
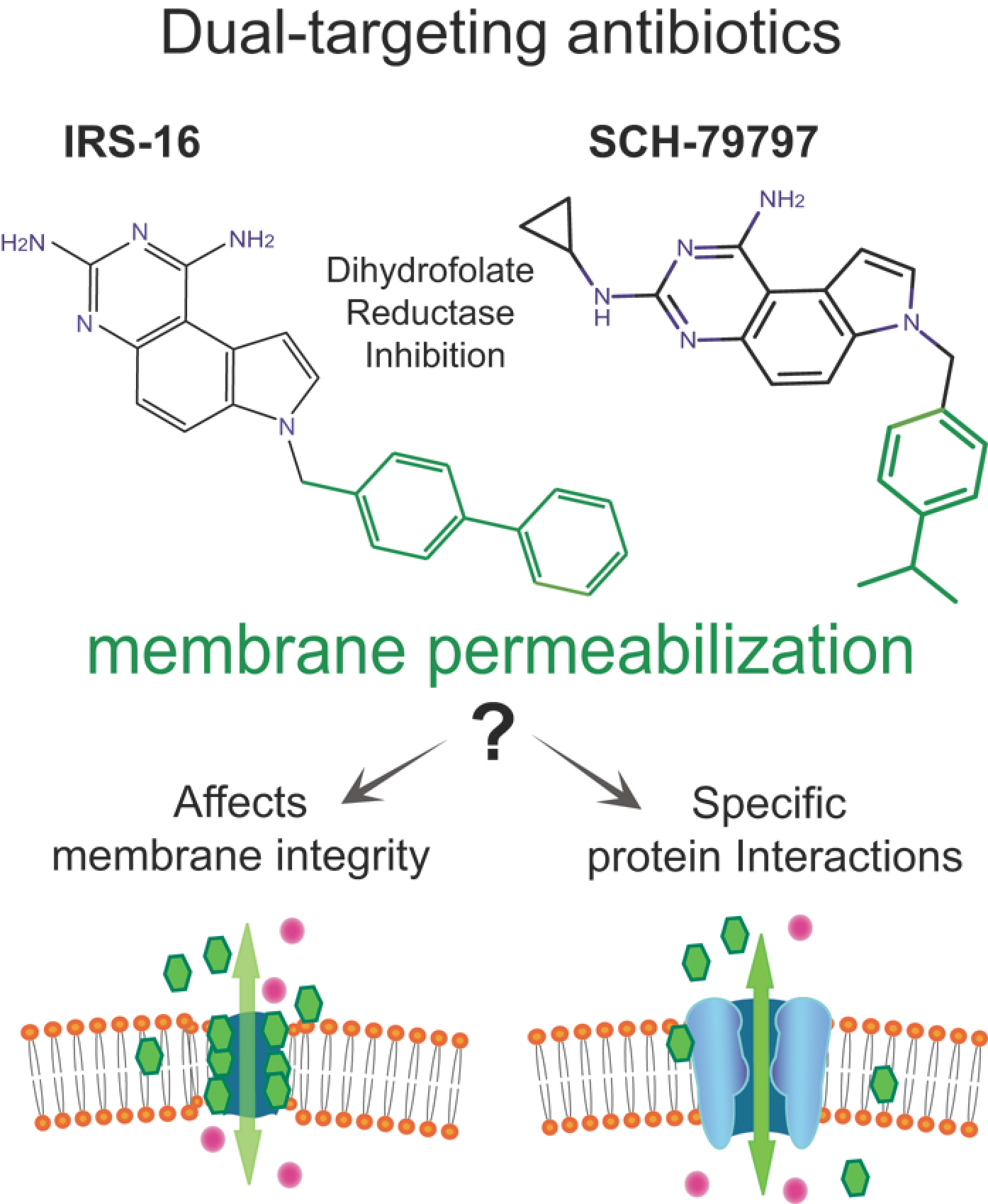
Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SCH-79797 Antibiotic Effects in E. coli Are MscL Dependent
2.2. IRS-16 Antibiotic Effects in E. coli Are MscL Dependent
2.3. Determination of the SCH-79797-Binding Site to MscL
3. Discussion
4. Materials and Methods
4.1. Strains and Cell Growth
4.2. In Vivo Assays
4.2.1. Growth Experiments
4.2.2. Viability Experiments
4.3. Electrophysiology
4.4. Molecular Modeling and Computational Analyses
4.4.1. Molecular Docking and MD Simulations
4.4.2. Free Energy Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination therapy for treatment of infections with gram-negative bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef] [Green Version]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Liu, R.; Shin, S.; Sinha, R.; Pogliano, J.; Pogliano, K.; Griffin, J.H.; Nizet, V.; Corriden, R. SCH79797 improves outcomes in experimental bacterial pneumonia by boosting neutrophil killing and direct antibiotic activity. J. Antimicrob. Chemother. 2018, 73, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.K., 2nd; Sheehan, J.P.; Bratton, B.P.; Moore, G.M.; Mateus, A.; Li, S.H.; Kim, H.; Rabinowitz, J.D.; Typas, A.; Savitski, M.M.; et al. A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell 2020, 181, 1518–1532.e14. [Google Scholar] [CrossRef]
- Blount, P.; Iscla, I. Life with Bacterial Mechanosensitive Channels, from Discovery to Physiology to Pharmacological Target. Microbiol. Mol. Biol. Rev. 2020, 84, e00055-19. [Google Scholar] [CrossRef]
- Cox, C.D.; Bavi, N.; Martinac, B. Bacterial Mechanosensors. Annu. Rev. Physiol. 2018, 80, 71–93. [Google Scholar] [CrossRef]
- Rojas, E.R.; Huang, K.C. Regulation of microbial growth by turgor pressure. Curr. Opin. Microbiol. 2018, 42, 62–70. [Google Scholar] [CrossRef]
- Levina, N.; Totemeyer, S.; Stokes, N.R.; Louis, P.; Jones, M.A.; Booth, I.R. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: Identification of genes required for MscS activity. EMBO J. 1999, 18, 1730–1737. [Google Scholar] [CrossRef]
- Cruickshank, C.C.; Minchin, R.F.; Le Dain, A.C.; Martinac, B. Estimation of the pore size of the large-conductance mechanosensitive ion channel of Escherichia coli. Biophys. J. 1997, 73, 1925–1931. [Google Scholar] [CrossRef] [Green Version]
- Perozo, E.; Cortes, D.M.; Sompornpisut, P.; Kloda, A.; Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature 2002, 418, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Wray, R.; Herrera, N.; Iscla, I.; Wang, J.; Blount, P. An agonist of the MscL channel affects multiple bacterial species and increases membrane permeability and potency of common antibiotics. Mol. Microbiol. 2019, 112, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wray, R.; Iscla, I.; Kovacs, Z.; Wang, J.; Blount, P. Novel compounds that specifically bind and modulate MscL: Insights into channel gating mechanisms. FASEB J. 2019, 33, 3180–3189. [Google Scholar] [CrossRef] [Green Version]
- Wray, R.; Wang, J.; Iscla, I.; Blount, P. Novel MscL agonists that allow multiple antibiotics cytoplasmic access activate the channel through a common binding site. PLoS ONE 2020, 15, e0228153. [Google Scholar] [CrossRef]
- Ou, X.; Blount, P.; Hoffman, R.J.; Kung, C. One face of a transmembrane helix is crucial in mechanosensitive channel gating. Proc. Natl. Acad. Sci. USA 1998, 95, 11471–11475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, J.A.; Dougherty, D.A. Generation and evaluation of a large mutational library from the Escherichia coli mechanosensitive channel of large conductance, MscL—Implications for channel gating and evolutionary design. J. Biol. Chem. 2003, 278, 21076–21082. [Google Scholar] [CrossRef] [Green Version]
- Strande, J.L.; Hsu, A.; Su, J.; Fu, X.; Gross, G.J.; Baker, J.E. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res. Cardiol. 2007, 102, 350–358. [Google Scholar] [CrossRef]
- Gobbetti, T.; Cenac, N.; Motta, J.P.; Rolland, C.; Martin, L.; Andrade-Gordon, P.; Steinhoff, M.; Barocelli, E.; Vergnolle, N. Serine protease inhibition reduces post-ischemic granulocyte recruitment in mouse intestine. Am. J. Pathol. 2012, 180, 141–152. [Google Scholar] [CrossRef]
- Sukharev, S.I.; Martinac, B.; Arshavsky, V.Y.; Kung, C. Two types of mechanosensitive channels in the Escherichia coli cell envelope: Solubilization and functional reconstitution. Biophys. J. 1993, 65, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Blount, P.; Sukharev, S.I.; Moe, P.C.; Martinac, B.; Kung, C. Mechanosensitive channels of bacteria. In Methods in Enzymology; Conn, P.M., Ed.; Ion Channels, Part C; Academic Press: San Diego, CA, USA, 1999; Volume 294, pp. 458–482. [Google Scholar]
- Blount, P.; Schroeder, M.J.; Kung, C. Mutations in a bacterial mechanosensitive channel change the cellular response to osmotic stress. J. Biol. Chem. 1997, 272, 32150–32157. [Google Scholar] [CrossRef] [Green Version]
- Iscla, I.; Levin, G.; Wray, R.; Reynolds, R.; Blount, P. Defining the physical gate of a mechanosensitive channel, MscL, by engineering metal-binding sites. Biophys. J. 2004, 87, 3172–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iscla, I.; Wray, R.; Wei, S.; Posner, B.; Blount, P. Streptomycin potency is dependent on MscL channel expression. Nat. Commun. 2014, 5, 4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, R.; Iscla, I.; Blount, P. Curcumin activation of a bacterial mechanosensitive channel underlies its membrane permeability and adjuvant properties. PLoS Pathog. 2021, 17, e1010198. [Google Scholar] [CrossRef] [PubMed]
- Wray, R.; Iscla, I.; Gao, Y.; Li, H.; Wang, J.; Blount, P. Dihydrostreptomycin Directly Binds to, Modulates, and Passes through the MscL Channel Pore. PLoS Biol. 2016, 14, e1002473. [Google Scholar] [CrossRef] [PubMed]
- Iscla, I.; Wray, R.; Blount, P. On the structure of the N-terminal domain of the MscL channel: Helical bundle or membrane interface. Biophys. J. 2008, 95, 2283–2291. [Google Scholar] [CrossRef] [Green Version]
- Iscla, I.; Wray, R.; Eaton, C.; Blount, P. Scanning MscL Channels with Targeted Post-Translational Modifications for Functional Alterations. PLoS ONE 2015, 10, e0137994. [Google Scholar] [CrossRef] [Green Version]
- Levin, G.; Blount, P. Cysteine scanning of MscL transmembrane domains reveals residues critical for mechanosensitive channel gating. Biophys. J. 2004, 86, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Parkes, A.L.; Yule, I.A. Hybrid antibiotics—Clinical progress and novel designs. Expert Opin. Drug Discov. 2016, 11, 665–680. [Google Scholar] [CrossRef]
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Kasson, P.M. Antibiotic Uptake Across Gram-Negative Outer Membranes: Better Predictions towards Better Antibiotics. ACS Infect. Dis. 2019, 5, 2096–2104. [Google Scholar] [CrossRef]
- Prajapati, J.D.; Kleinekathöfer, U.; Winterhalter, M. How to Enter a Bacterium: Bacterial Porins and the Permeation of Antibiotics. Chem. Rev. 2021, 121, 5158–5192. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Leus, I.V.; Weeks, J.W.; Wolloscheck, D.; Rybenkov, V.V.; Zgurskaya, H.I. Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria. mBio 2017, 8, e01172-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cama, J.; Henney, A.M.; Winterhalter, M. Breaching the Barrier: Quantifying Antibiotic Permeability across Gram-negative Bacterial Membranes. J. Mol. Biol. 2019, 431, 3531–3546. [Google Scholar] [CrossRef] [PubMed]
- Sidarta, M.; Baruah, L.; Wenzel, M. Roles of Bacterial Mechanosensitive Channels in Infection and Antibiotic Susceptibility. Pharmaceuticals 2022, 15, 770. [Google Scholar] [CrossRef]
- Wray, R.; Blount, P.; Wang, J.; Iscla, I. In Silico Screen Identifies a New Family of Agonists for the Bacterial Mechanosensitive Channel MscL. Antibiotics 2022, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Altman, R.B. Identifying druggable targets by protein microenvironments matching: Application to transcription factors. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, J.; Ou, X.; Zhang, M.; Li, Y.; Liu, Z. Mechanical coupling of the multiple structural elements of the large-conductance mechanosensitive channel during expansion. Proc. Natl. Acad. Sci. USA 2015, 112, 10726–10731. [Google Scholar] [CrossRef] [Green Version]
- Steinbacher, S.; Bass, R.; Strop, P.; Rees, D.C. Structures of the prokaryotic mechanosensitive channels MscL and MscS. In Mechanosensitive Ion Channels (a Volume in the Current Topics in Membranes Series); Hamill, O.P., Ed.; Elsievier Press: St. Louis, MO, USA, 2007; Volume 58, pp. 1–20. [Google Scholar]
- Iscla, I.; Wray, R.; Blount, P. The dynamics of protein-protein interactions between domains of MscL at the cytoplasmic-lipid interface. Channels 2012, 6, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Bavi, N.; Cortes, D.M.; Cox, C.D.; Rohde, P.R.; Liu, W.; Deitmer, J.W.; Bavi, O.; Strop, P.; Hill, A.P.; Rees, D.; et al. The role of MscL amphipathic N terminus indicates a blueprint for bilayer-mediated gating of mechanosensitive channels. Nat. Commun. 2016, 7, 11984. [Google Scholar]
- Blount, P.; Merlie, J.P. Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron 1989, 3, 349–357. [Google Scholar] [CrossRef]
- Cheng, W.W.L.; Arcario, M.J.; Petroff, J.T., 2nd. Druggable Lipid Binding Sites in Pentameric Ligand-Gated Ion Channels and Transient Receptor Potential Channels. Front. Physiol. 2022, 12, 798102. [Google Scholar] [CrossRef]
- Prince, A.; Sandhu, P.; Ror, P.; Dash, E.; Sharma, S.; Arakha, M.; Jha, S.; Akhter, Y.; Saleem, M. Lipid-II Independent Antimicrobial Mechanism of Nisin Depends On Its Crowding And Degree Of Oligomerization. Sci. Rep. 2016, 6, 37908. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, I.; Breukink, E.; van Kraaij, C.; Kuipers, O.P.; Bierbaum, G.; de Kruijff, B.; Sahl, H.G. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 2001, 276, 1772–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, S.S.; Dwibedy, S.K.; Padhy, I. Polymyxins, the last-resort antibiotics: Mode of action, resistance emergence, and potential solutions. J. Biosci. 2021, 46, 85. [Google Scholar] [CrossRef]
- Warren, G.H.; Gray, J.; Yurchenco, J.A. Effect of polymyxin on the lysis of Neisseria catarrhalis by lysozyme. J. Bacteriol. 1957, 74, 788–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, U.; Edwards, M.D.; Rasmussen, T.; Bartlett, W.; van West, P.; Booth, I.R. YbdG in Escherichia coli is a threshold-setting mechanosensitive channel with MscM activity. Proc. Natl. Acad. Sci. USA 2010, 107, 12664–12669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blount, P.; Moe, P. Bacterial mechanosensitive channels: Integrating physiology, structure and function. Trends Microbiol. 1999, 7, 420–424. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, A.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. Amber 18; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Wang, J.; Hou, T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 2012, 52, 1199–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wray, R.; Wang, J.; Blount, P.; Iscla, I. Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds. Antibiotics 2022, 11, 970. https://doi.org/10.3390/antibiotics11070970
Wray R, Wang J, Blount P, Iscla I. Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds. Antibiotics. 2022; 11(7):970. https://doi.org/10.3390/antibiotics11070970
Chicago/Turabian StyleWray, Robin, Junmei Wang, Paul Blount, and Irene Iscla. 2022. "Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds" Antibiotics 11, no. 7: 970. https://doi.org/10.3390/antibiotics11070970
APA StyleWray, R., Wang, J., Blount, P., & Iscla, I. (2022). Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds. Antibiotics, 11(7), 970. https://doi.org/10.3390/antibiotics11070970