
Microbial Genomics: Innovative Targets and Mechanisms
 ,
,
Abstract
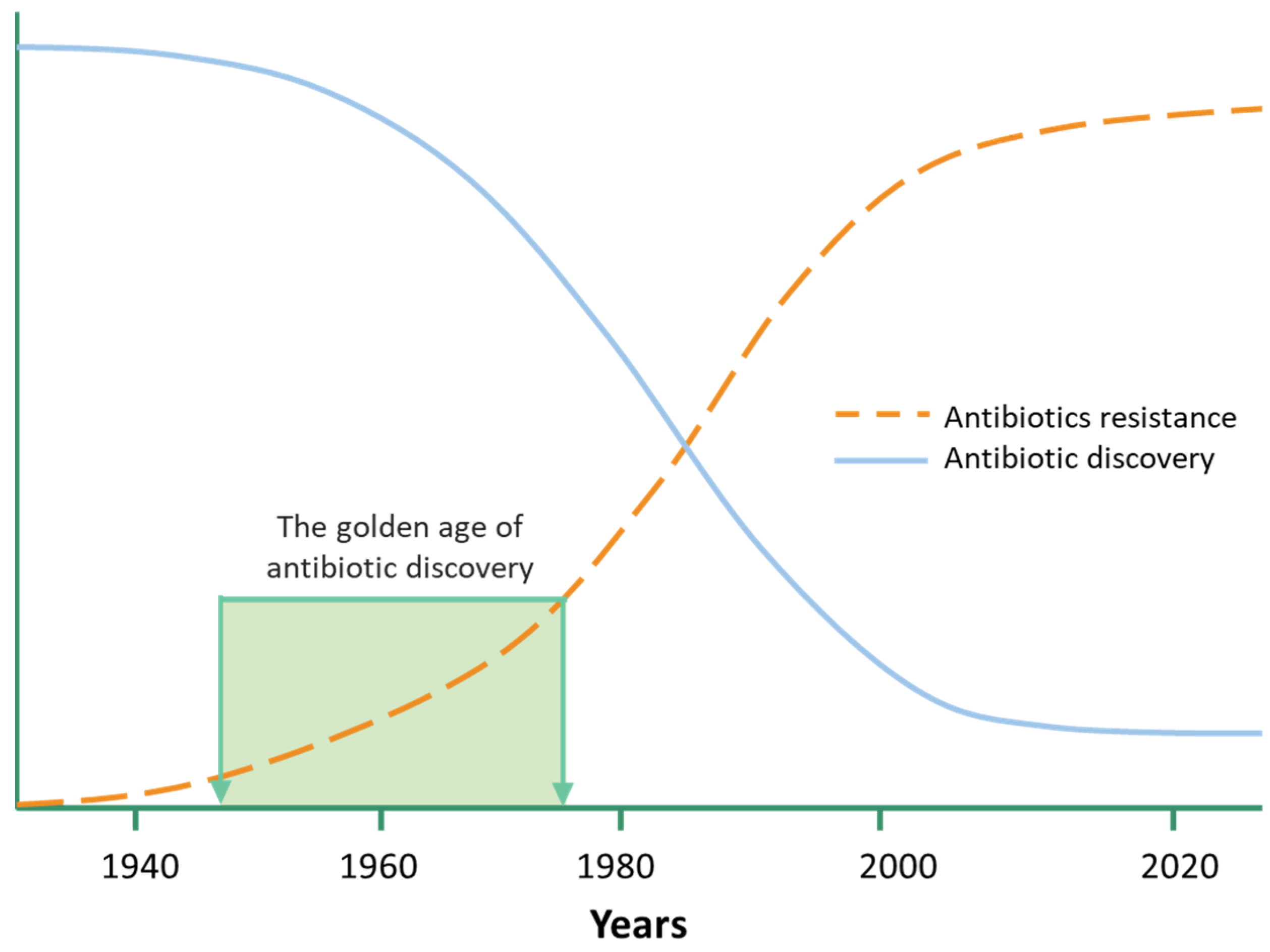
:1. Introduction
2. The Microbial Genomes Contribute to the Discovery of Novel Antimicrobial Agents
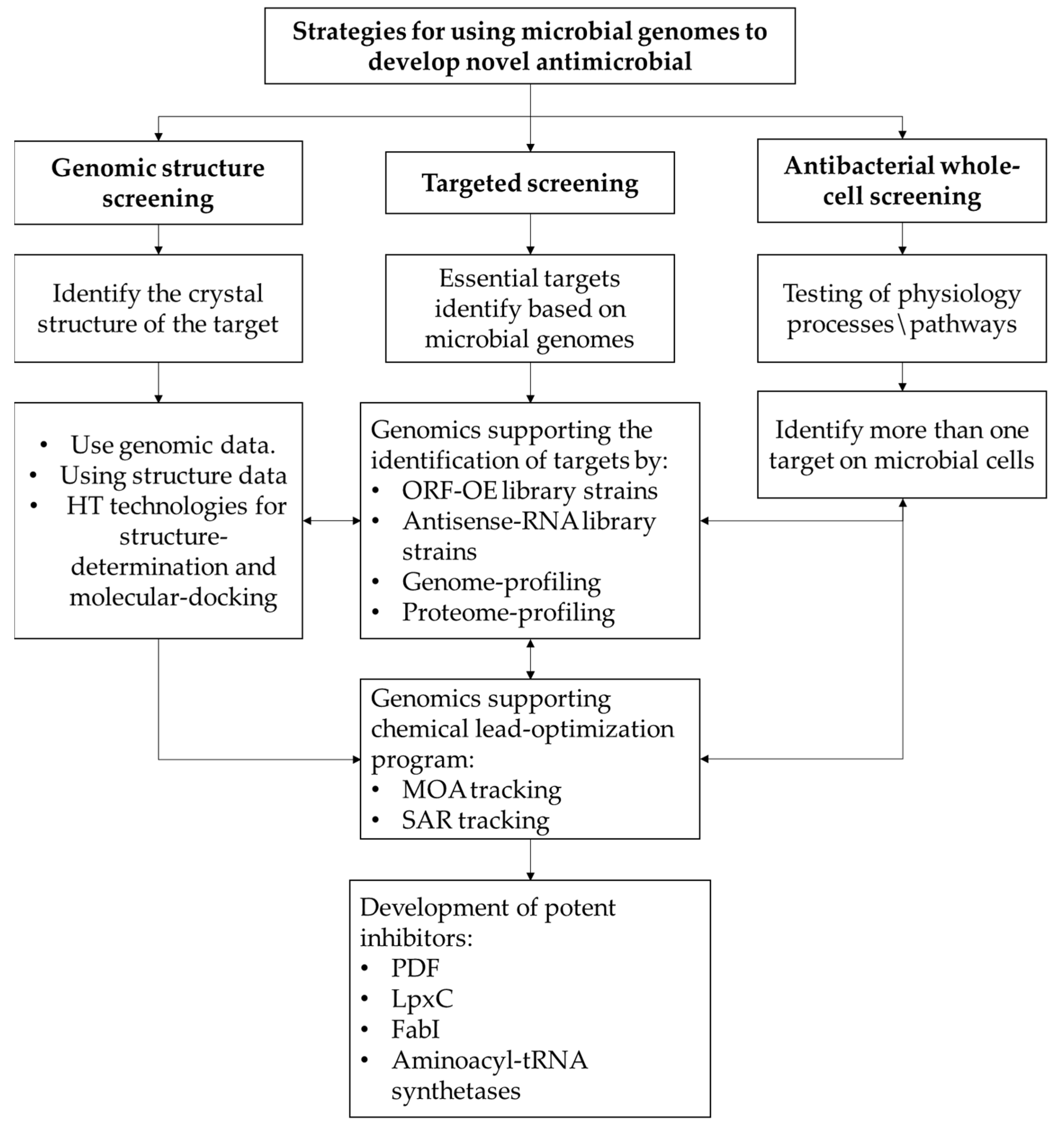
3. Pharmaceutical Companies’ Strategies for Using Bacterial Genomics to Discover Novel Antibacterial Agents
4. Innovative Targets and Mechanisms of Action to Develop Potent Antimicrobials
4.1. Cell Membrane Innovative Targets
4.1.1. LpxC Inhibitors
4.1.2. FabI Inhibitors
4.2. Protein Synthesis Inhibitor as an Innovative Target
4.2.1. Peptide Deformylase Inhibitors
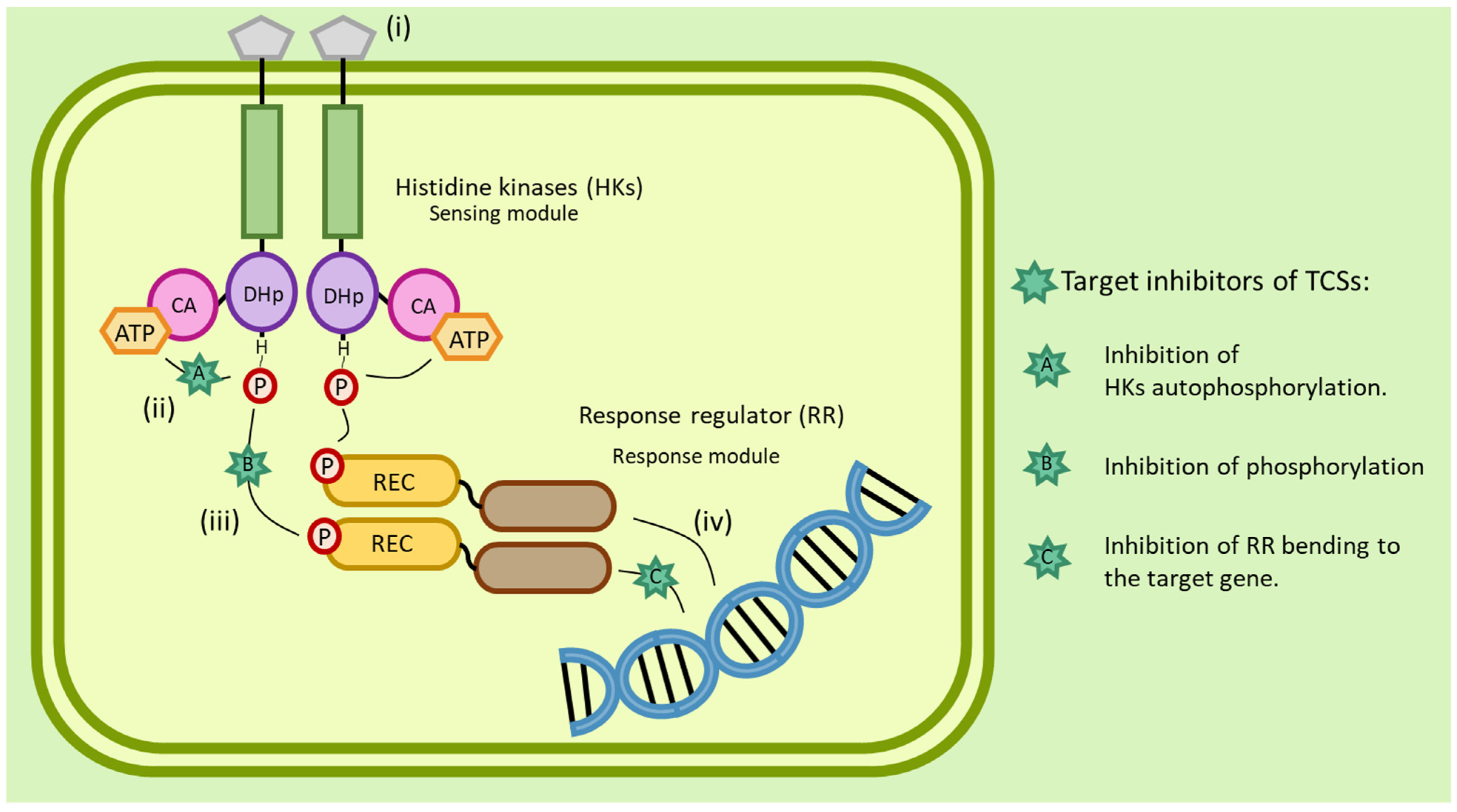
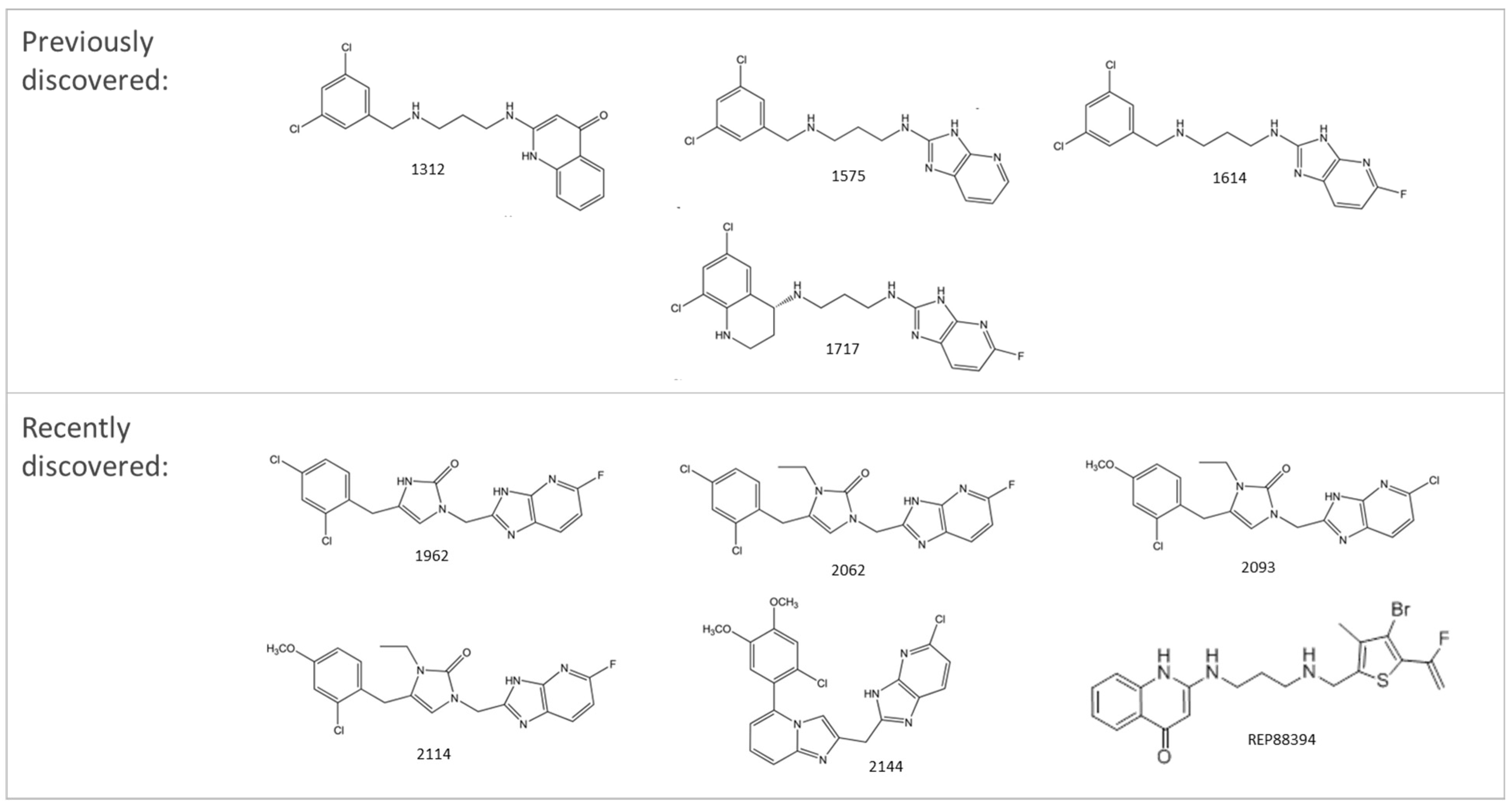
4.2.2. Histidine Kinases Inhibitors
4.2.3. Aminoacyl-tRNA Synthetases Inhibitors
4.2.4. Riboswitches as Target
5. Developing Targeted Antivirulence Therapeutics
- (A)
- VADs anti-adhesion:
- (B)
- VADs Quorum Sensing Inhibitors:
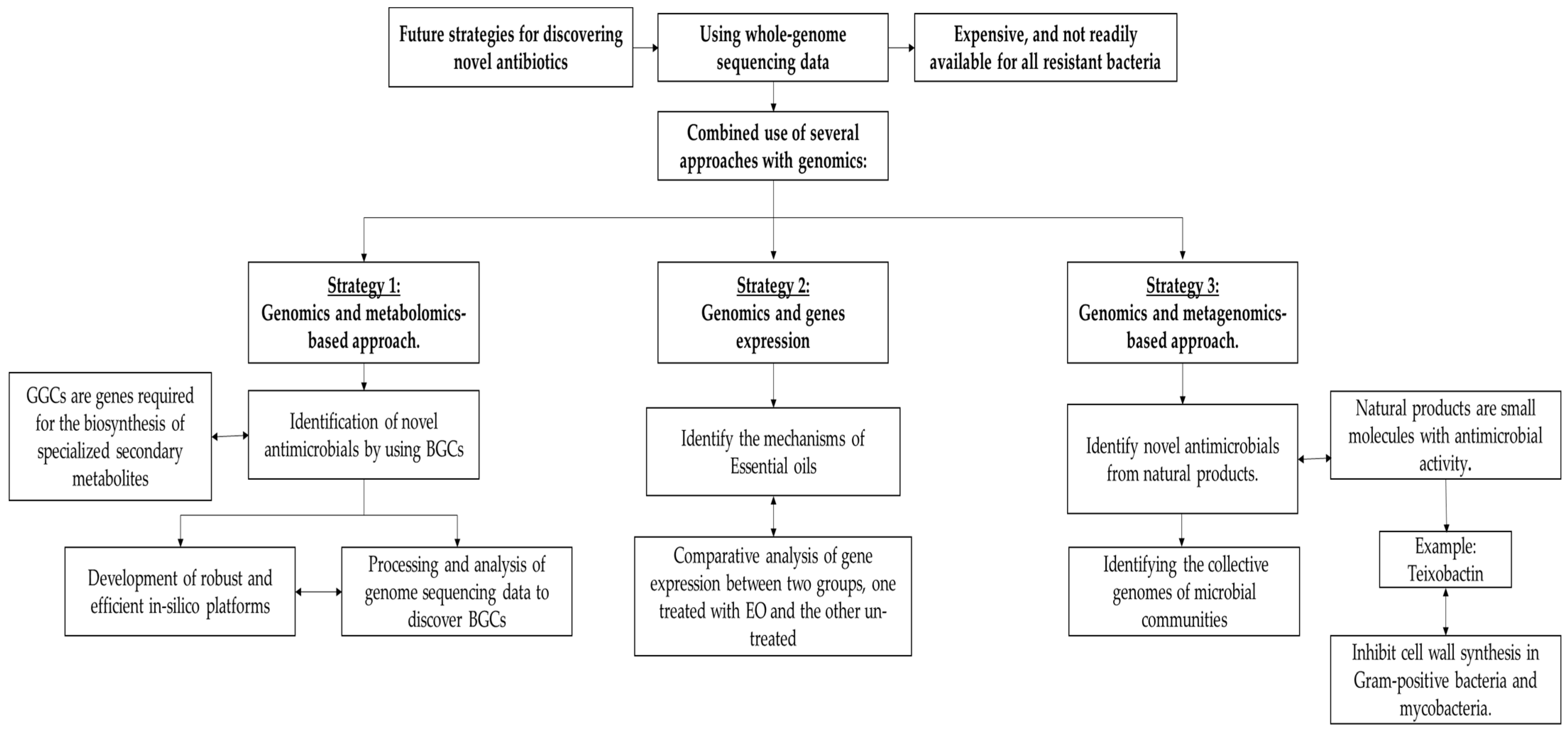
6. Future Strategies in Discovering Innovative Targets for Novel Antibiotics
6.1. Identification of Novel Antimicrobial by Using BGCs
6.2. Using Genomics to Identify the Mechanisms of Essential Oils
6.3. Using Genomics and Metagenomics to Identify Novel Antimicrobial Molecules from Microbial Communities
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walsh, F.; Amyes, S. Microbiology and Drug Resistance Mechanisms of Fully Resistant Pathogens. Curr. Opin. Microbiol. 2004, 7, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; McPherson, C.J.; Kuhn, M.; Carifa, A.; Mullins, L.; George, D.; Desbonnet, C.; Eidem, T.M.; Montgomery, J.I.; Brown, M.F.; et al. LpxC Inhibitors as New Antibacterial Agents and Tools for Studying Regulation of Lipid A Biosynthesis in Gram-Negative Pathogens. mBio 2014, 5, e01551-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, F.R.; Lee, S.W.; McConnell, M.J. Using Bacterial Genomes and Essential Genes for the Development of New Antibiotics. Biochem. Pharm. 2017, 134, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Threat Report 2013: Antibiotic/Antimicrobial Resistance; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2013. [Google Scholar]
- Chan, P.F.; Holmes, D.J.; Payne, D.J. Finding the Gems Using Genomic Discovery: Antibacterial Drug Discovery Strategies–the Successes and the Challenges. Drug Discov. Today: Ther. Strateg. 2004, 1, 519–527. [Google Scholar] [CrossRef]
- Bem, A.E.; Velikova, N.; Pellicer, M.T.; Baarlen, P.; van Marina, A.; Wells, J.M. Bacterial Histidine Kinases as Novel Antibacterial Drug Targets. ACS Chem. Biol. 2015, 10, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Hoch, J.A. Developing Inhibitors to Selectively Target Two-Component and Phosphorelay Signal Transduction Systems of Pathogenic Microorganisms. Curr. Med. Chem. 2004, 11, 765–773. [Google Scholar] [CrossRef]
- Coates, A.R.M.; Hu, Y. Novel Approaches to Developing New Antibiotics for Bacterial Infections. Br. J. Pharmacol. 2007, 152, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive Transposon Mutant Library of Pseudomonas Aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef] [Green Version]
- Mills, S.D. The Role of Genomics in Antimicrobial Discovery. J. Antimicrob. Chemother. 2003, 51, 749–752. [Google Scholar] [CrossRef]
- Ben Khedher, M.; Ghedira, K.; Rolain, J.-M.; Ruimy, R.; Croce, O. Application and Challenge of 3rd Generation Sequencing for Clinical Bacterial Studies. Int. J. Mol. Sci. 2022, 23, 1395. [Google Scholar] [CrossRef]
- Arriola, L.A.; Cooper, A.; Weyrich, L.S. Palaeomicrobiology: Application of Ancient DNA Sequencing to Better Understand Bacterial Genome Evolution and Adaptation. Front. Ecol. Evol. 2020, 8, 40. [Google Scholar] [CrossRef]
- Muzzi, A.; Masignani, V.; Rappuoli, R. The Pan-Genome: Towards a Knowledge-Based Discovery of Novel Targets for Vaccines and Antibacterials. Drug Discov. Today 2007, 12, 429–439. [Google Scholar] [CrossRef]
- Björkholm, B.; Lundin, A.; Sillén, A.; Guillemin, K.; Salama, N.; Rubio, C.; Gordon, J.I.; Falk, P.; Engstrand, L. Comparison of Genetic Divergence and Fitness between Two Subclones of Helicobacter Pylori. Infect. Immun. 2001, 69, 7832–7838. [Google Scholar] [CrossRef] [Green Version]
- Israel, D.A.; Salama, N.; Krishna, U.; Rieger, U.M.; Atherton, J.C.; Falkow, S.; Peek, R.M. Helicobacter Pylori Genetic Diversity within the Gastric Niche of a Single Human Host. Proc. Natl. Acad. Sci. USA 2001, 98, 14625–14630. [Google Scholar] [CrossRef] [Green Version]
- Fraser-Liggett, C.M. Insights on Biology and Evolution from Microbial Genome Sequencing. Genome Res. 2005, 15, 1603–1610. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, R.L.; Barrett, J.F. Antibacterial Drug Discovery--Then, Now and the Genomics Future. Biochem. Pharm. 2006, 71, 901–909. [Google Scholar] [CrossRef]
- Lai, P.-J.; Ng, E.-V.; Yang, S.-K.; Moo, C.-L.; Low, W.Y.; Yap, P.S.-X.; Lim, S.-H.E.; Lai, K.-S. Transcriptomic Analysis of Multi-Drug Resistant Escherichia Coli K-12 Strain in Response to Lavandula Angustifolia Essential Oil. 3 Biotech 2020, 10, 313. [Google Scholar] [CrossRef]
- Moir, D.T.; Shaw, K.J.; Hare, R.S.; Vovis, G.F. Genomics and Antimicrobial Drug Discovery. Antimicrob. Agents Chemother. 1999, 43, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A Genomic Perspective on Protein Families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Mushegian, A.R.; Koonin, E.V. A Minimal Gene Set for Cellular Life Derived by Comparison of Complete Bacterial Genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 10268–10273. [Google Scholar] [CrossRef]
- Arigoni, F.; Talabot, F.; Peitsch, M.; Edgerton, M.D.; Meldrum, E.; Allet, E.; Fish, R.; Jamotte, T.; Curchod, M.-L.; Loferer, H. A Genome-Based Approach for the Identification of Essential Bacterial Genes. Nat. Biotechnol. 1998, 16, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.M.; Beckett, R.P.; Brown, A.; Catlin, G.; Lobell, M.; Palan, S.; Thomas, W.; Whittaker, M.; Wood, S.; Salama, S.; et al. Antibiotic Activity and Characterization of BB-3497, a Novel Peptide Deformylase Inhibitor. Antimicrob. Agents Chemother. 2001, 45, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackbarth, C.J.; Chen, D.Z.; Lewis, J.G.; Clark, K.; Mangold, J.B.; Cramer, J.A.; Margolis, P.S.; Wang, W.; Koehn, J.; Wu, C.; et al. N-Alkyl Urea Hydroxamic Acids as a New Class of Peptide Deformylase Inhibitors with Antibacterial Activity. Antimicrob Agents Chemother 2002, 46, 2752–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Miller, W.H.; Berry, V.; Brosky, J.; Burgess, W.J.; Chen, E.; DeWolf, W.E.; Fosberry, A.P.; Greenwood, R.; Head, M.S.; et al. Discovery of a Novel and Potent Class of FabI-Directed Antibacterial Agents. Antimicrob. Agents Chemother. 2002, 46, 3118–3124. [Google Scholar] [CrossRef] [Green Version]
- Jarvest, R.L.; Berge, J.M.; Berry, V.; Boyd, H.F.; Brown, M.J.; Elder, J.S.; Forrest, A.K.; Fosberry, A.P.; Gentry, D.R.; Hibbs, M.J.; et al. Nanomolar Inhibitors of Staphylococcus Aureus Methionyl TRNA Synthetase with Potent Antibacterial Activity against Gram-Positive Pathogens. J. Med. Chem. 2002, 45, 1959–1962. [Google Scholar] [CrossRef]
- Beyer, D.; Kroll, H.-P.; Endermann, R.; Schiffer, G.; Siegel, S.; Bauser, M.; Pohlmann, J.; Brands, M.; Ziegelbauer, K.; Haebich, D.; et al. New Class of Bacterial Phenylalanyl-TRNA Synthetase Inhibitors with High Potency and Broad-Spectrum Activity. Antimicrob Agents Chemother 2004, 48, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Knowles, D.J.C.; Foloppe, N.; Matassova, N.B.; Murchie, A.I.H. The Bacterial Ribosome, a Promising Focus for Structure-Based Drug Design. Curr. Opin. Pharmacol. 2002, 2, 501–506. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Rosenberg, M. Genomic Approaches to Antibacterial Discovery. Methods Mol. Biol. 2004, 266, 231–259. [Google Scholar] [CrossRef]
- Landeta, C.; Mejia-Santana, A. Union Is Strength: Target-Based and Whole-Cell High-Throughput Screens in Antibacterial Discovery. J. Bacteriol. 2022, 204, e00477-21. [Google Scholar] [CrossRef]
- Bandow, J.E.; Brötz, H.; Leichert, L.I.O.; Labischinski, H.; Hecker, M. Proteomic Approach to Understanding Antibiotic Action. Antimicrob Agents Chemother 2003, 47, 948–955. [Google Scholar] [CrossRef] [Green Version]
- DeVito, J.A.; Mills, J.A.; Liu, V.G.; Agarwal, A.; Sizemore, C.F.; Yao, Z.; Stoughton, D.M.; Cappiello, M.G.; Barbosa, M.D.F.S.; Foster, L.A.; et al. An Array of Target-Specific Screening Strains for Antibacterial Discovery. Nat. Biotechnol. 2002, 20, 478–483. [Google Scholar] [CrossRef]
- Fischer, H.P.; Brunner, N.A.; Wieland, B.; Paquette, J.; Macko, L.; Ziegelbauer, K.; Freiberg, C. Identification of Antibiotic Stress-Inducible Promoters: A Systematic Approach to Novel Pathway-Specific Reporter Assays for Antibacterial Drug Discovery. Genome Res. 2004, 14, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Miller, J.H. Microbial Genomics—Challenges and Opportunities: The 9th International Conference on Microbial Genomes. J. Bacteriol. 2002, 184, 4327–4333. [Google Scholar] [CrossRef] [Green Version]
- Van Spriel, A.B.; van den Bogaart, G.; Cambi, A. Editorial: Membrane Domains as New Drug Targets. Front. Physiol. 2015, 6, 172. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [Green Version]
- Delcour, A.H. Outer Membrane Permeability and Antibiotic Resistance. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and Function of Lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A Comprehensive Classification System for Lipids1. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. LIPID A MODIFICATION SYSTEMS IN GRAM-NEGATIVE BACTERIA. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Serral, F.; Castello, F.A.; Sosa, E.J.; Pardo, A.M.; Palumbo, M.C.; Modenutti, C.; Palomino, M.M.; Lazarowski, A.; Auzmendi, J.; Ramos, P.I.P.; et al. From Genome to Drugs: New Approaches in Antimicrobial Discovery. Front. Pharmacol. 2021, 12, 1335. [Google Scholar] [CrossRef] [PubMed]
- MacNair, C.R.; Tsai, C.N.; Brown, E.D. Creative Targeting of the Gram-Negative Outer Membrane in Antibiotic Discovery. Ann. New York Acad. Sci. USA 2020, 1459, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, D.; Rosconi, F.; Sundaresh, B.; Rudmann, E.; Ramirez, L.M.N.; Nishimoto, A.T.; Wood, S.J.; Jana, B.; Buján, N.; Li, K.; et al. A Genome-Wide Atlas of Antibiotic Susceptibility Targets and Pathways to Tolerance. Nat. Commun. 2022, 13, 3165. [Google Scholar] [CrossRef]
- Zhou, P.; Barb, A.W. Mechanism and Inhibition of LpxC: An Essential Zinc-Dependent Deacetylase of Bacterial Lipid A Synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Caughlan, R.E.; Jones, A.K.; DeLucia, A.M.; Woods, A.L.; Xie, L.; Ma, B.; Barnes, S.W.; Walker, J.R.; Sprague, E.R.; Yang, X.; et al. Mechanisms Decreasing In Vitro Susceptibility to the LpxC Inhibitor CHIR-090 in the Gram-Negative Pathogen Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Mdluli, K.E.; Witte, P.R.; Kline, T.; Barb, A.W.; Erwin, A.L.; Mansfield, B.E.; McClerren, A.L.; Pirrung, M.C.; Tumey, L.N.; Warrener, P.; et al. Molecular Validation of LpxC as an Antibacterial Drug Target in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2178–2184. [Google Scholar] [CrossRef] [Green Version]
- Onishi, H.R.; Pelak, B.A.; Gerckens, L.S.; Silver, L.L.; Kahan, F.M.; Chen, M.-H.; Patchett, A.A.; Galloway, S.M.; Hyland, S.A.; Anderson, M.S.; et al. Antibacterial Agents That Inhibit Lipid A Biosynthesis. Science 1996, 274, 980–982. [Google Scholar] [CrossRef]
- Erwin, A.L. Antibacterial Drug Discovery Targeting the Lipopolysaccharide Biosynthetic Enzyme LpxC. Cold Spring Harb. Perspect Med. 2016, 6, a025304. [Google Scholar] [CrossRef] [Green Version]
- Krause, K.M.; Haglund, C.M.; Hebner, C.; Serio, A.W.; Lee, G.; Nieto, V.; Cohen, F.; Kane, T.R.; Machajewski, T.D.; Hildebrandt, D.; et al. Potent LpxC Inhibitors with In Vitro Activity against Multidrug-Resistant Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00977-19. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, The Gold Book, 4th ed.; Gold, V., Ed.; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 2019. [Google Scholar]
- McClerren, A.L.; Endsley, S.; Bowman, J.L.; Andersen, N.H.; Guan, Z.; Rudolph, J.; Raetz, C.R.H. A Slow, Tight-Binding Inhibitor of the Zinc-Dependent Deacetylase LpxC of Lipid A Biosynthesis with Antibiotic Activity Comparable to Ciprofloxacin. Biochemistry 2005, 44, 16574–16583. [Google Scholar] [CrossRef] [Green Version]
- Barb, A.W.; Jiang, L.; Raetz, C.R.H.; Zhou, P. Structure of the Deacetylase LpxC Bound to the Antibiotic CHIR-090: Time-Dependent Inhibition and Specificity in Ligand Binding. Proc. Natl. Acad. Sci. USA 2007, 104, 18433–18438. [Google Scholar] [CrossRef] [Green Version]
- Barb, A.W.; McClerren, A.L.; Snehelatha, K.; Reynolds, C.M.; Zhou, P.; Raetz, C.R.H. Inhibition of Lipid A Biosynthesis as the Primary Mechanism of CHIR-090 Antibiotic Activity in Escherichia Coli. Biochemistry 2007, 46, 3793–3802. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Ghouse, S.M.; Akunuri, R.; Madhavi, Y.V.; Chopra, S.; Nanduri, S. FabI (Enoyl Acyl Carrier Protein Reductase)-A Potential Broad Spectrum Therapeutic Target and Its Inhibitors. Eur. J. Med. Chem. 2020, 208, 112757. [Google Scholar] [CrossRef]
- Karchmer, A.; Hafkin, B. Is There a Future for FabI Inhibitors as Antibacterial Agents? Clin. Investig. 2013, 3, 707–709. [Google Scholar] [CrossRef]
- Livermore, D.M. Discovery Research: The Scientific Challenge of Finding New Antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.J.; Sohn, M.-J.; Lee, S.; Kim, W.-G. Meleagrin, a New FabI Inhibitor from Penicillium Chryosogenum with at Least One Additional Mode of Action. PLoS ONE 2013, 8, e78922. [Google Scholar] [CrossRef] [Green Version]
- Inflating Bacterial Cells by Increased Protein Synthesis. Mol. Syst. Biol. 2015, 11, 836. [CrossRef]
- Kadam, S. CHAPTER 8-Mechanism-Based Screens in the Discovery of Chemotherapeutic Antibacterials. In Discovery of Novel Natural Products with Therapeutic Potential; Gullo, V.P., Ed.; Newnes: Boston, MA, USA, 1994; ISBN 978-0-7506-9003-4. [Google Scholar]
- McCoy, L.S.; Xie, Y.; Tor, Y. Antibiotics That Target Protein Synthesis. WIREs RNA 2011, 2, 209–232. [Google Scholar] [CrossRef]
- Punina, N.V.; Makridakis, N.M.; Remnev, M.A.; Topunov, A.F. Whole-Genome Sequencing Targets Drug-Resistant Bacterial Infections. Hum. Genom. 2015, 9, 19. [Google Scholar] [CrossRef]
- Novac, O.; Guenier, A.; Pelletier, J. Inhibitors of Protein Synthesis Identified by a High Throughput Multiplexed Translation Screen. Nucleic Acids Res. 2004, 32, 902–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangshetti, J.N.; Khan, F.A.K.; Shinde, D.B. Peptide Deformylase: A New Target in Antibacterial, Antimalarial and Anticancer Drug Discovery. Curr. Med. Chem. 2015, 22, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Chen, D.; White, R.J.; Patel, D.V.; Yuan, Z. Bacterial Peptide Deformylase Inhibitors: A New Class of Antibacterial Agents. Curr. Med. Chem. 2005, 12, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Apfel, C.M.; Locher, H.; Evers, S.; Takács, B.; Hubschwerlen, C.; Pirson, W.; Page, M.G.P.; Keck, W. Peptide Deformylase as an Antibacterial Drug Target: Target Validation and Resistance Development. Antimicrob Agents Chemother 2001, 45, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kijjoa, A.; Sawangwong, P. Drugs and Cosmetics from the Sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Harwood, C.R.; Mouillon, J.-M.; Pohl, S.; Arnau, J. Secondary Metabolite Production and the Safety of Industrially Important Members of the Bacillus Subtilis Group. FEMS Microbiol. Rev. 2018, 42, 721–738. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Eisenreich, A.; Schneider, K.; Heinemeyer, I.; Morgenstern, B.; Voss, B.; Hess, W.R.; Reva, O.; et al. Comparative Analysis of the Complete Genome Sequence of the Plant Growth-Promoting Bacterium Bacillus Amyloliquefaciens FZB42. Nat. Biotechnol. 2007, 25, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, K.; Kizhakkekalam, V.K.; Joy, M.; Chakraborty, R.D. Moving Away from Traditional Antibiotic Treatment: Can Macrocyclic Lactones from Marine Macroalga-Associated Heterotroph Be the Alternatives? Appl. Microbiol. Biotechnol. 2020, 104, 7117–7130. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, R.; Chen, X.; Sun, X.; Yan, Y.; Shen, X.; Yuan, Q. Biosynthesis of Aromatic Polyketides in Microorganisms Using Type II Polyketide Synthases. Microb. Cell Factories 2020, 19, 110. [Google Scholar] [CrossRef]
- Miyanaga, A. Structure and Function of Polyketide Biosynthetic Enzymes: Various Strategies for Production of Structurally Diverse Polyketides. Biosci. Biotechnol. Biochem. 2017, 81, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Kaserer, A.O.; West, A.H. Chapter 78-Histidine Kinases in Two-Component Signaling Pathways. In Handbook of Cell Signaling, 2nd ed.; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press: San Diego, CA, USA, 2010; ISBN 978-0-12-374145-5. [Google Scholar]
- Singh, V.; Dhankhar, P.; Kumar, P. Chapter 26-Bacterial Histidine Kinases as Potential Antibacterial Drug Targets. In Protein Kinase Inhibitors; Hassan, M.d.I., Noor, S., Eds.; Academic Press: San Diego, CA, USA, 2022; ISBN 978-0-323-91287-7. [Google Scholar]
- Yoshida, T.; Phadtare, S.; Inouye, M. [7]-The Design and Development of Tar-EnvZ Chimeric Receptors. In Methods in Enzymology; Simon, M.I., Crane, B.R., Crane, A., Eds.; Two-Component Signaling Systems, Part B.; Academic Press: San Diego, CA, USA, 2007; Volume 423, pp. 166–183. [Google Scholar]
- Bhate, M.P.; Molnar, K.S.; Goulian, M.; DeGrado, W.F. Signal Transduction in Histidine Kinases: Insights from New Structures. Structure 2015, 23, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, L.E.; Zhulin, I.B. The MiST2 Database: A Comprehensive Genomics Resource on Microbial Signal Transduction. Nucleic Acids Res. 2010, 38, D401–D407. [Google Scholar] [CrossRef] [Green Version]
- Thomason, P.; Kay, R. Eukaryotic Signal Transduction via Histidine-Aspartate Phosphorelay. J. Cell Sci. 2000, 113, 3141–3150. [Google Scholar] [CrossRef]
- Takada, H.; Yoshikawa, H. Essentiality and Function of WalK/WalR Two-Component System: The Past, Present, and Future of Research. Biosci. Biotechnol. Biochem. 2018, 82, 741–751. [Google Scholar] [CrossRef]
- Rajput, A.; Seif, Y.; Choudhary, K.S.; Dalldorf, C.; Poudel, S.; Monk, J.M.; Palsson, B.O. Pangenome Analytics Reveal Two-Component Systems as Conserved Targets in ESKAPEE Pathogens. mSystems 2021, 6, e00981-20. [Google Scholar] [CrossRef]
- Chen, H.; Yu, C.; Wu, H.; Li, G.; Li, C.; Hong, W.; Yang, X.; Wang, H.; You, X. Recent Advances in Histidine Kinase-Targeted Antimicrobial Agents. Front. Chem. 2022, 10, 866392. [Google Scholar] [CrossRef]
- Boibessot, T.; Zschiedrich, C.P.; Lebeau, A.; Bénimèlis, D.; Dunyach-Rémy, C.; Lavigne, J.-P.; Szurmant, H.; Benfodda, Z.; Meffre, P. The Rational Design, Synthesis, and Antimicrobial Properties of Thiophene Derivatives That Inhibit Bacterial Histidine Kinases. J. Med. Chem. 2016, 59, 8830–8847. [Google Scholar] [CrossRef] [Green Version]
- Fakhruzzaman, M.; Inukai, Y.; Yanagida, Y.; Kino, H.; Igarashi, M.; Eguchi, Y.; Utsumi, R. Study on in Vivo Effects of Bacterial Histidine Kinase Inhibitor, Waldiomycin, in Bacillus Subtilis and Staphylococcus Aureus. J. Gen. Appl. Microbiol. 2015, 61, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Okajima, T.; Tochio, N.; Inukai, Y.; Shimizu, R.; Ueda, S.; Shinya, S.; Kigawa, T.; Fukamizo, T.; Igarashi, M.; et al. Angucycline Antibiotic Waldiomycin Recognizes Common Structural Motif Conserved in Bacterial Histidine Kinases. J. Antibiot 2017, 70, 251–258. [Google Scholar] [CrossRef]
- Lv, P.-C.; Zhu, H.-L. Aminoacyl-TRNA Synthetase Inhibitors as Potent Antibacterials. Curr. Med. Chem 2012, 19, 3550–3563. [Google Scholar] [CrossRef] [PubMed]
- Faghih, O.; Zhang, Z.; Ranade, R.M.; Gillespie, J.R.; Creason, S.A.; Huang, W.; Shibata, S.; Barros-Álvarez, X.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Development of Methionyl-TRNA Synthetase Inhibitors as Antibiotics for Gram-Positive Bacterial Infections. Antimicrob Agents Chemother 2017, 61, e00999-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francklyn, C.S.; Mullen, P. Progress and Challenges in Aminoacyl-TRNA Synthetase-Based Therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, S.; Nandi, N. Dynamics of the Catalytic Active Site of Isoleucyl TRNA Synthetase from Staphylococcus Aureus Bound with Adenylate and Mupirocin. J. Phys. Chem. B 2022, 126, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Haines, A.S.; Kendrew, S.G.; Crowhurst, N.; Stephens, E.R.; Connolly, J.; Hothersall, J.; Miller, C.E.; Collis, A.J.; Huckle, B.D.; Thomas, C.M. High Quality Genome Annotation and Expression Visualisation of a Mupirocin-Producing Bacterium. PLoS ONE 2022, 17, e0268072. [Google Scholar] [CrossRef]
- Klochko, V.V.; Zelena, L.B.; Kim, J.Y.; Avdeeva, L.V.; Reva, O.N. Prospects of a New Antistaphylococcal Drug Batumin Revealed by Molecular Docking and Analysis of the Complete Genome Sequence of the Batumin-Producer Pseudomonas Batumici UCM B-321. Int. J. Antimicrob Agents 2016, 47, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Gillespie, J.R.; Kelley, A.M.; Napuli, A.J.; Zhang, Z.; Kovzun, K.V.; Pefley, R.M.; Lam, J.; Zucker, F.H.; Van Voorhis, W.C.; et al. Selective Inhibitors of Methionyl-TRNA Synthetase Have Potent Activity against Trypanosoma Brucei Infection in Mice. Antimicrob. Agents Chemother. 2011, 55, 1982–1989. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Koh, C.Y.; Ranade, R.M.; Shibata, S.; Gillespie, J.R.; Hulverson, M.A.; Huang, W.; Nguyen, J.; Pendem, N.; Gelb, M.H.; et al. 5-Fluoroimidazo [4,5-b]Pyridine Is a Privileged Fragment That Conveys Bioavailability to Potent Trypanosomal Methionyl-TRNA Synthetase Inhibitors. ACS Infect. Dis. 2016, 2, 399–404. [Google Scholar] [CrossRef]
- Breaker, R.R. Riboswitches and the RNA World. Cold Spring Harb. Perspect Biol. 2012, 4, a003566. [Google Scholar] [CrossRef] [Green Version]
- Panchal, V.; Brenk, R. Riboswitches as Drug Targets for Antibiotics. Antibiotics 2021, 10, 45. [Google Scholar] [CrossRef]
- Pavlova, N.; Kaloudas, D.; Penchovsky, R. Riboswitch Distribution, Structure, and Function in Bacteria. Gene 2019, 708, 38–48. [Google Scholar] [CrossRef]
- Mandal, M.; Boese, B.; Barrick, J.E.; Winkler, W.C.; Breaker, R.R. Riboswitches Control Fundamental Biochemical Pathways in Bacillus Subtilis and Other Bacteria. Cell 2003, 113, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Penchovsky, R.; Stoilova, C.C. Riboswitch-Based Antibacterial Drug Discovery Using High-Throughput Screening Methods. Expert Opin. Drug Discov. 2013, 8, 65–82. [Google Scholar] [CrossRef]
- Breaker, R.R. Riboswitches and Translation Control. Cold Spring Harb Perspect Biol. 2018, 10, a032797. [Google Scholar] [CrossRef]
- Cross, A.S. What Is a Virulence Factor? Critical Care 2008, 12, 196. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Dhasmana, N.; Dubey, N.; Kumar, N.; Gangwal, A.; Gupta, M.; Singh, Y. Bacterial Virulence Factors: Secreted for Survival. Indian J. Microbiol. 2017, 57, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sintchenko, V.; Timms, V.; Sim, E.; Rockett, R.; Bachmann, N.; O’Sullivan, M.; Marais, B. Microbial Genomics as a Catalyst for Targeted Antivirulence Therapeutics. Front Med. 2021, 8, 641260. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-Virulence Strategies to Combat Bacteria-Mediated Disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Rentzsch, R.; Deneke, C.; Nitsche, A.; Renard, B.Y. Predicting Bacterial Virulence Factors–Evaluation of Machine Learning and Negative Data Strategies. Brief. Bioinform. 2020, 21, 1596–1608. [Google Scholar] [CrossRef]
- Maeda, T.; García-Contreras, R.; Pu, M.; Sheng, L.; Garcia, L.R.; Tomás, M.; Wood, T.K. Quorum Quenching Quandary: Resistance to Antivirulence Compounds. ISME J. 2012, 6, 493–501. [Google Scholar] [CrossRef]
- Imperi, F.; Fiscarelli, E.V.; Visaggio, D.; Leoni, L.; Visca, P. Activity and Impact on Resistance Development of Two Antivirulence Fluoropyrimidine Drugs in Pseudomonas Aeruginosa. Front. Cell. Infect. Microbiol. 2019, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Achtman, M.; Zhou, Z. Distinct Genealogies for Plasmids and Chromosome. PLoS Genet. 2014, 10, e1004874. [Google Scholar] [CrossRef] [Green Version]
- Lindh, M.V.; Sjöstedt, J.; Ekstam, B.; Casini, M.; Lundin, D.; Hugerth, L.W.; Hu, Y.O.O.; Andersson, A.F.; Andersson, A.; Legrand, C.; et al. Metapopulation Theory Identifies Biogeographical Patterns among Core and Satellite Marine Bacteria Scaling from Tens to Thousands of Kilometers. Environ. Microbiol. 2017, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesson, P. Metapopulations. In Encyclopedia of Biodiversity, 2nd ed.; Levin, S.A., Ed.; Academic Press: Waltham, MA, USA, 2013; ISBN 978-0-12-384720-1. [Google Scholar]
- Rockett, R.J.; Oftadeh, S.; Bachmann, N.L.; Timms, V.J.; Kong, F.; Gilbert, G.L.; Sintchenko, V. Genome-Wide Analysis of Streptococcus Pneumoniae Serogroup 19 in the Decade after the Introduction of Pneumococcal Conjugate Vaccines in Australia. Sci. Rep. 2018, 8, 16969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Hiley, L.; Octavia, S.; Tanaka, M.M.; Sintchenko, V.; Lan, R. Comparative Genomics of Australian and International Isolates of Salmonella Typhimurium: Correlation of Core Genome Evolution with CRISPR and Prophage Profiles. Sci. Rep. 2017, 7, 9733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, B.; Luyt, C.-E.; Stover, C.K.; Brubaker, J.O.; Chastre, J.; Jafri, H.S. New Strategies Targeting Virulence Factors of Staphylococcus Aureus and Pseudomonas Aeruginosa. Semin. Respir Crit Care Med. 2017, 38, 346–358. [Google Scholar] [CrossRef]
- Fasciano, A.C.; Shaban, L.; Mecsas, J. Promises and Challenges of the Type Three Secretion System Injectisome as an Antivirulence Target. EcoSal Plus 2019, 8, 16. [Google Scholar] [CrossRef]
- Kisiela, D.I.; Avagyan, H.; Friend, D.; Jalan, A.; Gupta, S.; Interlandi, G.; Liu, Y.; Tchesnokova, V.; Rodriguez, V.B.; Sumida, J.P.; et al. Inhibition and Reversal of Microbial Attachment by an Antibody with Parasteric Activity against the FimH Adhesin of Uropathogenic E. Coli. PLOS Pathog. 2015, 11, e1004857. [Google Scholar] [CrossRef] [Green Version]
- Mellini, M.; Di Muzio, E.; D’Angelo, F.; Baldelli, V.; Ferrillo, S.; Visca, P.; Leoni, L.; Polticelli, F.; Rampioni, G. In Silico Selection and Experimental Validation of FDA-Approved Drugs as Anti-Quorum Sensing Agents. Front. Microbiol. 2019, 10, 2355. [Google Scholar] [CrossRef]
- Guo, Z.; Li, X.; Li, J.; Yang, X.; Zhou, Y.; Lu, C.; Shen, Y. Licoflavonol Is an Inhibitor of the Type Three Secretion System of Salmonella Enterica Serovar Typhimurium. Biochem. Biophys. Res. Commun. 2016, 477, 998–1004. [Google Scholar] [CrossRef]
- Paluch, E.; Rewak-Soroczyńska, J.; Jędrusik, I.; Mazurkiewicz, E.; Jermakow, K. Prevention of Biofilm Formation by Quorum Quenching. Appl. Microbiol. Biotechnol. 2020, 104, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- García-Reyes, S.; Soberón-Chávez, G.; Cocotl-Yanez, M. 2020 The Third Quorum-Sensing System of Pseudomonas Aeruginosa: Pseudomonas Quinolone Signal and the Enigmatic PqsE Protein. J. Med. Microbiol. 2020, 69, 25–34. [Google Scholar] [CrossRef]
- Fleitas Martínez, O.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent Advances in Anti-Virulence Therapeutic Strategies With a Focus on Dismantling Bacterial Membrane Microdomains, Toxin Neutralization, Quorum-Sensing Interference and Biofilm Inhibition. Front. Cell. Infect. Microbiol. 2019, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Chbib, C. Impact of the Structure-Activity Relationship of AHL Analogues on Quorum Sensing in Gram-Negative Bacteria. Bioorganic Med. Chem. 2020, 28, 115282. [Google Scholar] [CrossRef]
- Udaondo, Z.; Matilla, M.A. Mining for Novel Antibiotics in the Age of Antimicrobial Resistance. Microb. Biotechnol. 2020, 13, 1702–1704. [Google Scholar] [CrossRef]
- Cattoir, V.; Felden, B. Future Antibacterial Strategies: From Basic Concepts to Clinical Challenges. J. Infect. Dis. 2019, 220, 350–360. [Google Scholar] [CrossRef]
- Williams, A.N.; Sorout, N.; Cameron, A.J.; Stavrinides, J. The Integration of Genome Mining, Comparative Genomics, and Functional Genetics for Biosynthetic Gene Cluster Identification. Front. Genet. 2020, 11, 600116. [Google Scholar] [CrossRef]
- Elizabeth, J.C.; Gerard, D.W. Biosynthetic Gene Clusters Guide Rational Antibiotic Discovery from Actinomycetes. Ph.D. Thesis, McMaster University, Hamilton, Canada, 2020. [Google Scholar]
- van Der Hooft, J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking Genomics and Metabolomics to Chart Specialized Metabolic Diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Srivastava, R.; Bharati, A.P.; Singh, A.K.; Sharma, A.; Das, S.; Tiwari, P.K.; Srivastava, A.K.; Chakdar, H.; Kashyap, P.L.; et al. Analysis of Biosynthetic Gene Clusters, Secretory, and Antimicrobial Peptides Reveals Environmental Suitability of Exiguobacterium Profundum PHM11. Front. Microbiol. 2022, 12, 4081. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.-M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete Genome Sequence of the Model Actinomycete Streptomyces Coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Della Sala, G.; Mangoni, A.; Costantino, V.; Teta, R. Identification of the Biosynthetic Gene Cluster of Thermoactinoamides and Discovery of New Congeners by Integrated Genome Mining and MS-Based Molecular Networking. Front. Chem. 2020, 8, 397. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. AntiSMASH 4.0—Improvements in Chemistry Prediction and Gene Cluster Boundary Identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Merwin, N.J.; Johnston, C.W.; Magarvey, N.A. PRISM 3: Expanded Prediction of Natural Product Chemical Structures from Microbial Genomes. Nucleic Acids Res. 2017, 45, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, P.S.X.; Yusoff, K.; Lim, S.-H.E.; Chong, C.-M.; Lai, K.-S. Membrane Disruption Properties of Essential Oils—A Double-Edged Sword? Processes 2021, 9, 595. [Google Scholar] [CrossRef]
- Yang, S.-K.; Tan, N.-P.; Chong, C.-W.; Abushelaibi, A.; Lim, S.-H.-E.; Lai, K.-S. The Missing Piece: Recent Approaches Investigating the Antimicrobial Mode of Action of Essential Oils. Evol Bioinform 2021, 17, 1176934320938391. [Google Scholar] [CrossRef]
- Roy, A.; Mahata, D.; Paul, D.; Korpole, S.; Franco, O.; Mandal, S. Purification, Biochemical Characterization and Self-Assembled Structure of a Fengycin-like Antifungal Peptide from Bacillus Thuringiensis Strain SM1. Front. Microbiol. 2013, 4, 332. [Google Scholar] [CrossRef] [Green Version]
- de Souza Cândido, E.; e Silva Cardoso, M.H.; Sousa, D.A.; Viana, J.C.; de Oliveira-Júnior, N.G.; Miranda, V.; Franco, O.L. The Use of Versatile Plant Antimicrobial Peptides in Agribusiness and Human Health. Peptides 2014, 55, 65–78. [Google Scholar] [CrossRef]
- Berglund, F.; Österlund, T.; Boulund, F.; Marathe, N.P.; Larsson, D.G.J.; Kristiansson, E. Identification and Reconstruction of Novel Antibiotic Resistance Genes from Metagenomes. Microbiome 2019, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- de Castro, A.P.; Fernandes, G.d.R.; Franco, O.L. Insights into Novel Antimicrobial Compounds and Antibiotic Resistance Genes from Soil Metagenomes. Front. Microbiol. 2014, 5, 489. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A New Antibiotic Kills Pathogens without Detectable Resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Mantravadi, P.K.; Kalesh, K.A.; Dobson, R.C.J.; Hudson, A.O.; Parthasarathy, A. The Quest for Novel Antimicrobial Compounds: Emerging Trends in Research, Development, and Technologies. Antibiotics 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Okada, B.K.; Seyedsayamdost, M.R. Antibiotic Dialogues: Induction of Silent Biosynthetic Gene Clusters by Exogenous Small Molecules. FEMS Microbiol. Rev. 2017, 41, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Donkor, E.S. Sequencing of Bacterial Genomes: Principles and Insights into Pathogenesis and Development of Antibiotics. Genes 2013, 4, 556–572. [Google Scholar] [CrossRef] [Green Version]
- Miesel, L.; Greene, J.; Black, T.A. Genetic Strategies for Antibacterial Drug Discovery. Nat. Rev. Genet. 2003, 4, 442–456. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LpxC Inhibitors | PF-5081090 | ACHN-975 | CHIR-090 |
|---|---|---|---|
| Chemical structure |  | 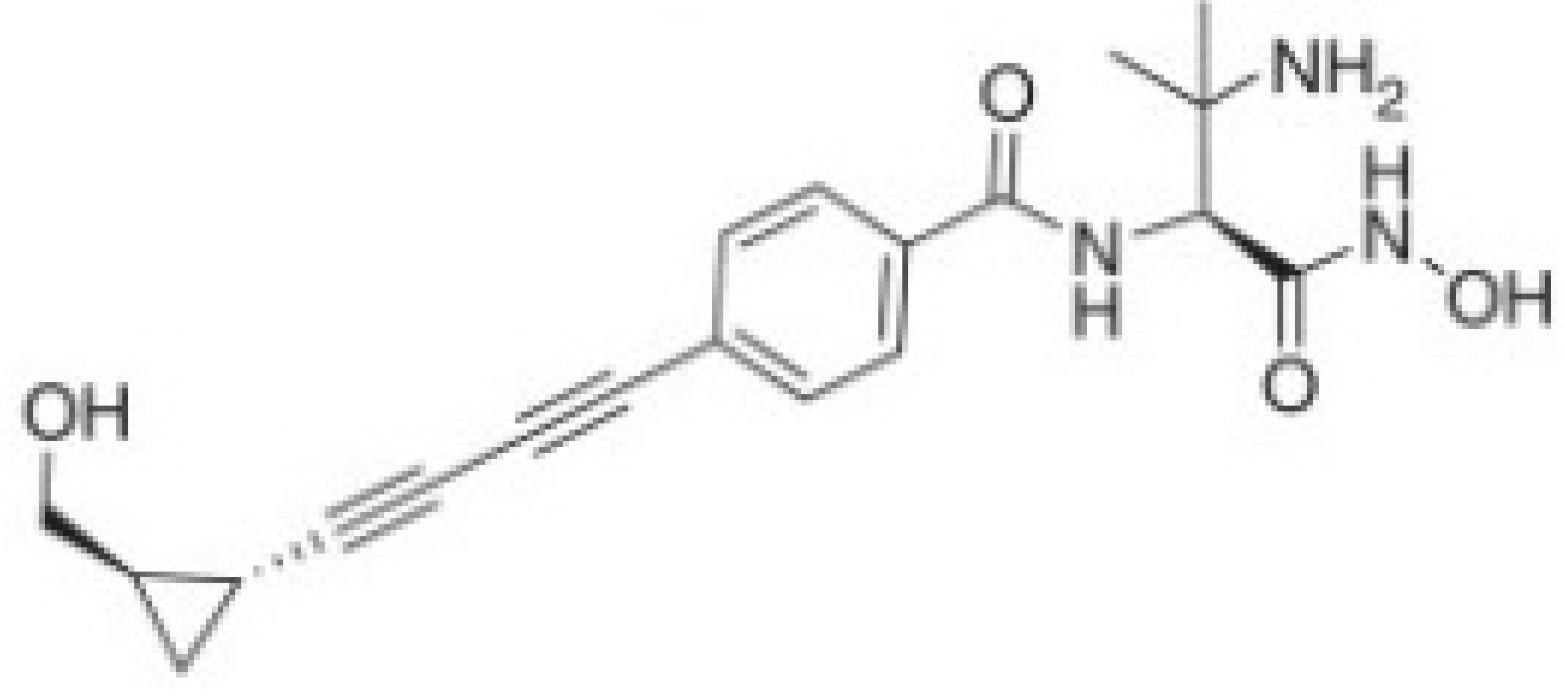 | 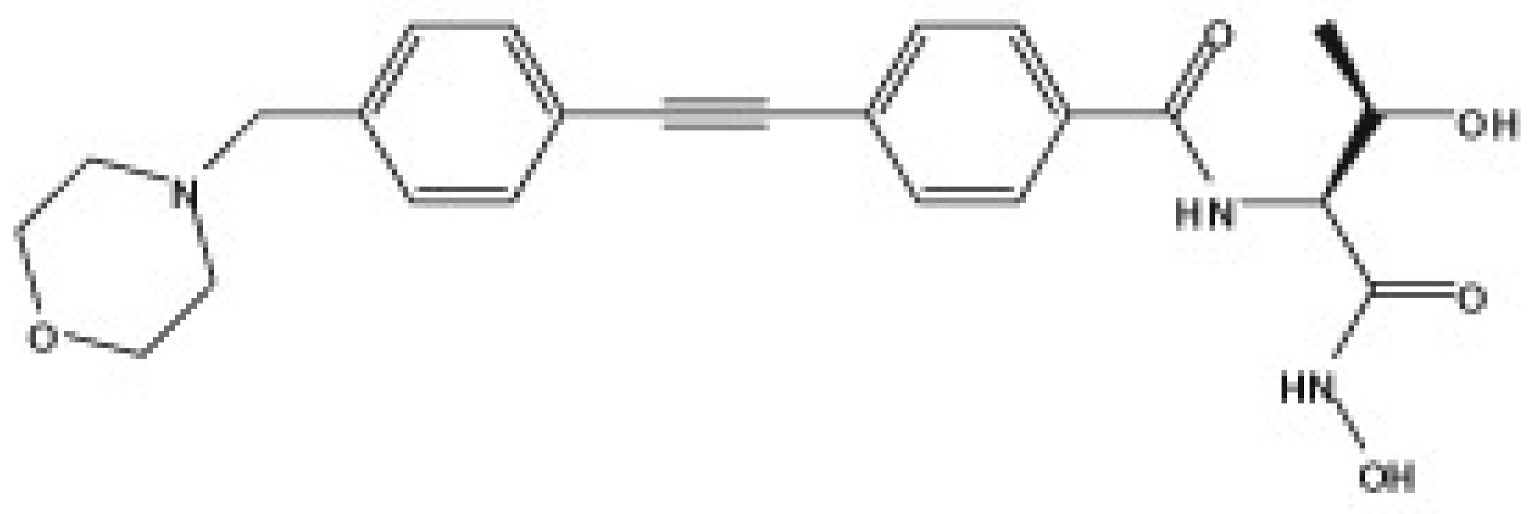 |
| IC50 (nM) | |||
| P. aeruginosa (138) | 1.1 | 0.05 | <2.1 |
| P. aeruginosa PAO1 WTe | 1.1 | 0.05 | <2.1 |
| P. aeruginosa PAO1 M62R | 2.1 | 0.5 | NT |
| K. pneumoniae (98) | 0.069 | ND | NT |
| E. coli (79) | NT | ND | NT |
| Enterobacter spp. (52)g | NT | ND | NT |
| Acinetobacter baumannii (31) | 183 | ND | NT |
| Burkholderia cepacia (30) | NT | ND | NT |
| Stenotrophomonas maltophilia (30) | NT | ND | NT |
| MIC90 (μg/mL) | |||
| P. aeruginosa (138) | 1 | ND | 4 |
| P. aeruginosa PAO1 WTe | 0.5 | 0.5 | 1 |
| P. aeruginosa PAO1 M62R | 0,5 | 0.5 | NT |
| K. pneumoniae | 1 | 2 | NT |
| E. coli | 0.25 | 0.5 | 0.25 |
| Enterobacter spp. | 0.25 | ND | 0.5 |
| Acinetobacter baumannii (31) | >64 | >64 | >64 |
| Burkholderia cepacia (30) | 0.5 | 16 | >64 |
| Stenotrophomonas maltophilia (30) | 2 | >16 | >64 |
| FabI inhibitors | Chemical structure | MIC (μg/mL) | IC50 (μM) | |
|---|---|---|---|---|
| S. aureus RN4220 | E. coli KCTC 1924 | [14C] acetate incorporation | ||
| Meleagrin |  | 64 | 32 | 64 |
| Compound 2 | 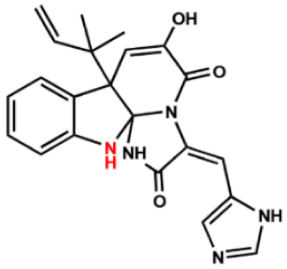 | 64 | 64 | 64 |
| Compound 3 | 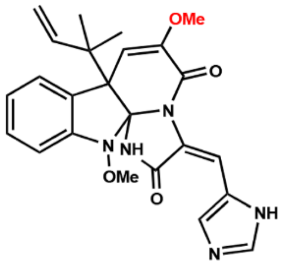 | 64 | 32 | 64 |
| Compound 4 |  | 64 | 32 | 64 |
| Compound 5 | 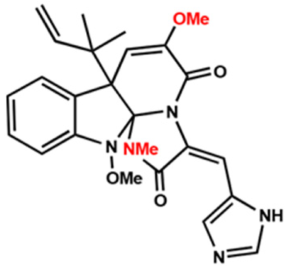 | 16 | 8 | 16 |
| Compound 6 | 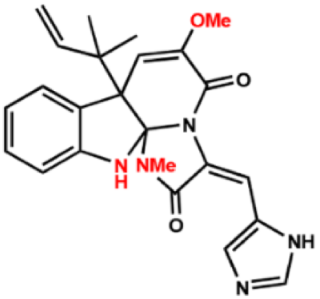 | 16 | 8 | 16 |
| Compound 7 | 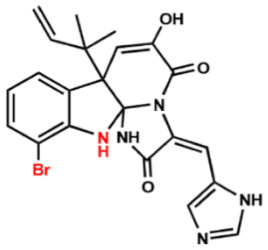 | >128 | >128 | >128 |
| Antibiotics | Pathogens | Zone of Inhibition (mm) | MIC (μg/mL) |
|---|---|---|---|
| (1) | MRSA | 21.00 a ± 0.05 | 3.00 |
| VREfs | 22.00 a ± 0.02 | 5.00 | |
| Pseudomonas aeruginosa | 17.00 b ± 0.03 | 3.17 | |
| Klebsiella pneumonia | 19.00 b ± 0.03 | 5.00 | |
| (2) | MRSA | 22.00 a ± 0.01 | 3.12 |
| VREfs | 25.00 b ± 0.04 | 1.50 | |
| Pseudomonas aeruginosa | 23.00 a ± 0.02 | 3.00 | |
| Klebsiella pneumonia | 26.00 b ± 0.02 | 3.00 | |
| (3) | MRSA | 30.00 a ± 0.01 | 1.50 |
| VREfs | 28.00 a ± 0.04 | 3.00 | |
| Pseudomonas aeruginosa | 25.00 b ± 0.02 | 1.50 | |
| Klebsiella pneumonia | 31.00 a ± 0.02 | 1.50 | |
| Ampicillin | MRSA | 10.00 a ± 0.02 | 12.50 |
| VREfs | 13.00 b ± 0.04 | 25.00 | |
| Pseudomonas aeruginosa | 7.00 c ± 0.01 | 12.50 | |
| Klebsiella pneumonia | 7.00 c ± 0.02 | 12.50 | |
| Chloramphenicol | MRSA | 14.00 a ± 0.02 | 6.25 |
| VREfs | 13.00 b ± 0.01 | 6.25 | |
| Pseudomonas aeruginosa | 11.00 a ± 0.02 | 16.00 | |
| Klebsiella pneumonia | 7.00 c ± 0.04 | 12.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkatheri, A.H.; Yap, P.S.-X.; Abushelaibi, A.; Lai, K.-S.; Cheng, W.-H.; Erin Lim, S.-H. Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics 2023, 12, 190. https://doi.org/10.3390/antibiotics12020190
Alkatheri AH, Yap PS-X, Abushelaibi A, Lai K-S, Cheng W-H, Erin Lim S-H. Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics. 2023; 12(2):190. https://doi.org/10.3390/antibiotics12020190
Chicago/Turabian StyleAlkatheri, Asma Hussain, Polly Soo-Xi Yap, Aisha Abushelaibi, Kok-Song Lai, Wan-Hee Cheng, and Swee-Hua Erin Lim. 2023. "Microbial Genomics: Innovative Targets and Mechanisms" Antibiotics 12, no. 2: 190. https://doi.org/10.3390/antibiotics12020190
APA StyleAlkatheri, A. H., Yap, P. S.-X., Abushelaibi, A., Lai, K.-S., Cheng, W.-H., & Erin Lim, S.-H. (2023). Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics, 12(2), 190. https://doi.org/10.3390/antibiotics12020190