
Antimicrobial and Cytotoxic Cyathane-Xylosides from Cultures of the Basidiomycete Dentipellis fragilis

 ,
,  and
and
Abstract
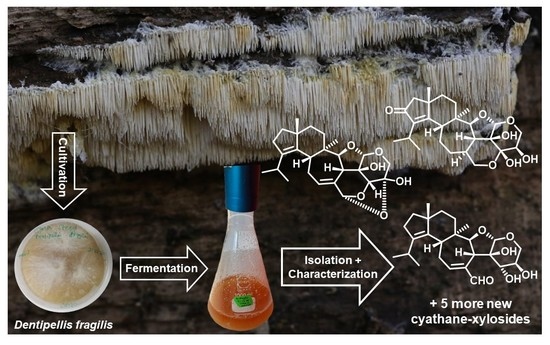
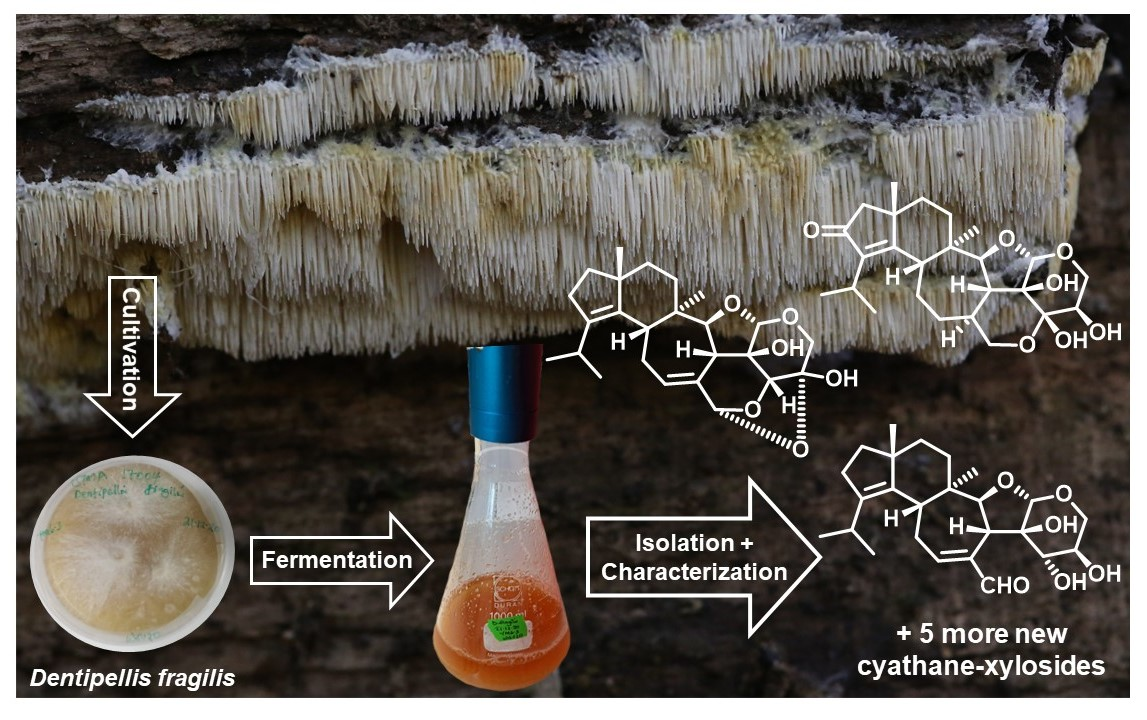
:
1. Introduction
2. Results and Discussion
2.1. Identity of the Producer Strain
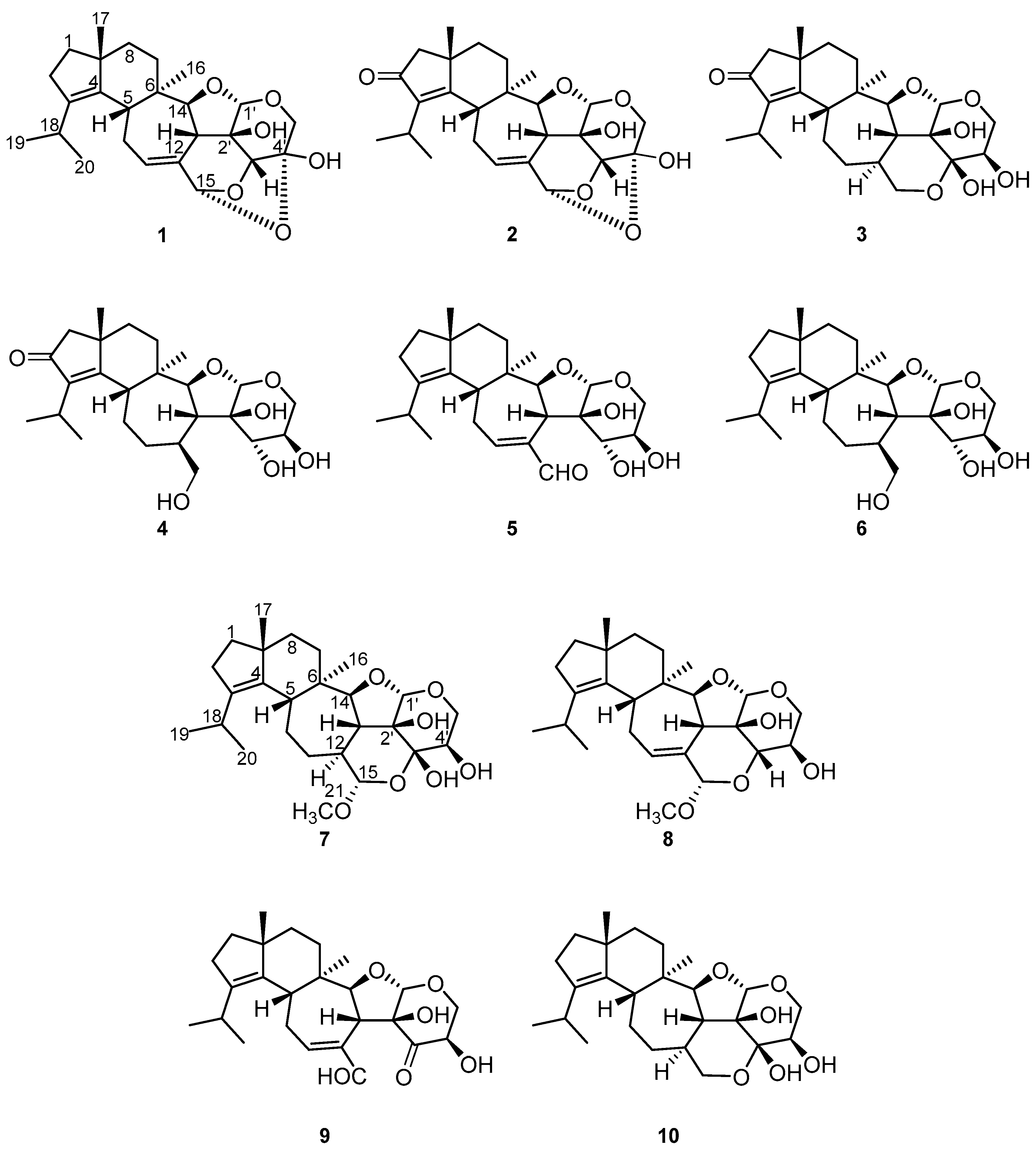
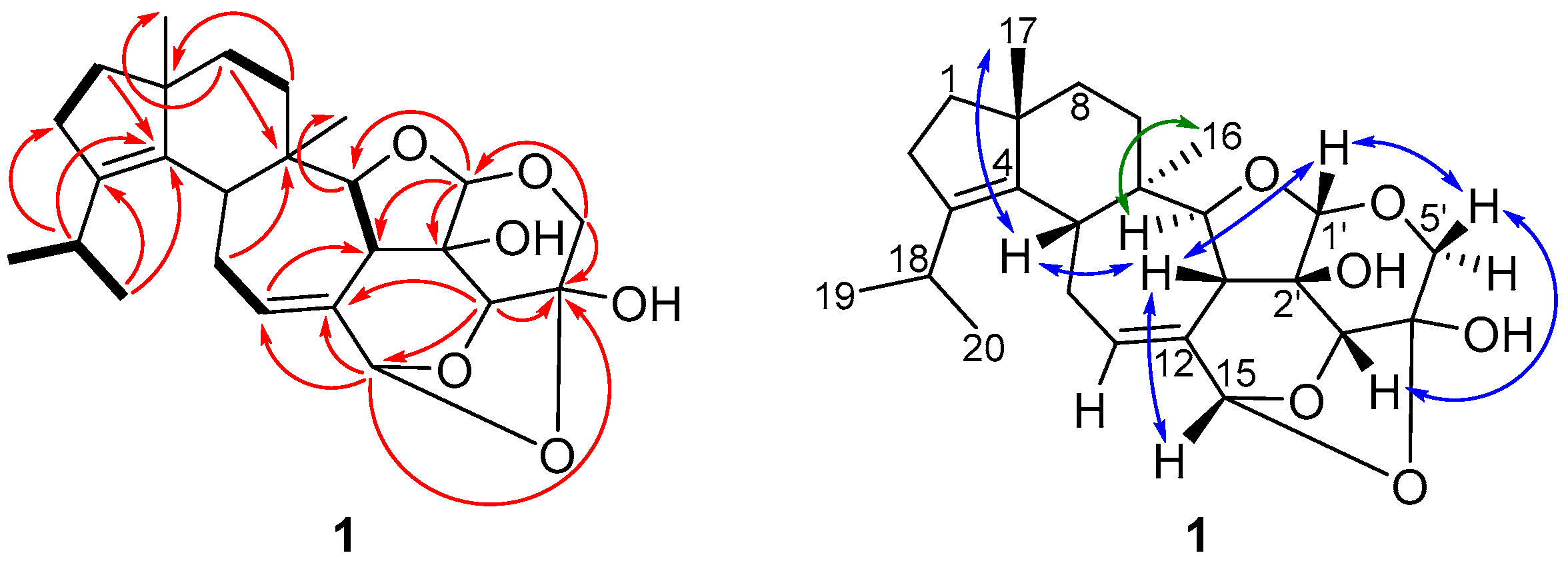
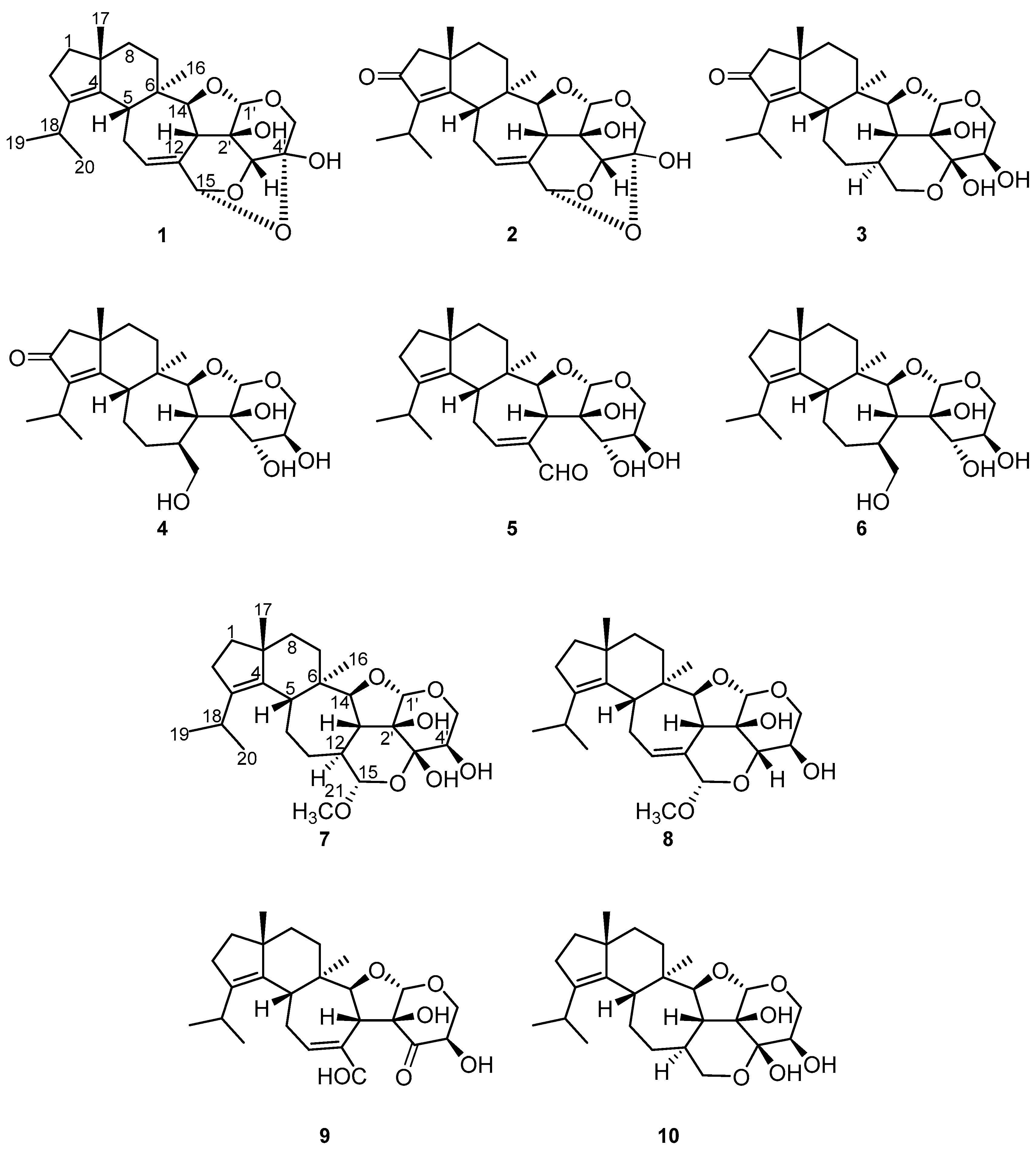
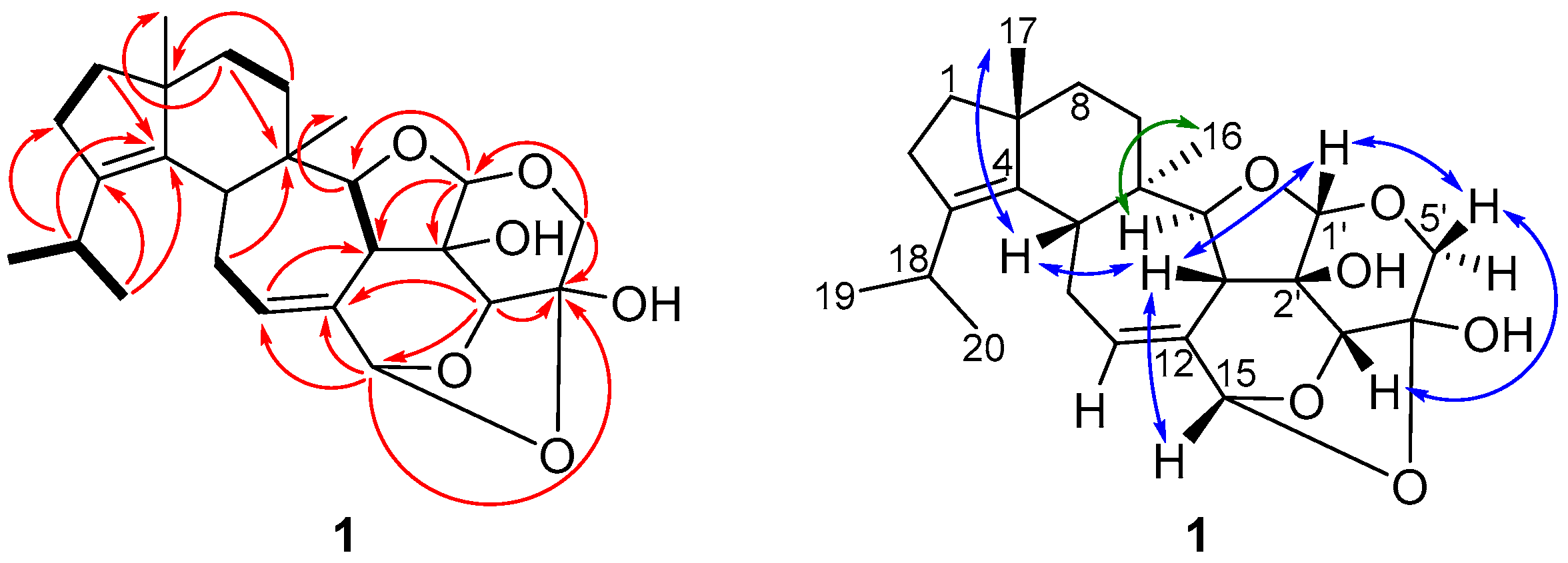
2.2. Structure Elucidation of Cyathane-Xyloside Compounds (1–10)
2.3. Biological Activity of Compounds 1–9
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation Scale-Up and Extraction of Metabolites
3.4. Isolation and Physico-Chemical Properties of Compounds
3.5. Antimicrobial Assays
3.6. Cytotoxicity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bills, G.F.; Gloer, J.B. Biologically active secondary metabolites from the Fungi. Microbiol. Spectr. 2016, 4, 1087–1119. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The amazing potential of fungi: 50 ways we can exploitfFungi industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar] [CrossRef] [Green Version]
- Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Muñoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biological and chemical diversity go hand in hand: Basidiomycota as source of new pharmaceuticals and agrochemicals. Biotechnol. Adv. 2019, 37, 107344. [Google Scholar] [CrossRef]
- Gressler, M.; Löhr, N.A.; Schäfer, T.; Lawrinowitz, S.; Seibold, P.S.; Hoffmeister, D. Mind the mushroom: Natural product biosynthetic genes and enzymes of Basidiomycota. Nat. Prod. Rep. 2021, 38, 702–722. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Chepkirui, C.; Decock, C.; Matasyoh, J.C.; Stadler, M. Heimiomycins A-C and calamenens from the African basidiomycete Heimiomyces sp. J. Nat. Prod. 2020, 83, 2501–2507. [Google Scholar] [CrossRef]
- Chepkirui, C.; Sum, W.C.; Cheng, T.; Matasyoh, J.C.; Decock, C.; Stadler, M. Aethiopinolones A-E, New pregnenolone type steroids from the East African basidiomycete Fomitiporia aethiopica. Molecules 2018, 23, 369. [Google Scholar] [CrossRef] [Green Version]
- Hyde, K.D.; Norphanphoun, C.; Chen, J.; Dissanayake, A.J.; Doilom, M.; Hongsanan, S.; Jayawardena, R.S.; Jeewon, R.; Perera, R.H.; Thongbai, B.; et al. Thailand’s amazing diversity: Up to 96% of fungi in Northern Thailand may be novel. Fungal Divers. 2018, 93, 215–239. [Google Scholar] [CrossRef]
- Sandargo, B.; Michehl, M.; Stadler, M.; Surup, F. Antifungal sesquiterpenoids, rhodocoranes, from submerged cultures of the Wrinkled Peach Mushroom, Rhodotus palmatus. J. Nat. Prod. 2020, 83, 720–724. [Google Scholar] [CrossRef]
- Zhou, L.W.; Dai, Y.C. Taxonomy and phylogeny of wood-inhabiting hydnoid species in Russulales: Two new genera, three new species and two new combinations. Mycologia 2013, 105, 636–649. [Google Scholar] [CrossRef]
- Donk, M.A. Notes on resupinate Hymenomycetes-VI. Persoonia-Mol. Phylogeny Evol. Fungi 1962, 2, 217–238. [Google Scholar]
- Niemelä, T.; Saarenoksa, R. Dentipellis fragilis and Steccherinum oreophilum: Finnish records of hydnaceous fungi. Karstenia 1985, 25, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Ha, L.S.; Ki, D.W.; Kim, J.Y.; Choi, D.C.; Lee, I.K.; Yun, B.S. Dentipellin, a new antibiotic from culture broth of Dentipellis fragilis. J. Antibiot. 2021, 74, 538–541. [Google Scholar] [CrossRef]
- Larsson, E.; Larsson, K.H. Phylogenetic relationships of russuloid basidiomycetes with emphasis on aphyllophoralean taxa. Mycologia 2003, 95, 1037–1065. [Google Scholar] [CrossRef]
- Anke, T.; Rabe, U.; Schu, P.; Eizenhöfer, T.; Schrage, M.; Steglich, W. Studies on the biosynthesis of striatal-type diterpenoids and the biological activity of herical. Z. Naturforsch. 2002, 57, 263–271. [Google Scholar] [CrossRef]
- Mudalungu, C.M.; Richter, C.; Wittstein, K.; Abdalla, M.A.; Matasyoh, J.C.; Stadler, M.; Süssmuth, R.D. Laxitextines A and B, cyathane xylosides from the tropical fungus Laxitextum incrustatum. J. Nat. Prod. 2016, 79, 894–898. [Google Scholar] [CrossRef]
- Bai, R.; Zhang, C.C.; Yin, X.; Wei, J.; Gao, J.M. Striatoids A-F, Cyathane diterpenoids with neurotrophic activity from cultures of the fungus Cyathus striatus. J. Nat. Prod. 2015, 78, 783–788. [Google Scholar] [CrossRef]
- Nitthithanasilp, S.; Intaraudom, C.; Boonyuen, N.; Suvannakad, R.; Pittayakhajonwut, P. Antimicrobial activity of cyathane derivatives from Cyathus subglobisporus BCC44381. Tetrahedron 2018, 74, 6907–6916. [Google Scholar] [CrossRef]
- Chen, L.; Yao, J.N.; Chen, H.P.; Zhao, Z.Z.; Li, Z.H.; Feng, T.; Liu, J.K. Hericinoids A–C, cyathane diterpenoids from culture of mushroom Hericium erinaceus. Phytochem. Lett. 2018, 27, 94–100. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Shirai, R.; Okamoto, K.; Ojima, F.; Sakamoto, H.; Ishiguro, Y.; Furukawa, S. Erinacines A, B and C, strong stimulators of Nerve Growth Factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1994, 35, 1569–1572. [Google Scholar] [CrossRef]
- Rupcic, Z.; Rascher, M.; Kanaki, S.; Köster, R.W.; Stadler, M.; Wittstein, K. Two new cyathane diterpenoids from mycelial cultures of the medicinal mushroom Hericium erinaceus and the rare species, Hericium flagellum. Int. J. Mol. Sci. 2018, 19, 740. [Google Scholar] [CrossRef] [Green Version]
- Hecht, H.J.; Höfle, G.; Steglich, W.; Anke, T.; Oberwinkler, F. Striatin A, B, and C: Novel diterpenoid antibiotics from Cyathus striatus; X-Ray crystal structure of striatin A. J. Chem. Soc. Chem. Commun. 1978, 1978, 665–666. [Google Scholar] [CrossRef]
- Tang, H.-Y.; Yin, X.; Zhang, C.-C.; Jia, Q.; Gao, J.-M. Structure Diversity, Synthesis, and Biological Activity of Cyathane Diterpenoids in Higher Fungi. Curr. Med. Chem. 2015, 22, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Noumeur, S.R.; Teponno, R.B.; Helaly, S.E.; Wang, X.W.; Harzallah, D.; Houbraken, J.; Crous, P.W.; Stadler, M. Diketopiperazines from Batnamyces globulariicola, gen. & sp. nov. (Chaetomiaceae), a fungus associated with roots of the medicinal plant Globularia alypum in Algeria. Mycol. Prog. 2020, 19, 589–603. [Google Scholar] [CrossRef]
- Becker, K.; Wessel, A.C.; Luangsa-Ard, J.J.; Stadler, M. Viridistratins A-C, Antimicrobial and cytotoxic benzo[j]fluoranthenes from stromata of Annulohypoxylon viridistratum (Hypoxylaceae, Ascomycota). Biomolecules 2020, 10, 805. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Simada, A.; Shizuki, K.; Mori, H.; Okamoto, K.; Sakamoto, H.; Furukawa, S. Erinacine D, a stimulator of NGF-synthesis, from the mycelia of Hericium erinaceum. Heterocycl. Commun. 1996, 2, 51–54. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Hosokawa, S.; Mori, H.; Sakamoto, H.; Ishiguro, Y.; Sakemi, S.; Bordner, J.; Kojima, N.; Furukawa, S. Erinacines E, F, and G, stimulators of Nerve Growth Factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1996, 37, 7399–7402. [Google Scholar] [CrossRef]
- Lee, E.W.; Shizuki, K.; Hosokawa, S.; Suzuki, M.; Suganuma, H.; Inakuma, T.; Li, J.; Ohnishi-Kameyama, M.; Nagata, T.; Furukawa, S.; et al. Two novel diterpenoids, erinacines H and I from the mycelia of Hericium erinaceum. Biosci. Biotechnol. Biochem. 2000, 64, 2402–2405. [Google Scholar] [CrossRef] [Green Version]
- Hofle, G.; Oberwinkler, F. The Striatins–New antibiotics from the basidiomycete Cyathus striatus (Huds. Ex Pers.) Willd. J. Antibiot. 1977, 30, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Kraisitudomsook, N.; Healy, R.A.; Smith, M.E. Molecular systematics and taxonomic overview of the Bird’s Nest fungi (Nidulariaceae). Fungal Biol. 2021, 125, 693–703. [Google Scholar] [CrossRef]
- Wijayawardene, N.N.; Hyde, K.; Dai, D.; Goto, B.T.; Saxena, R.; Erdoğdu, M.; Selçuk, F.; Rajeshkumar, K.C.; Aptroot, A.; Błaszkowski, J.; et al. Outline of Fungi and fungus-like taxa–2021. Mycosphere 2022, 13, 53–453. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the Number of Nucleotide Substitutions in the Control Region of Mitochondrial DNA in Humans and Chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 b | |||
|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| 1 | 38.5, CH2 | 1.56 (m) 1.65 (m) | 52.2, CH2 | 2.20 (d, 18.9) 2.24 (d, 18.9) | 52.4, CH2 | 2.20 (d, 18.9) 2.23 (d, 18.9) |
| 2 | 28.4, CH2 | 2.29 (m) | 207.8, C | 208.1, C | ||
| 3 | 139.8, C | 143.6, C | 142.7, C | |||
| 4 | 136.4, C | 174.7, C | 177.6, C | |||
| 5 | 42.5, CH | 2.61 (m) | 43.8, CH | 3.00 (d, 11.9) | 48.6, CH | 2.70 (d, 9.3) |
| 6 | 40.3, C | 42.3, C | 42.3, C | |||
| 7 | 27.1, CH2 | 1.52 (m) | 26.3, CH2 | 1.58 (m) 1.64 (m) | 27.4, CH2 | 1.62 (m) |
| 8 | 36.4, CH2 | 1.55 (m) | 37.3, CH2 | α: 1.57 (m) β: 1.89 (dd, 11.5, 4.4) | 38.0, CH2 | 1.56 (td, 13.4, 5.1) 1.87 (m) |
| 9 | 49.7, C | 42.5, C | 42.4, C | |||
| 10 | 29.5, CH2 | 2.56 (m) 2.61 (m) | 28.8, CH2 | 2.55 (dd, 17.9, 6.6) 2.77 (m) | 25.3, CH2 | 1.86 (m) 1.97 (m) |
| 11 | 127.4, CH | 5.60 (m) | 126.2, CH | 5.62 (m) | 33.7, CH2 | 0.98 (dd, 12.2, 1.6) 1.90 (m) |
| 12 | 134.0, C | 134.7, C | 40.4, CH | 1.88 (m) | ||
| 13 | 43.5, CH | 2.81 (m) | 43.7, CH | 2.83 (m) | 48.0, CH | 2.07 (dd, 11.2, 8.2) |
| 14 | 92.3, CH | 4.20 (d, 8.7) | 91.5, CH | 4.27 (d, 8.7) | 93.5, CH | 4.09 (d, 8.2) |
| 15 | 103.2, CH | 5.88 (s) | 102.9, CH | 5.90 (s) | 65.5, CH2 | 3.60 (t, 10.9) 3.83 (dd, 10.9, 4.6) |
| 16 | 17.0, CH3 | 1.02 (s) | 16.8, CH3 | 1.05 (s) | 18.9, CH3 | 1.03 (s) |
| 17 | 24.6, CH3 | 1.05 (s) | 24.4, CH3 | 1.24 (s) | 24.5, CH3 | 1.27 (s) |
| 18 | 27.0, CH | 2.80 (spt, 6.7) | 25.8, CH | 2.80 (spt, 6.9) | 25.9, CH | 2.74 (spt, 6.9) |
| 19 | 21.6, CH3 | 0.99 (d, 6.7) * | 19.8, CH3 | 1.22 (d, 6.9) | 19.6, CH3 | 1.22 (d, 6.9) |
| 20 | 21.8, CH3 | 0.97 (d, 6.7) * | 20.8, CH3 | 1.21 (d, 6.9) | 20.8, CH3 | 1.19(d, 6.9) |
| 1′ | 105.0, CH | 5.03 (s) | 105.0, CH | 5.06 (s) | 106.3, CH | 5.12 (s) |
| 2′ | 76.3, C | 76.3, C | 77.9, C | |||
| 3′ | 83.6, CH | 4.48 (s) | 83.7, CH | 4.50 (s) | 95.5, C | |
| 4′ | 96,8, C | 96.8, C | 70.4, CH | 3.92 (t, 5.3) | ||
| 5′ | 63.3, CH2 | β: 3.71 (d, 13.0) α: 3.98 (d, 13.0) | 63.4, CH2 | β: 3.72 (d, 13.1) α: 4.00 (d, 13.1) | 65.0, CH2 | β: 3.69 (dd, 11.7, 5.5) α: 4.04 (dd, 11.7, 5.1) |
| 2′-OH | + | |||||
| 3′-OH | 4.78 (br s) | |||||
| 4′-OH | + | |||||
| No. | 4 b | 5 a | 6 a | |||
|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| 1 | 52.3, CH2 | 2.18 (d, 18.7) 2.23 (d, 18.7) | 38.3, CH2 | 1.58 (m) 1.65 (m) | 38.3, CH2 | 1.55 (m) 1.63 (m) |
| 2 | 208.1, C | 28.4, CH2 | 2.30 (m) | 28.2, CH2 | 2.28 (m) | |
| 3 | 142.8, C | 139.7, C | 138.5, C | |||
| 4 | 177.8, C | 136.3, C | 138.3, C | |||
| 5 | 47.2, CH | 2.56 (d, 9.0) | 42.5, CH | 2.42 (d, 11.6) | 45.4, CH | 2.18 (m) |
| 6 | 45.1, C | 42.5, C | 43.3, C | |||
| 7 | 27.3, CH2 | 1.69 (m) | 26.6, CH2 | 1.64 (m) | 28.0, CH2 | 1.59 (m) |
| 8 | 37.9, CH2 | 1.54 (m) 1.88 (m) | 36.4, CH2 | 1.56 (m) | 36.7, CH2 | 1.53 (m) |
| 9 | 42.1, C | 49.6, C | 49.3, C | |||
| 10 | 24.4, CH2 | 1.80 (m) 1.86 (m) | 29.6, CH2 | 2.76 (m) 2.87 (m) | 24.8, CH2 | 1.84 (m) 1.67 (m) |
| 11 | 35.6, CH2 | 1.45 (m) 1.92 (m) | 161.3, CH | 7.08 (m) | 35.3, CH2 | 1.41 (m) 1.86 (m) |
| 12 | 39.6, CH | 2.46 (m) | 143.4, C | 39.4, CH | 2.42 (m) | |
| 13 | 46.7, CH | 2.47 (m) | 46.6, CH | 3.37 (m) | 46.9, CH | 2.45 (dd, 9.2, 10.7) |
| 14 | 86.0, CH | 3.98 (d, 8.8) | 85.8, CH | 4.19 (d, 10.5) | 86.7, CH | 3.92 (d, 9.2) |
| 15 | 66.1, CH2 | 3.56 (dd, 11.7, 3.1) 4.17 (dd, 11.7, 3.1) | 197.1, C | 9.41 (s) | 66.6, CH2 | 4.11 (dd, 11.0, 1.8) 3.55 (br d, 11.0) |
| 16 | 18.7, CH3 | 0.97 (s) | 17.3, CH3 | 1.00 (s) | 18.8, CH3 | 0.93 (s) |
| 17 | 24.5, CH3 | 1.28 (s) | 24.6, CH3 | 1.05 (s) | 24.9, CH3 | 1.08 (s) |
| 18 | 25.8, CH | 2.75 (spt, 6.9) | 27.1, CH | 2.75 (m) | 27.0, CH | 2.76 (spt, 6.8) |
| 19 | 19.5, CH3 | 1.21 (d, 6.9) | 21.5, CH3 | 1.01 (6.9) | 21.4, CH3 | 0.96 (d, 6.8) |
| 20 | 20.8, CH3 | 1.18 (d, 6.9) | 21.9, CH3 | 0.99 (6.9) | 21.8, CH3 | 0.95 (d, 6.8) |
| 1′ | 102.2, CH | 5.05 (s) | 102.0, CH | 5.16 (s) | 102.2, CH | 5.04 (br d, 1.1) |
| 2′ | 73.8, C | 74.0, C | 73.7, C | |||
| 3′ | 69.2, CH | 4.28 (m) | 70.1, CH | 3.90 (br s) | 69.3, CH | 4.24 (m) |
| 4′ | 68.7, CH | 3.86 (m) | 68.4, CH | 3.56 (m) | 68.8, CH | 3.83 (m) |
| 5′ | 59.7, CH2 | β: 3.73 (br d, 12.9) α: 4.28 (br d, 12.9) | 59.8, CH2 | 3.66 (d, 11.8) * 4.22 (d, 11.8) * | 59.8, CH2 | 3.72 (d, 12.9) * 4.26 (d, 12.9) * |
| 15-OH | 2.90 (br s) | |||||
| 2′-OH | 5.65 (br s) | 5.00 (s) | ||||
| 3′-OH | 1.79 (br s) | 2.54 (d, 10.7) | ||||
| 4′-OH | 3.76 (m) | 3.43 (br d, 6.4) | ||||
| No. | 7 b | 8 c | ||
|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| 1 | 38.3, CH2 | 1.57 (m) 1.63 (m) | 39.7, CH2 | 1.58 (m) 1.65 (m) |
| 2 | 28.1, CH2 | 2.28 (m) | 29.2, CH2 | 2.32 (m) |
| 3 | 138.5, C | 140.4, C | ||
| 4 | 138.4, C | 139.0, C | ||
| 5 | 47.1, CH | 2.27 (m) | 45.1, CH | 2.55 (d, 11.8 Hz)) |
| 6 | 40.3, C | 41.5, C | ||
| 7 | 28.3, CH2 | 1.54 (m) | 28.4, CH2 | 1.46 (ddd, 13.5, 4.6, 1.8) 1.74 (td, 13.5, 4.6) |
| 8 | 36.6, CH2 | 1.50 (m) 1.54 (m) | 37.8, CH2 | 1.51 (ddd, 13.5, 4.6, 1.8) 1.58 (m) |
| 9 | 49.5, C | 50.9, C | ||
| 10 | 25.1, CH2 | 1.92 (dddd, 14.6, 4.5, 3.2) + 1.68 (m) | 29.7, CH2 | 2.43 (m) 2.61 (m) |
| 11 | 35.9, CH2 | 1.08 (m) 2.19 (m) | 132.0, CH | 6.01 (m) |
| 12 | 43.5, CH | 1.76 (m) | 134.9, C | |
| 13 | 46.0, CH | 2.17 (m) | 46.8, CH | 2.89 (m) |
| 14 | 93.3, CH | 4.01 (d, 8.6) | 92.4, CH | 4.13 (d, 9.9) |
| 15 | 104.6, CH | 4.50 (d, 8.4) | 99.8, CH | 5.17 (m) |
| 16 | 18.9, CH3 | 0.99 (s) | 18.1, CH3 | 1.00 (s) |
| 17 | 25.0, CH3 | 1.08 (s) | 25.2, CH3 | 1.07 (s) |
| 18 | 27.1, CH | 2.73 (spt, 6.7) | 28.4, CH | 2.86 (spt, 6.8) |
| 19 | 21.3, CH3 | 0.97 (t, 6.7) | 22.0, CH3 | 1.01 (d, 6.8) |
| 20 | 21.8, CH3 | 0.95 (t, 6.7) | 22.4, CH3 | 0.99 (d, 6.8) |
| 21 | 56.5, CH3 | 3.53 (s) | 56.4, CH3 | 3.55 (s) |
| 1′ | 106.5, CH | 4.90 (s) | 108.1, CH | 4.97 (s) |
| 2′ | 80.5, C | 81.2, C | ||
| 3′ | 96.1, C | 84.5, CH | 3.79 (d, 9.8) | |
| 4′ | 69.8, CH | 4.26 (dd, 10.7, 5.4) | 68.4, CH | 3.94 (td, 9.8, 5.0) |
| 5′ | 63.0, CH2 | β: 3.36 (dd, 11.6, 10.7) α: 3.83 (dd, 11.6, 5.4) | 66.2, CH2 | β: 3.13 (dd, 11.4, 9.8) α: 3.69 (dd, 11.4, 5.0) |
| 2′-OH | 3.16 (br s) | |||
| 3′-OH | 4.72 (s) | |||
| 4′-OH | * | |||
| Microorganism | MIC (µg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Ref. | |
| Bacillus subtilis (DSM 10) | 1.0 | 66.6 | n.i. | 16.7 | 16.7 | ND | ND | ND | 1.0 | 4.2 a |
| Mycolicibacterium smegmatis (ATCC 700084) | 66.6 | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 33.3 | 1.7 b |
| Staphylococcus aureus (DSM 346) | 4.2 | 66.6 | n.i. | 33.3 | 16.7 | ND | ND | ND | 2.1 | 0.2 a |
| Actinobacter baumannii (DSM 30008) | n.i. | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 66.6 | 0.3 c |
| Chromobacterium violaceum (DSM 30191) | n.i. | n.i. | n.i. | n.i. | 66.6 | ND | ND | ND | 16.6 | 0.8 a |
| Escherichia coli (DSM 1116) | n.i. | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 66.6 | 3.3 a |
| Pseudomonas aeruginosa (DSM PA14) | n.i. | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | n.i. | 0.4 d |
| Microorganism | MIC (µg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Ref. | |
| Candida albicans (DSM 1665) | n.i. | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 33.3 | 8.3 |
| Pichia anomala (DSM 6766) | 66.6 | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 33.3 | 8.3 |
| Rhodotorula glutinis (DSM 10134) | 16.7 | n.i. | n.i. | 66.6 | 66.6 | ND | ND | ND | 1.0 | 2.1 |
| Schizosaccharomyces pombe (DSM 70572) | 33.3 | n.i. | n.i. | n.i. | n.i. | ND | ND | ND | 4.2 | 4.2 |
| Mucor hiemalis (DSM 2656) | 16.7 | n.i. | n.i. | 66.6 | 33.3 | ND | ND | ND | 2.1 | 4.2 |
| Cell line | IC50 (µM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Ref. | |
| L929 (ACC2) | 5.8 | n.c. | n.c. | 48.9 | 34.7 | n.c. | 14.9 | 10.0 | 0.8 | 4.7 × 10−5 |
| KB3.1 (ACC158) | 2.2 | 51.0 | n.c. | 42.2 | 15.7 | n.c. | 10.3 | 2.0 | 0.4 | 3.3 × 10−5 |
| MCF-7 (A115) | 0.7 | ND | ND | ND | ND | ND | ND | ND | 0.1 | 3.0 × 10−5 |
| A549 (ACC107) | 2.8 | ND | ND | ND | ND | ND | ND | ND | 0.1 | 9.5 × 10−5 |
| PC-3 (ACC465) | 2.3 | ND | ND | ND | ND | ND | ND | ND | 0.4 | 1.5 × 10−4 |
| SKOV-3 | 1.9 | ND | ND | ND | ND | ND | ND | ND | 0.1 | 5.1 × 10−5 |
| A431 (ACC91) | 15.8 | ND | ND | ND | ND | ND | ND | ND | 1.0 | 6.7 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sum, W.C.; Mitschke, N.; Schrey, H.; Wittstein, K.; Kellner, H.; Stadler, M.; Matasyoh, J.C. Antimicrobial and Cytotoxic Cyathane-Xylosides from Cultures of the Basidiomycete Dentipellis fragilis. Antibiotics 2022, 11, 1072. https://doi.org/10.3390/antibiotics11081072
Sum WC, Mitschke N, Schrey H, Wittstein K, Kellner H, Stadler M, Matasyoh JC. Antimicrobial and Cytotoxic Cyathane-Xylosides from Cultures of the Basidiomycete Dentipellis fragilis. Antibiotics. 2022; 11(8):1072. https://doi.org/10.3390/antibiotics11081072
Chicago/Turabian StyleSum, Winnie Chemutai, Nico Mitschke, Hedda Schrey, Kathrin Wittstein, Harald Kellner, Marc Stadler, and Josphat Clement Matasyoh. 2022. "Antimicrobial and Cytotoxic Cyathane-Xylosides from Cultures of the Basidiomycete Dentipellis fragilis" Antibiotics 11, no. 8: 1072. https://doi.org/10.3390/antibiotics11081072
APA StyleSum, W. C., Mitschke, N., Schrey, H., Wittstein, K., Kellner, H., Stadler, M., & Matasyoh, J. C. (2022). Antimicrobial and Cytotoxic Cyathane-Xylosides from Cultures of the Basidiomycete Dentipellis fragilis. Antibiotics, 11(8), 1072. https://doi.org/10.3390/antibiotics11081072