google Scholar] [CrossRef]
- Auffret, N.; Anghel, R.; Brisse, S.; Rey, B.; Schenesse, D.; Moquet, O. Elizabethkingia Anophelis Meningitis in a Traveler Returning from the Americas. Med. Mal. Infect. 2020, 51, 503–505. [Google Scholar] [CrossRef] [PubMed]
- González, L.J.; Vila, A.J. Carbapenem Resistance in Elizabethkingia Meningoseptica Is Mediated by Metallo-β-Lactamase BlaB. Antimicrob. Agents Chemother. 2012, 56, 1686–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossolini, G.M.; Franceschini, N.; Lauretti, L.; Caravelli, B.; Riccio, M.L.; Galleni, M.; Frère, J.M.; Amicosante, G. Cloning of a Chryseobacterium (Flavobacterium) Meningosepticum Chromosomal Gene (BlaA(CME)) Encoding an Extended-Spectrum Class a β- Lactamase Related to the Bacteroides Cephalosporinases and the VEB-1 and PER β-Lactamases. Antimicrob. Agents Chemother. 1999, 43, 2193–2199. [Google Scholar] [CrossRef] [Green Version]
- Bellais, S.; Aubert, D.; Naas, T.; Nordmann, P. Molecular and Biochemical Heterogeneity of Class B Carbapenem- Hydrolyzing β-Lactamases in Chryseobacterium Meningosepticum. Antimicrob. Agents Chemother. 2000, 44, 1878–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, K.L.; Cheng, B.; Lin, R.T.P.; Teo, J.W.P. Elizabethkingia Anophelis Is the Dominant Elizabethkingia Species Found in Blood Cultures in Singapore. J. Clin. Microbiol. 2018, 56, e01445-17. [Google Scholar] [CrossRef] [Green Version]
- Han, M.S.; Kim, H.; Lee, Y.; Kim, M.; Ku, N.S.; Choi, J.Y.; Yong, D.; Jeong, S.H.; Lee, K.; Chong, Y. Relative Prevalence and Antimicrobial Susceptibility of Clinical Isolates of Elizabethkingia Species Based on 16S RRNA Gene Sequencing. J. Clin. Microbiol. 2017, 55, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.N.; Lai, C.H.; Yang, C.H.; Huang, Y.H.; Lin, H.H. Clinical Manifestations, Molecular Characteristics, Antimicrobial Susceptibility Patterns and Contributions of Target Gene Mutation to Fluoroquinolone Resistance in Elizabethkingia Anophelis. J. Antimicrob. Chemother. 2018, 73, 2497–2502. [Google Scholar] [CrossRef]
- Doijad, S.; Ghosh, H.; Glaeser, S.; Kämpfer, P.; Chakraborty, T. Taxonomic Reassessment of the Genus Elizabethkingia Using Whole-Genome Sequencing: Elizabethkingia Endophytica Kämpfer et Al. 2015 Is a Later Subjective Synonym of Elizabethkingia Anophelis Kämpfer et Al. 2011. Int. J. Syst. Evol. Microbiol. 2016, 66, 4555–4559. [Google Scholar] [CrossRef]
- Kämpfer, P.; Busse, H.J.; McInroy, J.A.; Glaeser, S.P. Elizabethkingia Endophytica Sp. Nov., Isolated from Zea Mays and Emended Description of Elizabethkingia Anophelis Kämpfer et Al. 2011. Int. J. Syst. Evol. Microbiol. 2015, 65, 2187–2193. [Google Scholar] [CrossRef] [Green Version]
- Guillon, H.; Eb, F.; Mammeri, H. Characterization of CSP-1, a Novel Extended-Spectrum β-Lactamase Produced by a Clinical Isolate of Capnocytophaga Sputigena. Antimicrob. Agents Chemother. 2010, 54, 2231–2234. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, Q.; Gong, X.; Liu, Y.; Zheng, F. RanB, a Putative ABC-Type Multidrug Efflux Transporter Contributes to Aminoglycosides Resistance and Organic Solvents Tolerance in Riemerella Anatipestifer. Vet. Microbiol. 2020, 243, 108641. [Google Scholar] [CrossRef] [PubMed]
- Cianciotto, N.P.; Fields, B.S. Legionella Pneumophila Mip Gene Potentiates Intracellular Infection of Protozoa and Human Macrophages. Proc. Natl. Acad. Sci. USA 1992, 89, 5188–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimer, A.; Seufert, F.; Weiwad, M.; Ebert, J.; Bzdyl, N.M.; Kahler, C.M.; Sarkar-Tyson, M.; Holzgrabe, U.; Rudel, T.; Kozjak-Pavlovic, V. Inhibitors of Macrophage Infectivity Potentiator-like PPIases Affect Neisserial and Chlamydial Pathogenicity. Int. J. Antimicrob. Agents 2016, 48, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Mancl, J.M.; Zhao, M.; Tang, W.J. Structural Analysis of Mycobacterium Tuberculosis M13 Metalloprotease Zmp1 Open States. Structure 2021, 29, 709–720.e3. [Google Scholar] [CrossRef] [PubMed]
- Burnard, D.; Gore, L.; Henderson, A.; Ranasinghe, A.; Bergh, H.; Cottrell, K.; Sarovich, D.S.; Price, E.P.; Paterson, D.L.; Harris, P.N.A. Comparative Genomics and Antimicrobial Resistance Profiling of Elizabethkingia Isolates Reveal Nosocomial Transmission and in Vitro Susceptibility to Fluoroquinolones, Tetracyclines, and Trimethoprim-Sulfamethoxazole. J. Clin. Microbiol. 2020, 58, e00730-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, H.; Lin, N.; Zhang, Y.; Huang, N.; Walker, E.D.; Ming, D.; Chen, S.; Hu, S. The Antibiotic Resistance and Pathogenicity of a Multidrug-Resistant Elizabethkingia Anophelis Isolate. Microbiologyopen 2019, 8, e804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahia, A.C.; Dong, Y.; Blumberg, B.J.; Mlambo, G.; Tripathi, A.; Benmarzouk-Hidalgo, O.J.; Chandra, R.; Dimopoulos, G. Exploring Anopheles Gut Bacteria for Plasmodium Blocking Activity. Environ. Microbiol. 2014, 16, 2980–2994. [Google Scholar] [CrossRef] [Green Version]
- Terenius, O.; Lindh, J.M.; Eriksson-Gonzales, K.; Bussière, L.; Laugen, A.T.; Bergquist, H.; Titanji, K.; Faye, I. Midgut Bacterial Dynamics in Aedes Aegypti. FEMS Microbiol. Ecol. 2012, 80, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Bagdasarian, M.; Walker, E.D. Elizabethkingia Anophelis: Molecular Manipulation and Interactions with Mosquito Hosts. Appl. Environ. Microbiol. 2015, 81, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.L.; Ramachandran, A.; Torres, N.J.; Nicholson, A.C.; Whitney, A.M.; Bell, M.; Villarma, A.; Humrighouse, B.W.; Sheth, M.; Dowd, S.E.; et al. The Draft Genomes of Elizabethkingia Anophelis of Equine Origin Are Genetically Similar to Three Isolates from Human Clinical Specimens. PLoS ONE 2018, 13, e0200731. [Google Scholar] [CrossRef]
- Bruehl, G.W. Soilborne Plant Pathogens; Macmillan Pub. Co.: New York, NY, USA; Collier Macmillan: London, UK, 1987; 368p. [Google Scholar]
- Soil Born Human Diseases—Publications Office of the EU. Available online: https://op.europa.eu/en/publication-detail/-/publication/ee3d1682-0ea4-483d-9a3d-92e83812b6c9/language-en (accessed on 9 February 2022).
- Adgamov, R.R.; Timchenko, N.F.; Zaitseva, E.A.; Pushkareva, V.I.; Kolbasov, D.V.; Egorova, I.Y.; Pukhovskaya, N.M.; Musatov, Y.S.; Ivanov, L.I.; Ermolaeva, S.A. Ecological and Genetic Mechanisms of Development of Epidemiologically Significant Strains of Sapronosis Causative Agents. Biol. Bull. Rev. 2013, 3, 125–138. [Google Scholar] [CrossRef]
- Pushkareva, V.I.; Ermolaeva, S.A. Listeria Monocytogenes Virulence Factor Listeriolysin O Favors Bacterial Growth in Co-Culture with the Ciliate Tetrahymena Pyriformis, Causes Protozoan Encystment and Promotes Bacterial Survival inside Cysts. BMC Microbiol. 2010, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matz, C.; Kjelleberg, S. Off the Hook—How Bacteria Survive Protozoan Grazing. Trends Microbiol. 2005, 13, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cho, J.H.; Song, M.; Cho, J.H.; Kim, S.; Kim, E.S.; Keum, G.B.; Kim, H.B.; Lee, J.H. Evaluating the Prevalence of Foodborne Pathogens in Livestock Using Metagenomics Approach. J. Microbiol. Biotechnol. 2021, 31, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Franco-Frías, E.; Mercado-Guajardo, V.; Merino-Mascorro, A.; Pérez-Garza, J.; Heredia, N.; León, J.S.; Jaykus, L.A.; Dávila-Aviña, J.; García, S. Analysis of Bacterial Communities by 16S RRNA Gene Sequencing in a Melon-Producing Agro-Environment. Microb. Ecol. 2021, 82, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Rasch, J.; Krüger, S.; Fontvieille, D.; Ünal, C.M.; Michel, R.; Labrosse, A.; Steinert, M. Legionella-Protozoa-Nematode Interactions in Aquatic Biofilms and Influence of Mip on Caenorhabditis Elegans Colonization. Int. J. Med. Microbiol. 2016, 306, 443–451. [Google Scholar] [CrossRef]
- Trifi, A.; Abdellatif, S.; Abdennebi, C.; Daly, F.; Nasri, R.; Touil, Y.; Ben Lakhal, S. Appropriateness of Empiric Antimicrobial Therapy with Imipenem/Colistin in Severe Septic Patients: Observational Cohort Study. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 39. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Owen, C.; Kirby, L. Activation of a Cryptic Streptomycin-resistance Gene in the Bacteroides Erm Transposon, Tn4551. Mol. Microbiol. 1992, 6, 2287–2297. [Google Scholar] [CrossRef]
- Coyne, S.; Rosenfeld, N.; Lambert, T.; Courvalin, P.; Périchon, B. Overexpression of Resistance-Nodulation-Cell Division Pump AdeFGH Confers Multidrug Resistance in Acinetobacter Baumannii. Antimicrob. Agents Chemother. 2010, 54, 4389–4393. [Google Scholar] [CrossRef] [Green Version]
- Warburton, P.J.; Ciric, L.; Lerner, A.; Seville, L.A.; Roberts, A.P.; Mullany, P.; Allan, E. TetAB(46), a Predicted Heterodimeric ABC Transporter Conferring Tetracycline Resistance in Streptococcus Australis Isolated from the Oral Cavity. J. Antimicrob. Chemother. 2013, 68, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Teo, J.W.P.; Tan, T.M.C.; Poh, C.L. Genetic Determinants of Tetracycline Resistance in Vibrio Harveyi. Antimicrob. Agents Chemother. 2002, 46, 1038–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.N.; Lai, C.H.; Yang, C.H.; Huang, Y.H.; Lin, H.H. Genomic Features, Phylogenetic Relationships, and Comparative Genomics of Elizabethkingia Anophelis Strain EM361-97 Isolated in Taiwan. Sci. Rep. 2017, 7, 14317. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.T.; Lai, C.H.; Huang, Y.H.; Yang, C.H.; Lin, J.N. Comparative Analysis of Gradient Diffusion and Disk Diffusion with Agar Dilution for Susceptibility Testing of Elizabethkingia Anophelis. Antibiotics 2021, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S Ribosomal DNA Amplification for Phylogenetic Study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertels, F.; Silander, O.K.; Pachkov, M.; Rainey, P.B.; Van Nimwegen, E. Automated Reconstruction of Whole-Genome Phylogenies from Short-Sequence Reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A Web Server for Whole-Genome Comparison and Annotation of Orthologous Clusters across Multiple Species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Antibiotic | Interpretation | Zone Diameter, mm |
|---|---|---|---|
| Aminoglycosides | Amikacin | R | 0 |
| Gentamicin | R | 0 | |
| Kanamycin | R | 0 | |
| Neomycin | R | 0 | |
| Tobramycin | R | 0 | |
| ß-lactams: Penicillins | Penicillin | R | 0 |
| Ampicillin | R | 0 | |
| Ticarcillin | R | 0 | |
| Piperacillin | R | 14 | |
| Amoxicillin-Clavulanate | R | 0 | |
| Ticarcillin-Clavulanate | R | 13 | |
| Piperacillin-Tazobactam | R | 16 | |
| ß-lactams: Cephalosporins | Ceftazidime | R | 0 |
| Cefotaxime | R | 0 | |
| Cefepime | R | 10 | |
| ß-lactams: Carbapenems | Imipenem | R | 0 |
| Meropenem | R | 0 | |
| ß-lactams: Monobactam | Aztreonam | R | 0 |
| Fluoroquinolones | Ciprofloxacin | S | 32 |
| Levofloxacin | S | 30 | |
| Enrofloxacin | I | 28 | |
| Nalidixic acid | R | 18 | |
| Macrolides | Clarithromycin | R | 20 |
| Erythromycin | R | 19 | |
| Tylosin | R | 13 | |
| Other | Linezolid | S | 22 |
| Rifampicin | S | 20 | |
| Tetracycline | R | 18 | |
| Trimethoprim | R | 13 | |
| Trimethoprim-Sulfametoxazol | R | 0 | |
| Chloramphenicol | R | 0 | |
| Clindamycin | R | 17 | |
| Polymyxin | R | 0 |
| Query Genome | Reference Genome | DDH (f2), % |
|---|---|---|
| ML-44 | JM-87 | 93.5 |
| ML-44 | F3201 | 93.2 |
| ML-44 | OSUVM2 | 90.5 |
| ML-44 | FDAARGOS_198 | 79.7 |
| ML-44 | CSID_3015183678 | 79.3 |
| ML-44 | NUHP2 | 78.3 |
| ML-44 | LDVH-AR107 | 78 |
| ML-44 | R26 | 77.3 |
| ML-44 | As1 | 77 |
| № | Strain Name | Source | Region/Country | Collection Date | WGS Status | WGS GenBank Accession No. |
|---|---|---|---|---|---|---|
| 1 | As1 | Mosquito (A. gambiae) | Pennsylvania, USA | 2013 | Draft | LFKT01 |
| 2 | CSID_3015183678 | Human patient | Wisconsin, USA | 2016 | Complete genome | CP014805.2 |
| 3 | CSID_3015183681 | Human patient | Wisconsin, USA | 2016 | Complete genome | CP015068.2 |
| 4 | F3201 | Human host | Kuwait | 1982 | Complete genome | CP016374.1 |
| 5 | FDAARGOS 134 | Human patient | Washington, D.C., USA | 2014 | Complete genome | CP014021.1 |
| 6 | FDAARGOS 198 | Human patient | Sweden | Missing | Complete genome | CP023010.2 |
| 7 | JM-87 | Sweet corn (Zea mays) | Alabama, USA | 2011 | Complete genome | CP016372.1 |
| 8 | LDVH-AR107 | Common carp (Cyprinus carpio) | Montpellier, France | 2004 | Draft | FTPG01 |
| 9 | NUH1 | Human patient | Singapore | 2012 | Draft | ASYH01 |
| 10 | NUH4 | Human patient | Singapore | 2012 | Draft | ASYI01 |
| 11 | NUHP2 | Human patient | Singapore | 2012 | Draft | ASYF01 |
| 12 | OSUVM1 | Equine Stall | Oklahoma, USA | 2016 | Draft | PJMA01 |
| 13 | OSUVM2 | Horse (Equus caballus) | Oklahoma, USA | 2016 | Draft | PJLZ01 |
| 14 | R26 | Mosquito (A. gambiae) | Stockholm, Sweden | 2005 | Complete genome | CP023401.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andriyanov, P.A.; Zhurilov, P.A.; Kashina, D.D.; Tutrina, A.I.; Liskova, E.A.; Razheva, I.V.; Kolbasov, D.V.; Ermolaeva, S.A. Antimicrobial Resistance and Comparative Genomic Analysis of Elizabethkingia anophelis subsp. endophytica Isolated from Raw Milk. Antibiotics 2022, 11, 648. https://doi.org/10.3390/antibiotics11050648
Andriyanov PA, Zhurilov PA, Kashina DD, Tutrina AI, Liskova EA, Razheva IV, Kolbasov DV, Ermolaeva SA. Antimicrobial Resistance and Comparative Genomic Analysis of Elizabethkingia anophelis subsp. endophytica Isolated from Raw Milk. Antibiotics. 2022; 11(5):648. https://doi.org/10.3390/antibiotics11050648
Chicago/Turabian StyleAndriyanov, Pavel A., Pavel A. Zhurilov, Daria D. Kashina, Anastasia I. Tutrina, Elena A. Liskova, Irina V. Razheva, Denis V. Kolbasov, and Svetlana A. Ermolaeva. 2022. "Antimicrobial Resistance and Comparative Genomic Analysis of Elizabethkingia anophelis subsp. endophytica Isolated from Raw Milk" Antibiotics 11, no. 5: 648. https://doi.org/10.3390/antibiotics11050648