
Design and Evaluation of Short Bovine Lactoferrin-Derived Antimicrobial Peptides against Multidrug-Resistant Enterococcus faecium

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Minimal Inhibitory Concentration (MIC) Assay
2.3. MIC Assay of Designed Peptides in Various Salt Concentrations
2.4. Prevention of E. faecium Biofilm Formation
2.5. Disruption of E. faecium Established Biofilms
2.6. Confocal Microscopy of Peptide-Treated E. faecium Established Biofilms
2.7. Quantitative Polymerase Chain Reaction (qPCR)
2.8. E. faecium Persister Cell and Time-Kill Assays
2.9. Persister Cell Membrane Permeability Assay
2.10. Membrane Depolarization
2.11. Laurdan-Based Membrane Fluidity Assay
2.12. Propidium Iodide-Based Membrane Permeability
2.13. Growth Inhibition Assay
2.14. ATP Release Assay
2.15. Molecular Dynamics Simulation
2.16. Metabolomics Analysis
2.17. Hemolysis of Human Red Blood Cells (hRBCs)
2.18. Mammalian Cell Cytotoxicity Assays
2.19. Ex Vivo Porcine Skin Infection Assay
2.20. Statistical Analysis
3. Results
3.1. Design and Bactericidal Activity of Short LfcinB6-Derived Peptides
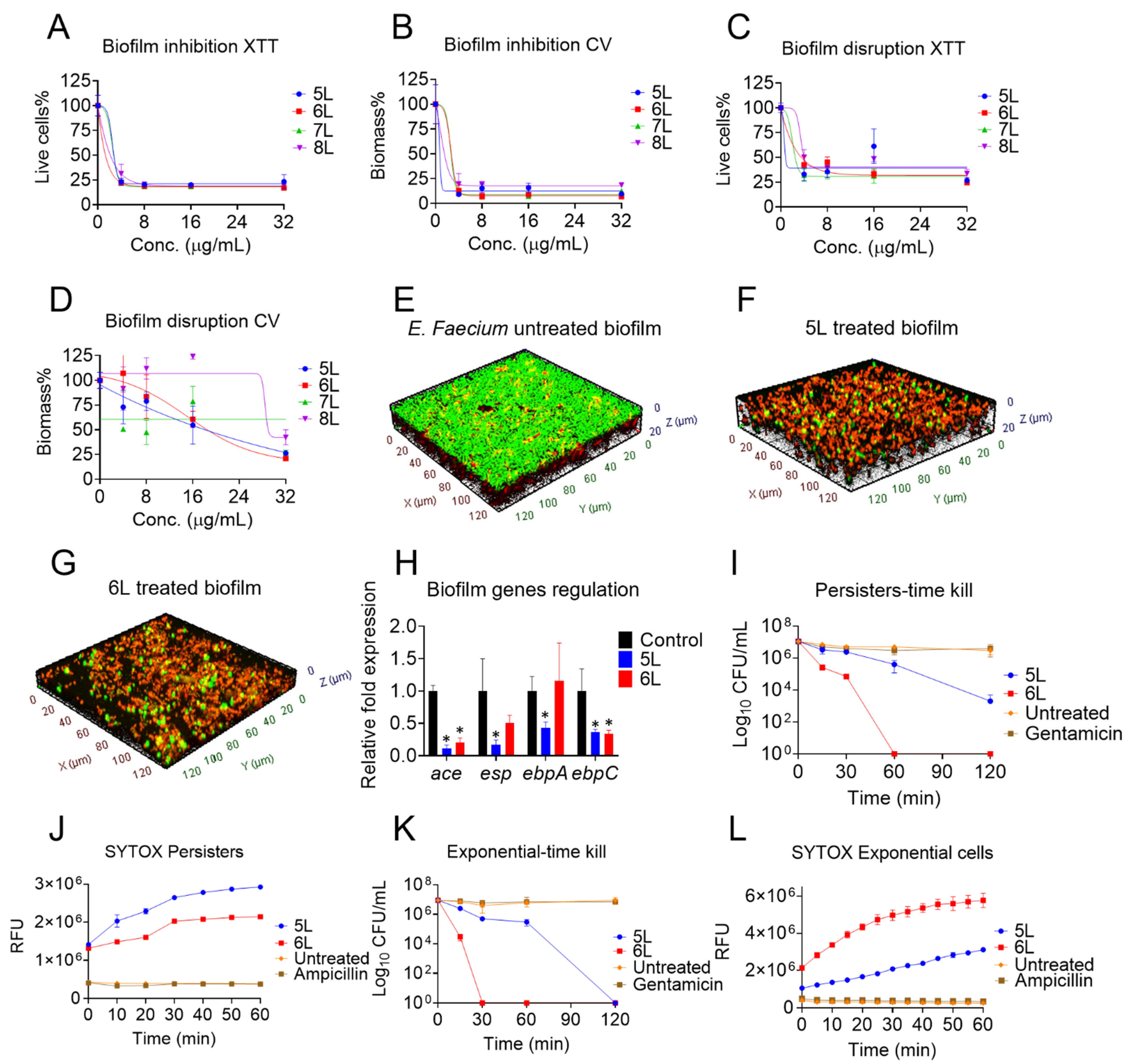
3.2. Antibiofilm Effects of LfcinB6-Derived Peptides
3.3. Antipersister Effects of LfcinB6 Peptides
3.4. Mechanism of Action (MOA) of LfcinB6 Peptides
3.5. MD Simulation of Peptide 5L in E. faecium Membrane Mimetic Model
3.6. Metabolomic Changes in E. faecium Physiology in Presence of LfcinB6 Peptide
3.7. Cytotoxicity Analysis of LfcinB6 Peptides
3.8. Ex Vivo Antibacterial Efficacy of LfcinB6-Derived Peptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Tyne, D.; Manson, A.L.; Huycke, M.M.; Karanicolas, J.; Earl, A.M.; Gilmore, M.S. Impact of Antibiotic Treatment and Host Innate Immune Pressure on Enterococcal Adaptation in the Human Bloodstream. Sci. Transl. Med. 2019, 11, eaat8418. [Google Scholar] [CrossRef] [PubMed]
- Hancock, L.; Murray, B.; Sillanpaa, J. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. In Cell Wall Components and Structures; Glimore, M.S., Clewell, D.B., Ike, Y., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Arias, C.A.; Contreras, G.A.; Murray, B.E. Management of Multidrug-Resistant Enterococcal Infections. Clin. Microbiol. Infect. 2010, 16, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Jabbari Shiadeh, S.M.; Pormohammad, A.; Hashemi, A.; Lak, P. Global Prevalence of Antibiotic Resistance in Blood-Isolated Enterococcus Faecalis and Enterococcus Faecium: A Systematic Review and Meta-Analysis. Infect. Drug Resist. 2019, 12, 2713–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC’s Antibiotic Resistance Threats in the United States, 2019. Available online: https://www.Cdc.Gov/Drugresistance/Biggest-Threats.Html#van (accessed on 10 June 2022).
- Weiner-Lastinger, L.M.; Abner, S.; Edwards, J.R.; Kallen, A.J.; Karlsson, M.; Magill, S.S.; Pollock, D.; See, I.; Soe, M.M.; Walters, M.S.; et al. Antimicrobial-Resistant Pathogens Associated with Adult Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network, 2015–2017. Infect. Control Hosp. Epidemiol. 2020, 41, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Willems, R.J.L.; Friedrich, A.W.; Rossen, J.W.A.; Bathoorn, E. Enterococcus Faecium: From Microbiological Insights to Practical Recommendations for Infection Control and Diagnostics. Antimicrob. Resist. Infect. Control 2020, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance Collaborators. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Langdon, A.; Crook, N.; Dantas, G. The Effects of Antibiotics on the Microbiome throughout Development and Alternative Approaches for Therapeutic Modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Melander, R.J.; Zurawski, D.V.; Melander, C. Narrow-Spectrum Antibacterial Agents. Medchemcomm 2018, 9, 12–21. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host Defense Antimicrobial Peptides as Antibiotics: Design and Application Strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. The Importance of Amino Acid Composition in Natural AMPs: An Evolutional, Structural, and Functional Perspective. Front. Immunol. 2012, 3, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Mishra, B.; Khader, R.; Felix, L.; Mylonakis, E. Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus Aureus. Antibiotics 2021, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of Drug Toxicity and Relevance to Pharmaceutical Development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Huertas, N.D.J.; Monroy, Z.J.R.; Medina, R.F.; Castañeda, J.E.G. Antimicrobial Activity of Truncated and Polyvalent Peptides Derived from the FKCRRQWQWRMKKGLA Sequence against Escherichia Coli ATCC 25922 and Staphylococcus Aureus ATCC 25923. Molecules 2017, 22, 987. [Google Scholar] [CrossRef] [Green Version]
- Ajish, C.; Yang, S.; Kumar, S.D.; Kim, E.Y.; Min, H.J.; Lee, C.W.; Shin, S.-H.; Shin, S.Y. A Novel Hybrid Peptide Composed of LfcinB6 and KR-12-A4 with Enhanced Antimicrobial, Anti-Inflammatory and Anti-Biofilm Activities. Sci. Rep. 2022, 12, 4365. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Y.; Yang, K.; Li, R.; Xiao, X.; Wang, Z. Antagonizing Vancomycin Resistance in Enterococcus by Surface Localized Antimicrobial Display-Derived Peptides. ACS Infect. Dis. 2020, 6, 761–767. [Google Scholar] [CrossRef]
- Wu, C.-L.; Peng, K.-L.; Yip, B.-S.; Chih, Y.-H.; Cheng, J.-W. Boosting Synergistic Effects of Short Antimicrobial Peptides With Conventional Antibiotics Against Resistant Bacteria. Front. Microbiol. 2021, 12, 747760. [Google Scholar] [CrossRef]
- Luo, X.; Ye, X.; Ding, L.; Zhu, W.; Yi, P.; Zhao, Z.; Gao, H.; Shu, Z.; Li, S.; Sang, M.; et al. Fine-Tuning of Alkaline Residues on the Hydrophilic Face Provides a Non-Toxic Cationic α-Helical Antimicrobial Peptide Against Antibiotic-Resistant ESKAPE Pathogens. Front. Microbiol. 2021, 12, 684591. [Google Scholar] [CrossRef]
- NIH Clinical Trials. Available online: https://clinicaltrials.gov/ (accessed on 10 June 2022).
- Mishra, B.; Leishangthem, G.D.; Gill, K.; Singh, A.K.; Das, S.; Singh, K.; Xess, I.; Dinda, A.; Kapil, A.; Patro, I.K.; et al. A Novel Antimicrobial Peptide Derived from Modified N-Terminal Domain of Bovine Lactoferrin: Design, Synthesis, Activity against Multidrug-Resistant Bacteria and Candida. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 677–686. [Google Scholar] [CrossRef] [Green Version]
- León-Calvijo, M.A.; Leal-Castro, A.L.; Almanzar-Reina, G.A.; Rosas-Pérez, J.E.; García-Castañeda, J.E.; Rivera-Monroy, Z.J. Antibacterial Activity of Synthetic Peptides Derived from Lactoferricin against Escherichia Coli ATCC 25922 and Enterococcus Faecalis ATCC 29212. Biomed Res. Int. 2015, 2015, 453826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, K.; Tomita, M.; Giehl, T.J.; Ellison, R.T. Antibacterial Activity of Lactoferrin and a Pepsin-Derived Lactoferrin Peptide Fragment. Infect. Immun. 1993, 61, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionysius, D.A.; Milne, J.M. Antibacterial Peptides of Bovine Lactoferrin: Purification and Characterization. J. Dairy Sci. 1997, 80, 667–674. [Google Scholar] [CrossRef]
- Vega, S.C.; Martínez, D.A.; Chalá, M.D.S.; Vargas, H.A.; Rosas, J.E. Design, Synthesis and Evaluation of Branched RRWQWR-Based Peptides as Antibacterial Agents Against Clinically Relevant Gram-Positive and Gram-Negative Pathogens. Front. Microbiol. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, C.; Jain, V.; Mishra, P.; Prasad, K.N. Clinical and Laboratory Standards Institute versus European Committee for Antimicrobial Susceptibility Testing Guidelines for Interpretation of Carbapenem Antimicrobial Susceptibility Results for Escherichia Coli in Urinary Tract Infection (UTI). J. Lab. Physicians 2018, 10, 289–293. [Google Scholar] [CrossRef]
- Felix, L.; Mishra, B.; Khadar, R.; Ganeshan, N.; Mylonakis, E. In Vitro and in Vivo Bactericidal and Antibiofilm Efficacy of Alpha Mangostin against Staphylococcus Aureus Persister Cells. Front. Cell. Infect. Microbiol. 2022, 12, 898794. [Google Scholar] [CrossRef]
- Mishra, B.; Lakshmaiah Narayana, J.; Lushnikova, T.; Wang, X.; Wang, G. Low Cationicity Is Important for Systemic in Vivo Efficacy of Database-Derived Peptides against Drug-Resistant Gram-Positive Pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 13517–13522. [Google Scholar] [CrossRef] [Green Version]
- Michiels, J.E.; van den Bergh, B.; Verstraeten, N.; Fauvart, M.; Michiels, J. In Vitro Emergence of High Persistence upon Periodic Aminoglycoside Challenge in the ESKAPE Pathogens. Antimicrob. Agents Chemother. 2016, 60, 4630–4637. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Zou, G.; Kim, H.; Kang, M.; Ahn, S.; Heo, H.Y.; Kim, J.-S.; Lim, K.-M.; Ausubel, F.M.; Mylonakis, E.; et al. Antimicrobial Activity of the Membrane-Active Compound NTZDpa Is Enhanced at Low PH. Biomed. Pharmacother. 2022, 150, 112977. [Google Scholar] [CrossRef]
- Mishra, N.N.; Bayer, A.S.; Tran, T.T.; Shamoo, Y.; Mileykovskaya, E.; Dowhan, W.; Guan, Z.; Arias, C.A. Daptomycin Resistance in Enterococci Is Associated with Distinct Alterations of Cell Membrane Phospholipid Content. PLoS ONE 2012, 7, e43958. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Bruininks, B.M.H.; Jefferies, D.; Cesar Telles de Souza, P.; Lee, J.; Patel, D.S.; Marrink, S.J.; Qi, Y.; Khalid, S.; Im, W. CHARMM-GUI Martini Maker for Modeling and Simulation of Complex Bacterial Membranes with Lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic Programming of Distinct Cancer Stem Cells Promotes Metastasis of Pancreatic Ductal Adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Pasupuleti, M.; Mörgelin, M.; Davoudi, M.; Alenfall, J.; Chalupka, A.; Malmsten, M. Boosting Antimicrobial Peptides by Hydrophobic Oligopeptide End Tags. J. Biol. Chem. 2009, 284, 17584–17594. [Google Scholar] [CrossRef] [Green Version]
- Rice, L.B.; Carias, L.L.; Rudin, S.; Lakticová, V.; Wood, A.; Hutton-Thomas, R. Enterococcus Faecium Low-Affinity Pbp5 Is a Transferable Determinant. Antimicrob. Agents Chemother. 2005, 49, 5007–5012. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Wang, G. Ab Initio Design of Potent Anti-MRSA Peptides Based on Database Filtering Technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef] [Green Version]
- Midura-Nowaczek, K.; Markowska, A. Antimicrobial Peptides and Their Analogs: Searching for New Potential Therapeutics. Perspect. Med. Chem. 2014, 6, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanram, H.; Bhattacharjya, S. ‘Lollipop’-Shaped Helical Structure of a Hybrid Antimicrobial Peptide of Temporin B-Lipopolysaccharide Binding Motif and Mapping Cationic Residues in Antibacterial Activity. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 1362–1372. [Google Scholar] [CrossRef]
- Brosig, B.; Langosch, D. The Dimerization Motif of the Glycophorin A Transmembrane Segment in Membranes: Importance of Glycine Residues. Protein Sci. 1998, 7, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammons, M.C.; Copié, V. Mini-Review: Lactoferrin: A Bioinspired, Anti-Biofilm Therapeutic. Biofouling 2013, 29, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, P.M.; Zhou, N.; Shan, X.; Arrowsmith, C.H.; Vogel, H.J. Three-Dimensional Solution Structure of Lactoferricin B, an Antimicrobial Peptide Derived from Bovine Lactoferrin. Biochemistry 1998, 37, 4288–4298. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Peptide Database. Available online: https://aps.unmc.edu/ (accessed on 10 June 2022).
- Ostorhazi, E.; Hoffmann, R.; Herth, N.; Wade, J.D.; Kraus, C.N.; Otvos, L. Advantage of a Narrow Spectrum Host Defense (Antimicrobial) Peptide Over a Broad Spectrum Analog in Preclinical Drug Development. Front. Chem. 2018, 6, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanan, R.; Li, X.; Lim, K.; Mohanram, H.; Peng, L.; Mishra, B.; Basu, A.; Lee, J.-M.; Bhattacharjya, S.; Leong, S.S.J. Design of Short Membrane Selective Antimicrobial Peptides Containing Tryptophan and Arginine Residues for Improved Activity, Salt-Resistance, and Biocompatibility. Biotechnol. Bioeng. 2014, 111, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister Cells, Dormancy and Infectious Disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef]
- Gebreyohannes, G.; Nyerere, A.; Bii, C.; Sbhatu, D.B. Challenges of Intervention, Treatment, and Antibiotic Resistance of Biofilm-Forming Microorganisms. Heliyon 2019, 5, e02192. [Google Scholar] [CrossRef] [Green Version]
- Quintieri, L.; Zühlke, D.; Fanelli, F.; Caputo, L.; Liuzzi, V.C.; Logrieco, A.F.; Hirschfeld, C.; Becher, D.; Riedel, K. Proteomic Analysis of the Food Spoiler Pseudomonas Fluorescens ITEM 17298 Reveals the Antibiofilm Activity of the Pepsin-Digested Bovine Lactoferrin. Food Microbiol. 2019, 82, 177–193. [Google Scholar] [CrossRef]
- García-Borjas, K.A.; Ceballos-Olvera, I.; Luna-Castro, S.; Peña-Avelino, Y. Bovine Lactoferrin Can Decrease the In Vitro Biofilm Production and Show Synergy with Antibiotics Against Listeria and Escherichia Coli Isolates. Protein Pept. Lett. 2021, 28, 101–107. [Google Scholar] [CrossRef]
- Avery, T.M.; Boone, R.L.; Lu, J.; Spicer, S.K.; Guevara, M.A.; Moore, R.E.; Chambers, S.A.; Manning, S.D.; Dent, L.; Marshall, D.; et al. Analysis of Antimicrobial and Antibiofilm Activity of Human Milk Lactoferrin Compared to Bovine Lactoferrin against Multidrug Resistant and Susceptible Acinetobacter Baumannii Clinical Isolates. ACS Infect. Dis. 2021, 7, 2116–2126. [Google Scholar] [CrossRef]
- Ramamourthy, G.; Vogel, H.J. Antibiofilm Activity of Lactoferrin-Derived Synthetic Peptides against Pseudomonas Aeruginosa PAO1. Biochem. Cell Biol. 2021, 99, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Kadry, A.A.; Tawfik, A.; Abu El-Asrar, A.A.; Shibl, A.M. Reduction of Mucoid Staphylococcus Epidermidis Adherence to Intraocular Lenses by Selected Antimicrobial Agents. Chemotherapy 1999, 45, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.J.; Edwards, J.R.; Culver, D.H.; Gaynes, R.P. Nosocomial Infections in Combined Medical-Surgical Intensive Care Units in the United States. Infect. Control Hosp. Epidemiol. 2000, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Feng, L.; Zhang, L.; He, J.; An, T.; Fu, X.; Li, C.; Zhao, X.; Zhai, Y.; Li, H.; et al. Antimicrobial Resistance, Virulence Genes, and Biofilm Formation Capacity Among Enterococcus Species From Yaks in Aba Tibetan Autonomous Prefecture, China. Front. Microbiol. 2020, 11, 1250. [Google Scholar] [CrossRef]
- Rich, R.L.; Kreikemeyer, B.; Owens, R.T.; LaBrenz, S.; Narayana, S.V.; Weinstock, G.M.; Murray, B.E.; Höök, M. Ace Is a Collagen-Binding MSCRAMM from Enterococcus Faecalis. J. Biol. Chem. 1999, 274, 26939–26945. [Google Scholar] [CrossRef] [Green Version]
- Sava, I.G.; Heikens, E.; Huebner, J. Pathogenesis and Immunity in Enterococcal Infections. Clin. Microbiol. Infect. 2010, 16, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of Short Cationic Antibacterial Peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Epand, R.F.; Epand, R.M.; Wang, G. Structural Location Determines Functional Roles of the Basic Amino Acids of KR-12, the Smallest Antimicrobial Peptide from Human Cathelicidin LL-37. RSC Adv. 2013, 3, 19560–19571. [Google Scholar] [CrossRef] [Green Version]
- Zarena, D.; Mishra, B.; Lushnikova, T.; Wang, F.; Wang, G. The π Configuration of the WWW Motif of a Short Trp-Rich Peptide Is Critical for Targeting Bacterial Membranes, Disrupting Preformed Biofilms, and Killing Methicillin-Resistant Staphylococcus Aureus. Biochemistry 2017, 56, 4039–4043. [Google Scholar] [CrossRef]
- saint Jean, K.D.; Henderson, K.D.; Chrom, C.L.; Abiuso, L.E.; Renn, L.M.; Caputo, G.A. Effects of Hydrophobic Amino Acid Substitutions on Antimicrobial Peptide Behavior. Probiotics Antimicrob. Proteins 2018, 10, 408–419. [Google Scholar] [CrossRef]
- Romo, T.D.; Bradney, L.A.; Greathouse, D.V.; Grossfield, A. Membrane Binding of an Acyl-Lactoferricin B Antimicrobial Peptide from Solid-State NMR Experiments and Molecular Dynamics Simulations. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 2019–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Han, F.; Xie, Y.; Wang, Y. Comparative Antimicrobial Activity and Mechanism of Action of Bovine Lactoferricin-Derived Synthetic Peptides. Biometals 2011, 24, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.-H.; Ho, Y.-H.; Chuang, Y.-C.; Chen, P.-C.; Chen, C.-S. Identification of Lactoferricin B Intracellular Targets Using an Escherichia Coli Proteome Chip. PLoS ONE 2011, 6, e28197. [Google Scholar] [CrossRef] [Green Version]
- Leng, W.; Wu, X.; Shi, T.; Xiong, Z.; Yuan, L.; Jin, W.; Gao, R. Untargeted Metabolomics on Skin Mucus Extract of Channa Argus against Staphylococcus Aureus: Antimicrobial Activity and Mechanism. Foods 2021, 10, 2995. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhou, Z.; Gu, J.; Yang, J.; Deng, J. Reducing the Periplasmic Glutathione Content Makes Escherichia Coli Resistant to Trimethoprim and Other Antimicrobial Drugs. Microbiol. Spectr. 2021, 9, e0074321. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-Targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline-Arginine-Rich Peptide, and Lactoferricin B, by Using Escherichia Coli Proteome Microarrays. Mol. Cell. Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Carias, L.L.; Rudin, S.D.; Donskey, C.J.; Rice, L.B. Genetic Linkage and Cotransfer of a Novel, VanB-Containing Transposon (Tn5382) and a Low-Affinity Penicillin-Binding Protein 5 Gene in a Clinical Vancomycin-Resistant Enterococcus Faecium Isolate. J. Bacteriol. 1998, 180, 4426–4434. [Google Scholar] [CrossRef] [Green Version]
- Donabedian, S.M.; Chow, J.W.; Boyce, J.M.; McCabe, R.E.; Markowitz, S.M.; Coudron, P.E.; Kuritza, A.; Pierson, C.L.; Zervos, M.J. Molecular Typing of Ampicillin-Resistant, Non-Beta-Lactamase-Producing Enterococcus Faecium Isolates from Diverse Geographic Areas. J. Clin. Microbiol. 1992, 30, 2757–2761. [Google Scholar] [CrossRef] [Green Version]
- Thorisdottir, A.S.; Carias, L.L.; Marshall, S.H.; Green, M.; Zervos, M.J.; Giorgio, C.; Mermel, L.A.; Boyce, J.M.; Medeiros, A.A.; Fraimow, H.; et al. IS6770, an Enterococcal Insertion-like Sequence Useful for Determining the Clonal Relationship of Clinical Enterococcal Isolates. J. Infect. Dis. 1994, 170, 1539–1548. [Google Scholar] [CrossRef]
- Garsin, D.A.; Sifri, C.D.; Mylonakis, E.; Qin, X.; Singh, K.V.; Murray, B.E.; Calderwood, S.B.; Ausubel, F.M. A Simple Model Host for Identifying Gram-Positive Virulence Factors. Proc. Natl. Acad. Sci. USA 2001, 98, 10892–10897. [Google Scholar] [CrossRef] [Green Version]
- Sahm, D.F.; Kissinger, J.; Gilmore, M.S.; Murray, P.R.; Mulder, R.; Solliday, J.; Clarke, B. In Vitro Susceptibility Studies of Vancomycin-Resistant Enterococcus Faecalis. Antimicrob. Agents Chemother. 1989, 33, 1588–1591. [Google Scholar] [CrossRef] [Green Version]
- Dunny, G.M.; Brown, B.L.; Clewell, D.B. Induced Cell Aggregation and Mating in Streptococcus Faecalis: Evidence for a Bacterial Sex Pheromone. Proc. Natl. Acad. Sci. USA 1978, 75, 3479–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huycke, M.M.; Spiegel, C.A.; Gilmore, M.S. Bacteremia Caused by Hemolytic, High-Level Gentamicin-Resistant Enterococcus Faecalis. Antimicrob. Agents Chemother. 1991, 35, 1626–1634. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.; Takeuchi, F.; Kuroda, M.; Yuzawa, H.; Aoki, K.; Oguchi, A.; Nagai, Y.; Iwama, N.; Asano, K.; Naimi, T.; et al. Genome and Virulence Determinants of High Virulence Community-Acquired MRSA. Lancet 2002, 359, 1819–1827. [Google Scholar] [CrossRef]
- Vankerckhoven, V.; van Autgaerden, T.; Vael, C.; Lammens, C.; Chapelle, S.; Rossi, R.; Jabes, D.; Goossens, H. Development of a Multiplex PCR for the Detection of Asa1, GelE, CylA, Esp, and Hyl Genes in Enterococci and Survey for Virulence Determinants among European Hospital Isolates of Enterococcus Faecium. J. Clin. Microbiol. 2004, 42, 4473–4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duprè, I.; Zanetti, S.; Schito, A.M.; Fadda, G.; Sechi, L.A. Incidence of Virulence Determinants in Clinical Enterococcus Faecium and Enterococcus Faecalis Isolates Collected in Sardinia (Italy). J. Med. Microbiol. 2003, 52, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Hashem, Y.A.; Amin, H.M.; Essam, T.M.; Yassin, A.S.; Aziz, R.K. Biofilm Formation in Enterococci: Genotype-Phenotype Correlations and Inhibition by Vancomycin. Sci. Rep. 2017, 7, 5733. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence a | NC b | Hph% c | Hy d | Hm e | rT f (min) | MIC (µg/mL) |
|---|---|---|---|---|---|---|---|
| EF C68 g | |||||||
| 1L | RRWQWRLLLL-NH2 | 3 | 60 | 0.805 | 0.168 | 18.083 | 16 |
| 2L | RRWLWRL-NH2 | 3 | 57 | NP | NP | 13.321 | >32 |
| 3L | LRWLWRRRWLWR-NH2 | 5 | 58 | 0.754 | 0.146 | 16.145 | 16 |
| 4L | RLWLWRRRWLWR-NH2 | 5 | 58 | 0.754 | 0.219 | 18.377 | 4 |
| 5L | RRWLWLRRWLWR-NH2 | 5 | 58 | 0.754 | 0.333 | 17.522 | 4 |
| 6L | RRWLWRLRWLWR-NH2 | 5 | 58 | 0.754 | 0.425 | 18.603 | 4 |
| 7L | RRWLWRRLWLWR-NH2 | 5 | 58 | 0.754 | 0.214 | 16.290 | 4 |
| 8L | RRWLWRRRWLWL-NH2 | 5 | 58 | 0.754 | 0.075 | 21.235 | 4 |
| Amp | N.P. | N.P. | N.P. | N.P. | N.P. | N.P. | >32 |
| Van | N.P. | N.P. | N.P. | N.P. | N.P. | N.P. | >32 |
| Peptide | MIC (µg/mL) | ||||
|---|---|---|---|---|---|
| Media Only | + NaCl (150 mM) | + CaCl2 (2.5 mM) | + ZnSO4 (8 µM) | + MgSO4 (1 mM) | |
| 1L | 16 | 4 | 32 | 4 | 8 |
| 2L | >32 | N.A. | N.A. | N.A. | N.A. |
| 3L | 16 | 16 | >32 | 4 | 8 |
| 4L | 4 | 8 | >32 | 4 | 8 |
| 5L | 4 | 4 | 16 | 4 | 8 |
| 6L | 4 | 4 | 8 | 4 | 8 |
| 7L | 4 | 4 | 8 | 2 | 4 |
| 8L | 4 | 4 | 16 | 2 | 4 |
| Amp a | >32 | >32 | >32 | >32 | >32 |
| Peptide | MIC (µg/mL) | |||||
|---|---|---|---|---|---|---|
| D14 | D24 | D25 | D29 | E007 | WC176 | |
| 5L | 16 | 8 | 4 | 16 | 4 | 8 |
| 6L | 8 | 8 | 4 | 8 | 4 | 8 |
| 7L | 8 | 4 | 4 | 8 | 4 | 16 |
| 8L | 16 | 8 | 8 | 32 | 4 | 16 |
| Ampicillin | ≥32 | >32 | >32 | >32 | >32 | >32 |
| Vancomycin | 2 | 1 | 1 | 1 | 1 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, B.; Felix, L.; Basu, A.; Kollala, S.S.; Chhonker, Y.S.; Ganesan, N.; Murry, D.J.; Mylonakis, E. Design and Evaluation of Short Bovine Lactoferrin-Derived Antimicrobial Peptides against Multidrug-Resistant Enterococcus faecium. Antibiotics 2022, 11, 1085. https://doi.org/10.3390/antibiotics11081085
Mishra B, Felix L, Basu A, Kollala SS, Chhonker YS, Ganesan N, Murry DJ, Mylonakis E. Design and Evaluation of Short Bovine Lactoferrin-Derived Antimicrobial Peptides against Multidrug-Resistant Enterococcus faecium. Antibiotics. 2022; 11(8):1085. https://doi.org/10.3390/antibiotics11081085
Chicago/Turabian StyleMishra, Biswajit, LewisOscar Felix, Anindya Basu, Sai Sundeep Kollala, Yashpal Singh Chhonker, Narchonai Ganesan, Daryl J. Murry, and Eleftherios Mylonakis. 2022. "Design and Evaluation of Short Bovine Lactoferrin-Derived Antimicrobial Peptides against Multidrug-Resistant Enterococcus faecium" Antibiotics 11, no. 8: 1085. https://doi.org/10.3390/antibiotics11081085