
Fragment-Based Lead Discovery Strategies in Antimicrobial Drug Discovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Metalloenzymes
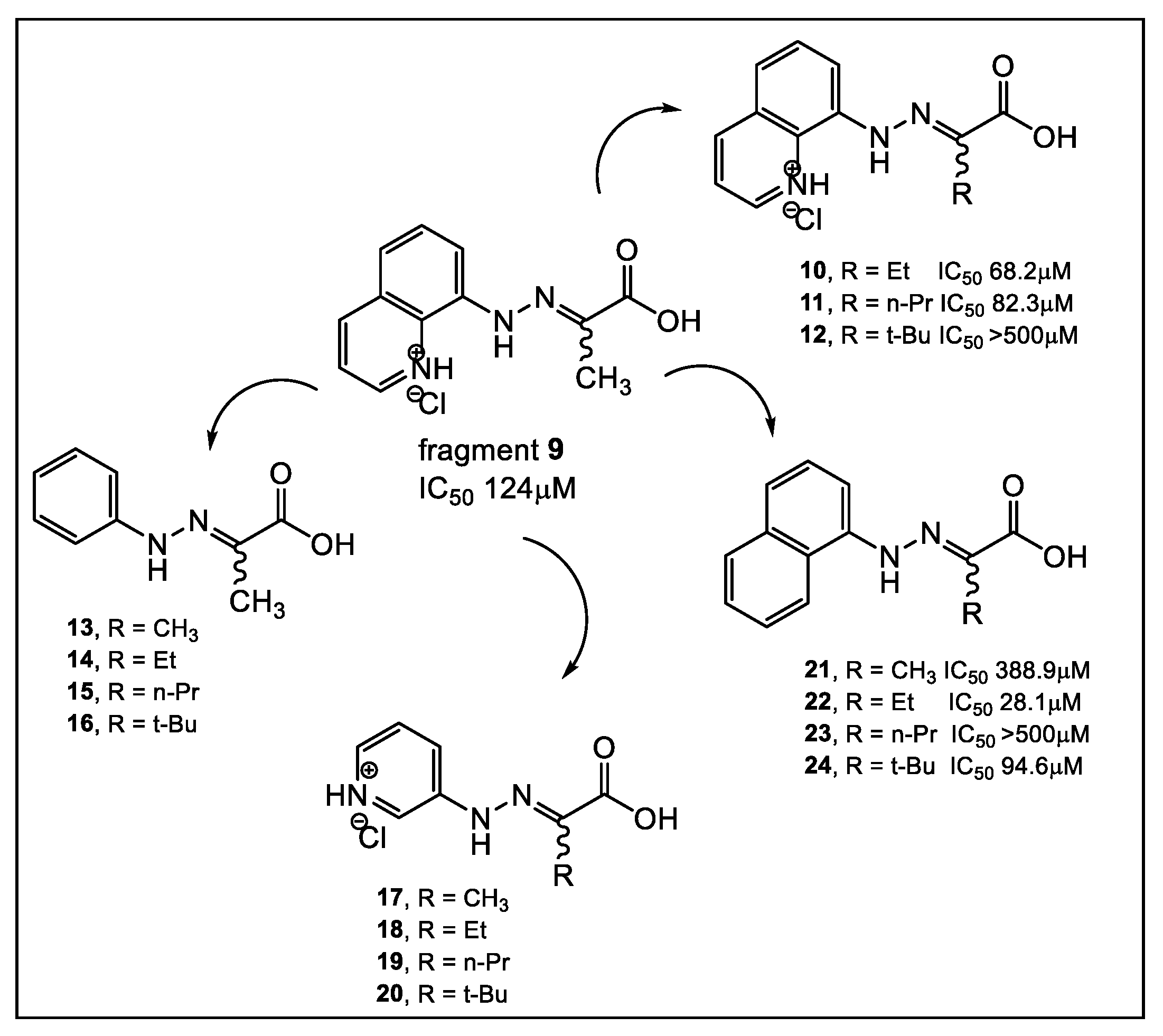
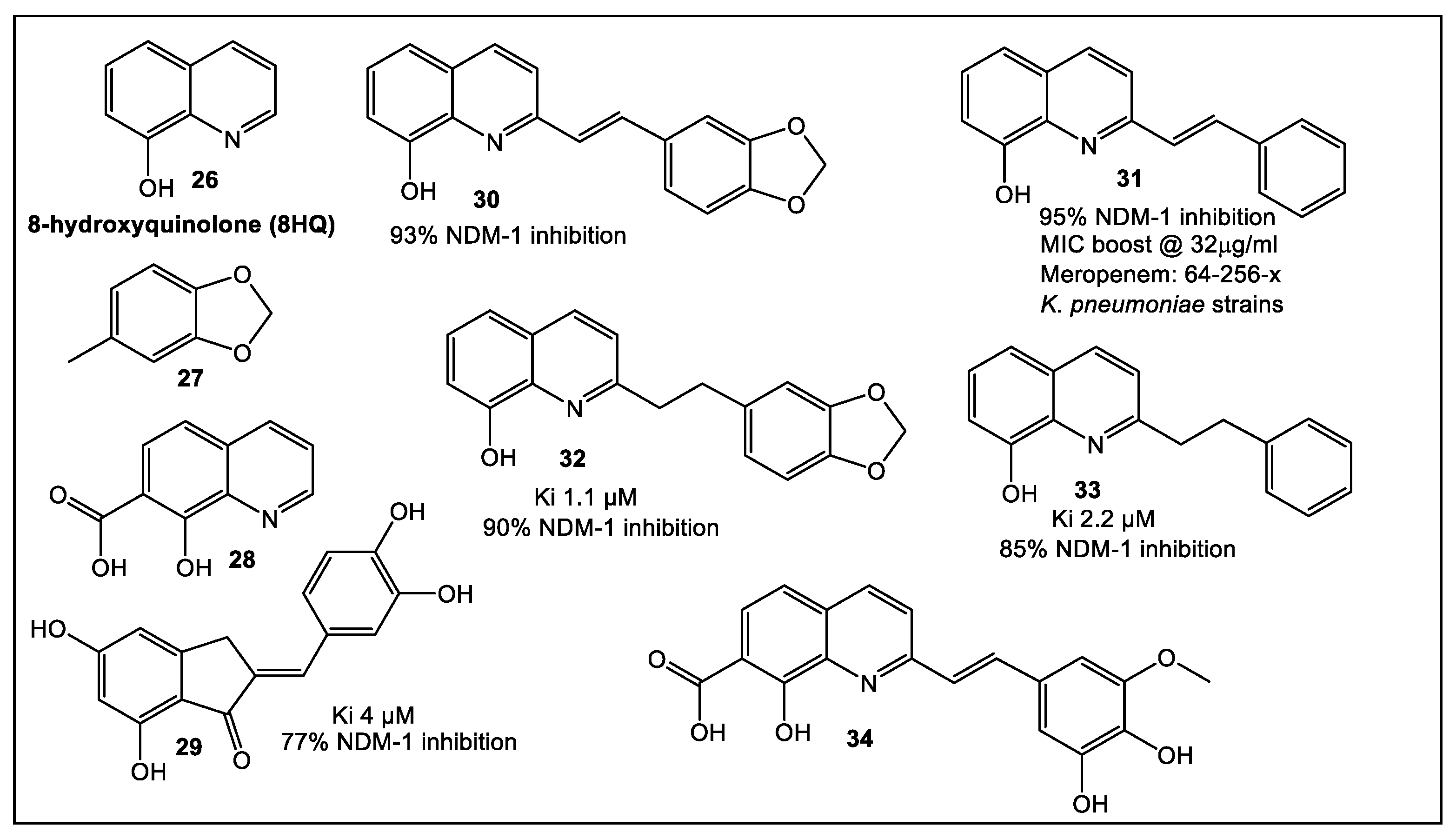
2.1. Lactamases—Focus on NDM-1
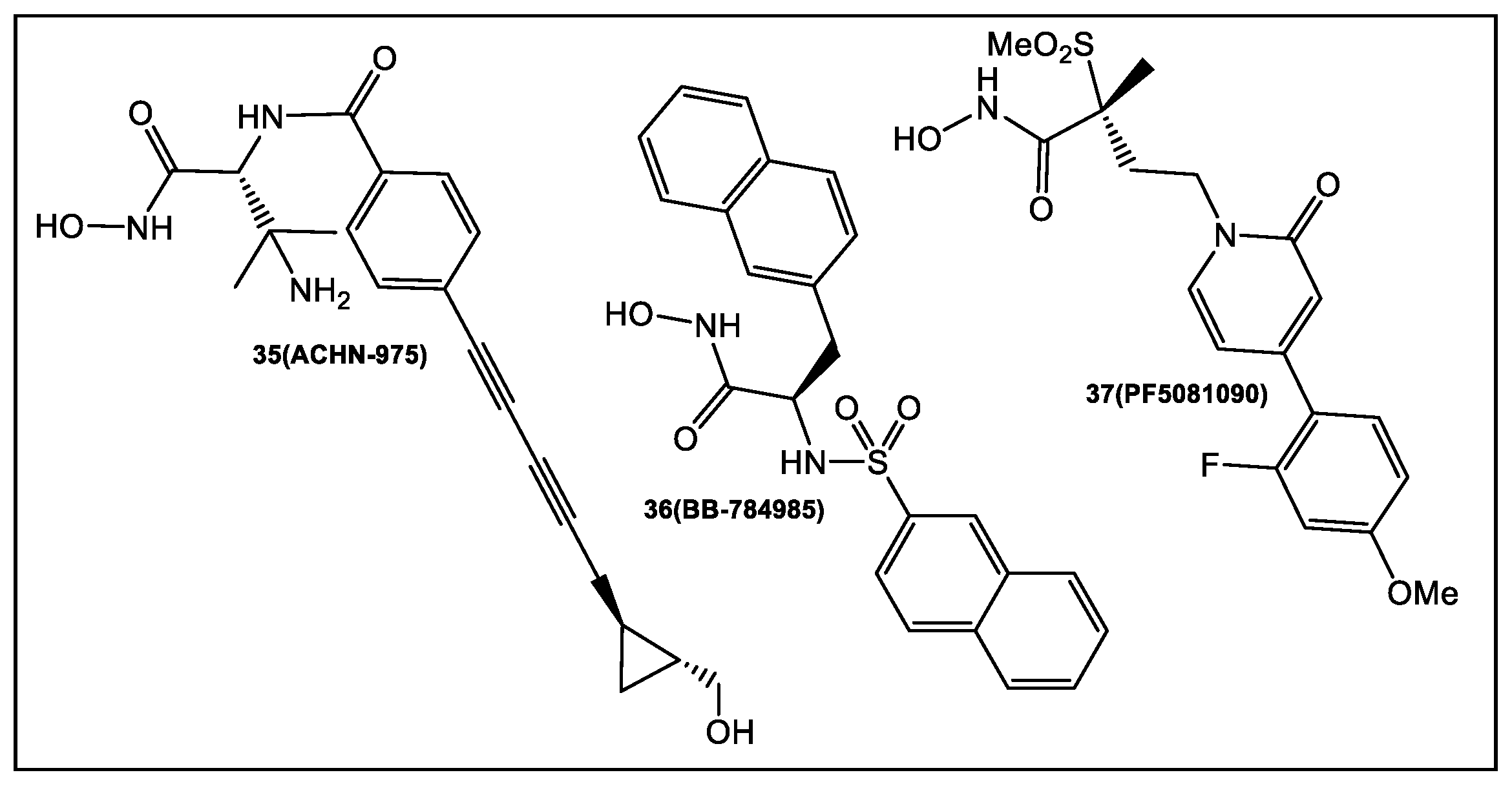
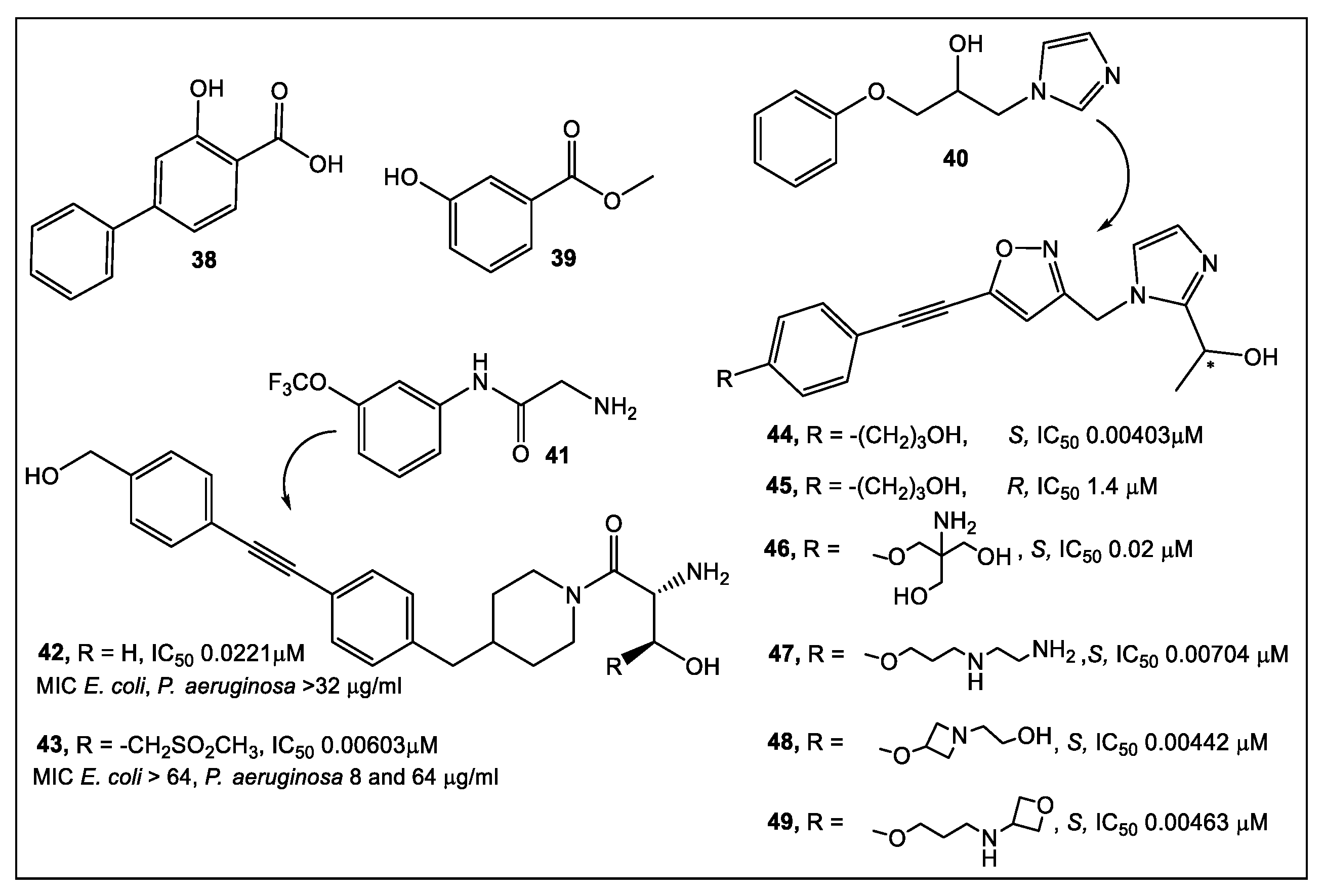
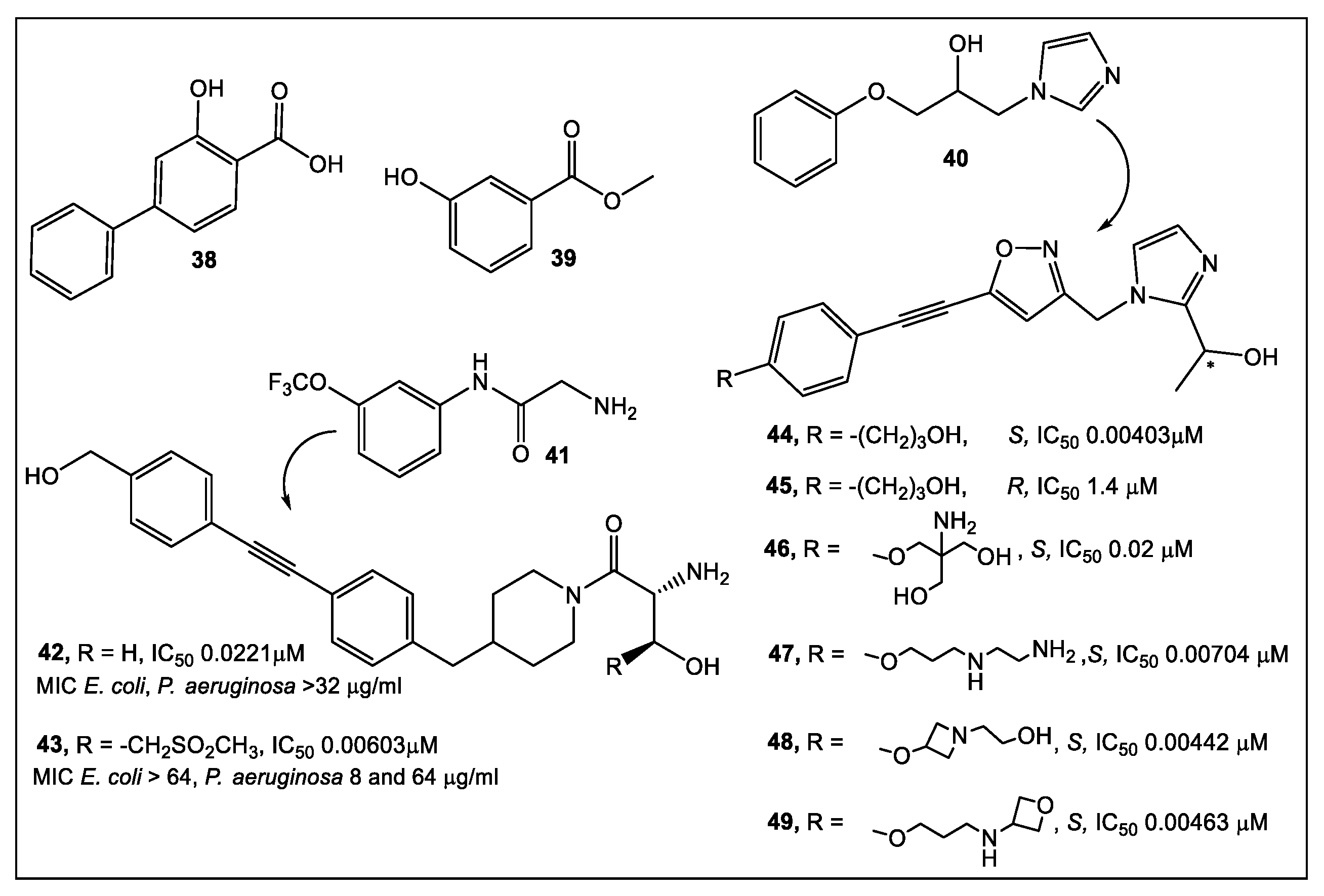
2.2. UDP-3-O-acyl-N-acetylglucosamine Deacetylase (LpxC)
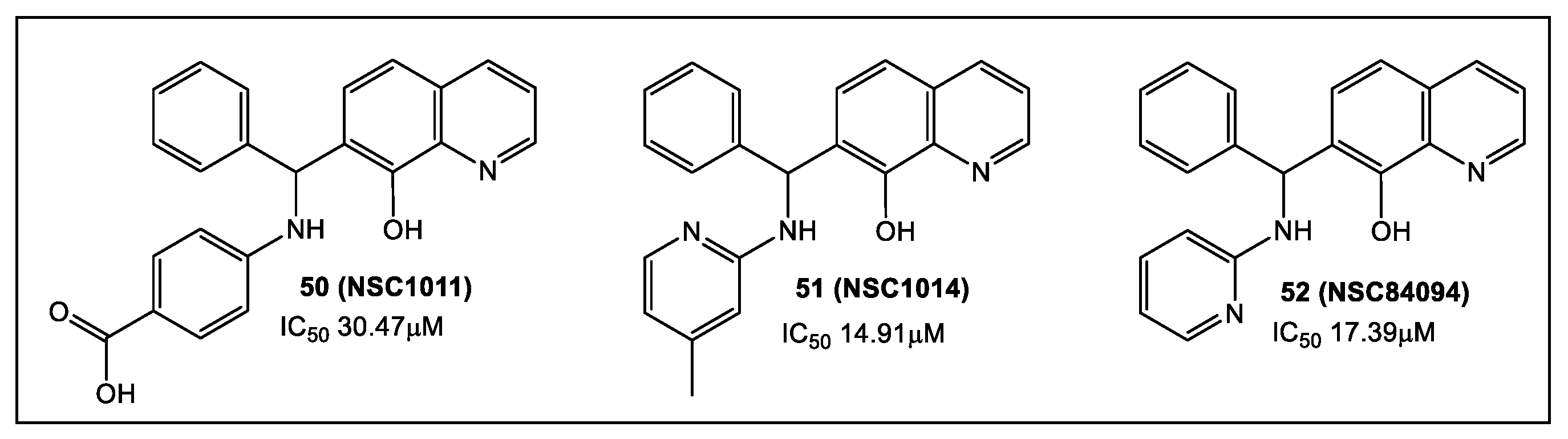
2.3. Botulinum Neurotoxins’ Metalloprotease
3. Cell Wall Enzymes
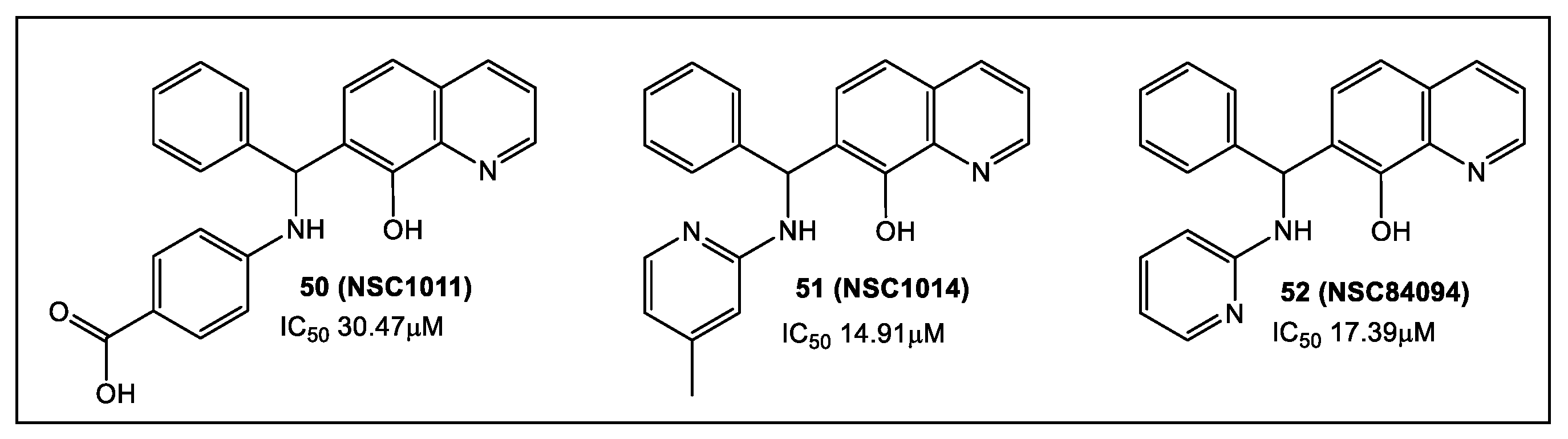
3.1. Glycosidases: N-Acetylglucosaminidases
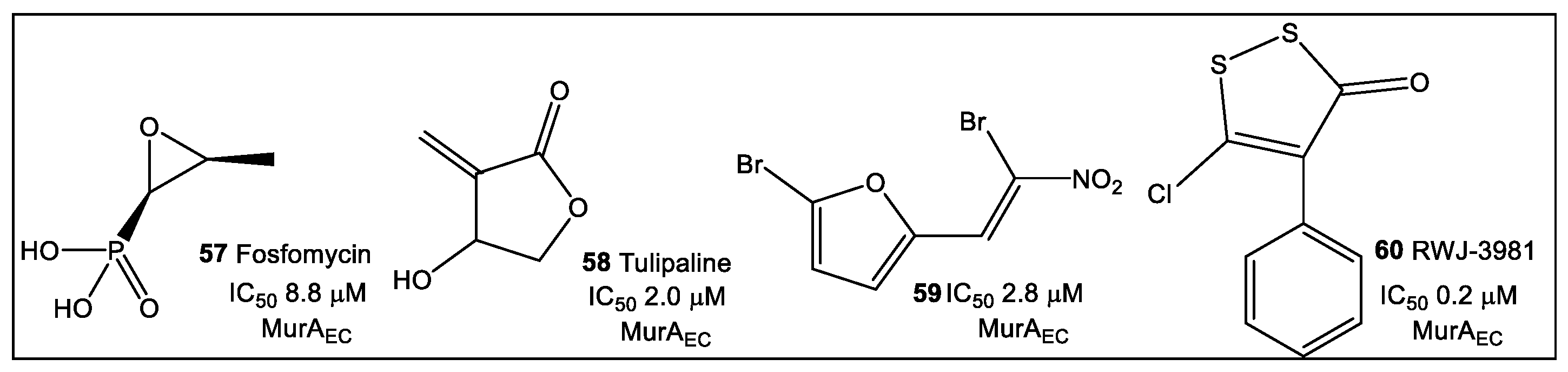
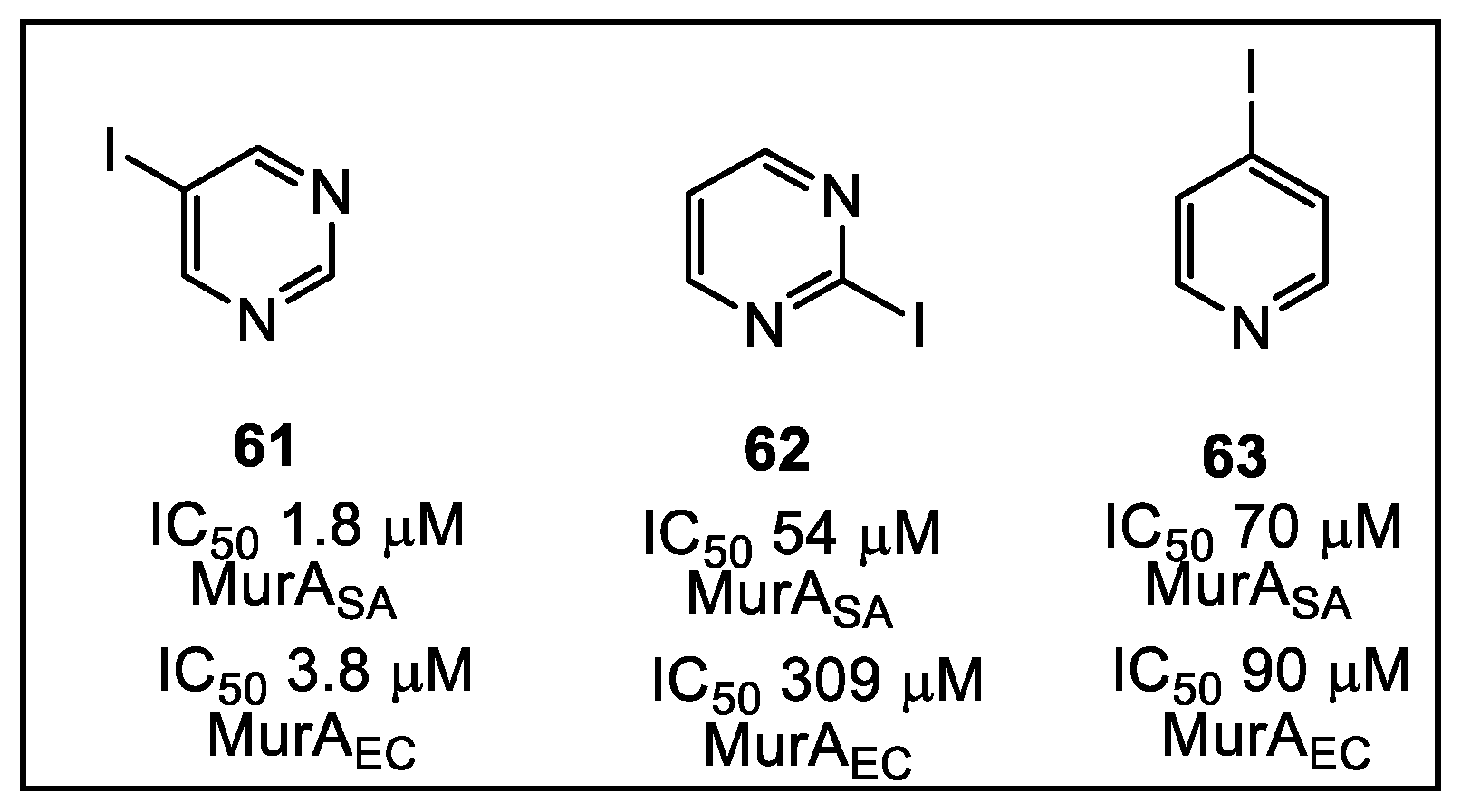
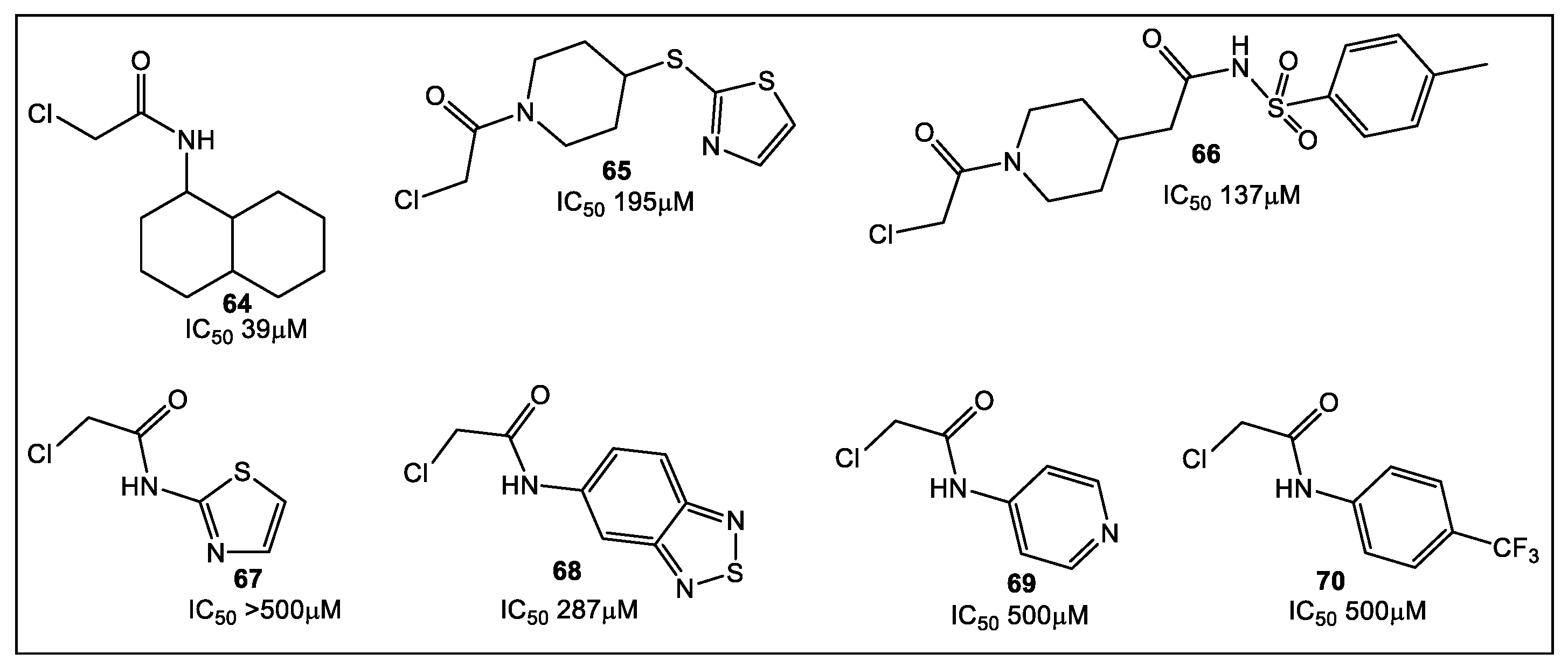
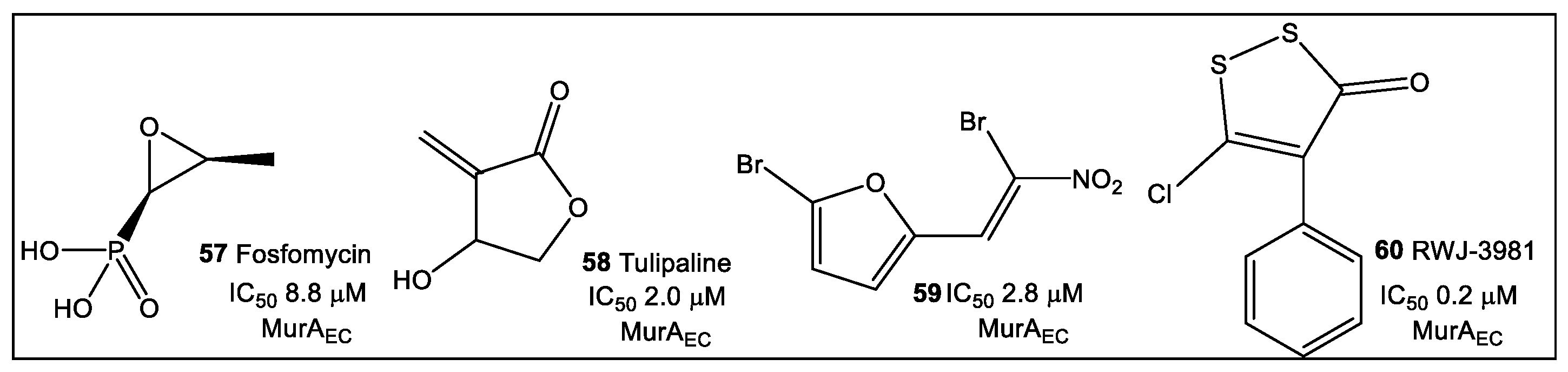
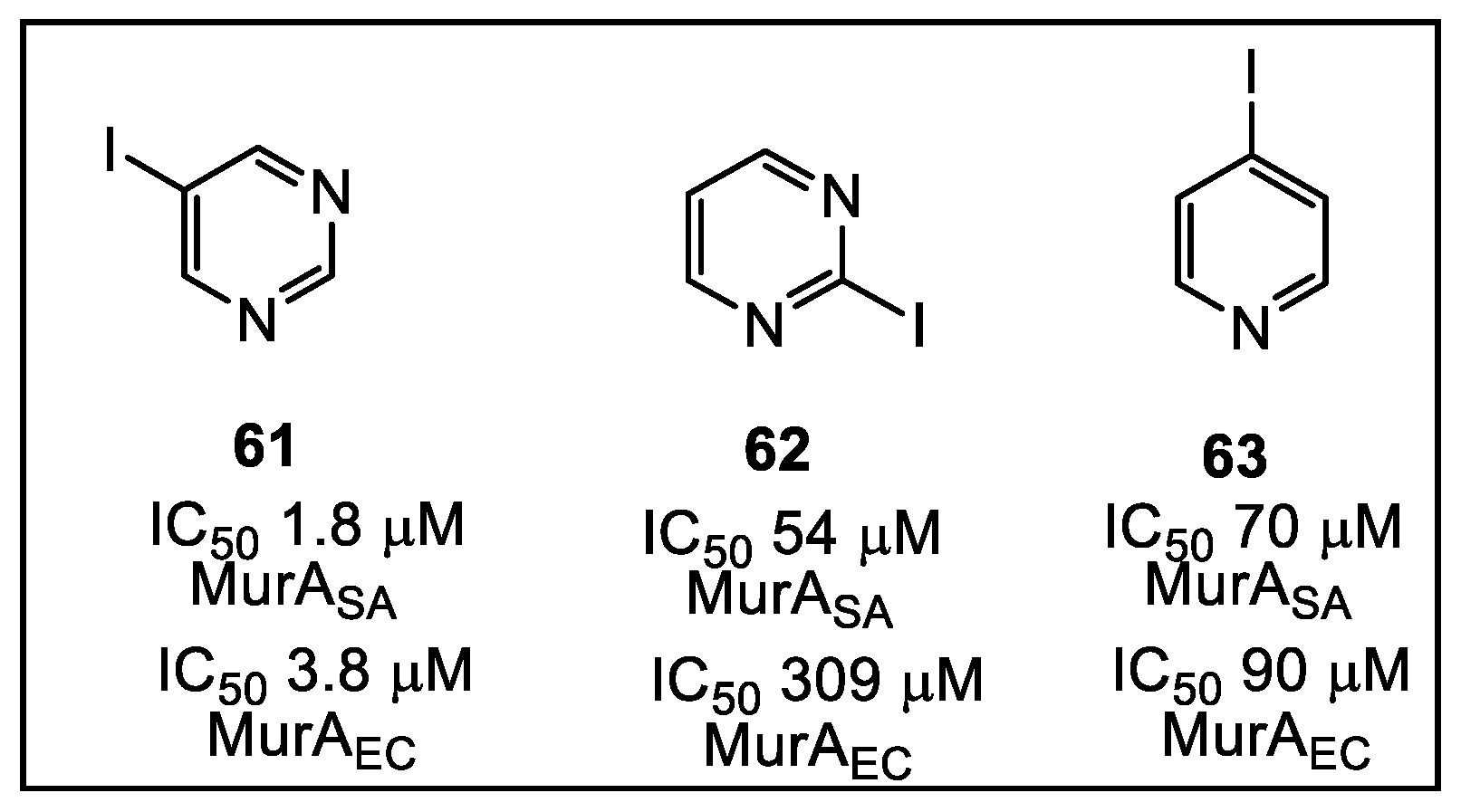
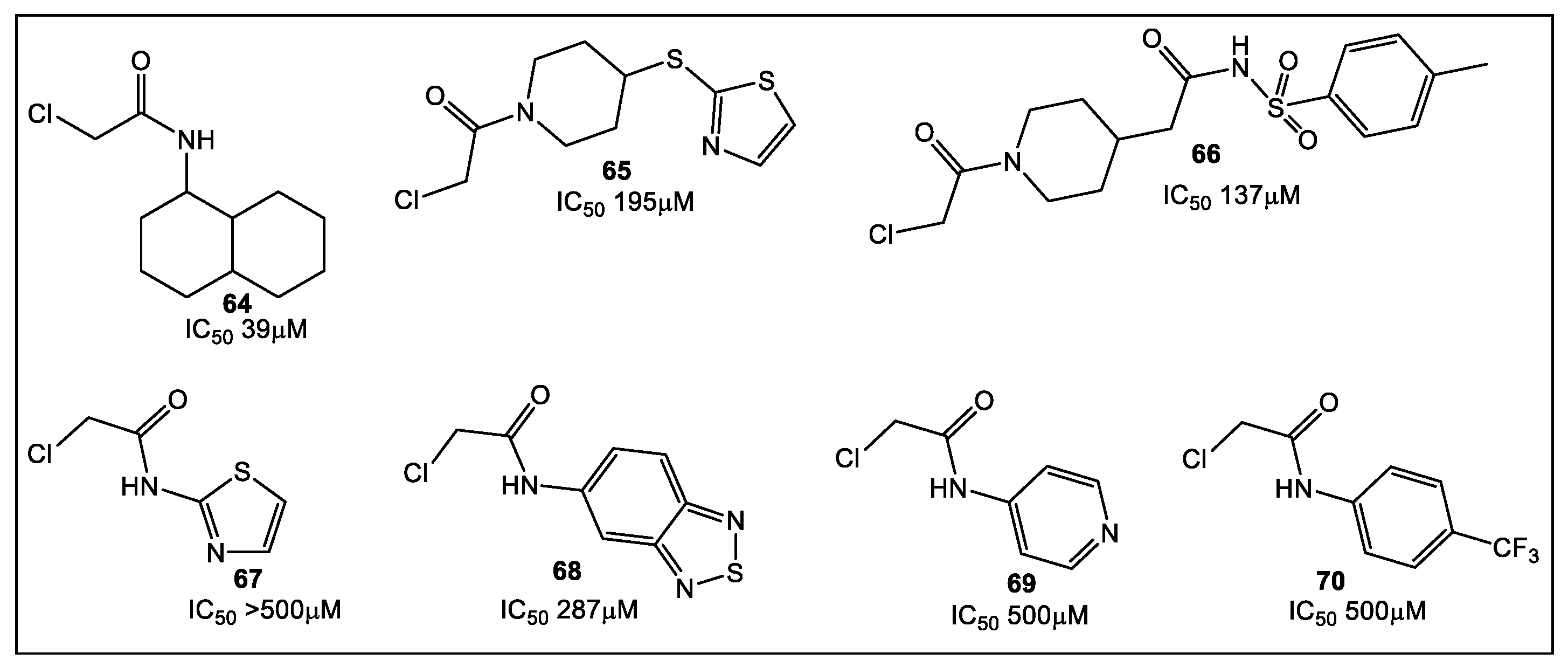
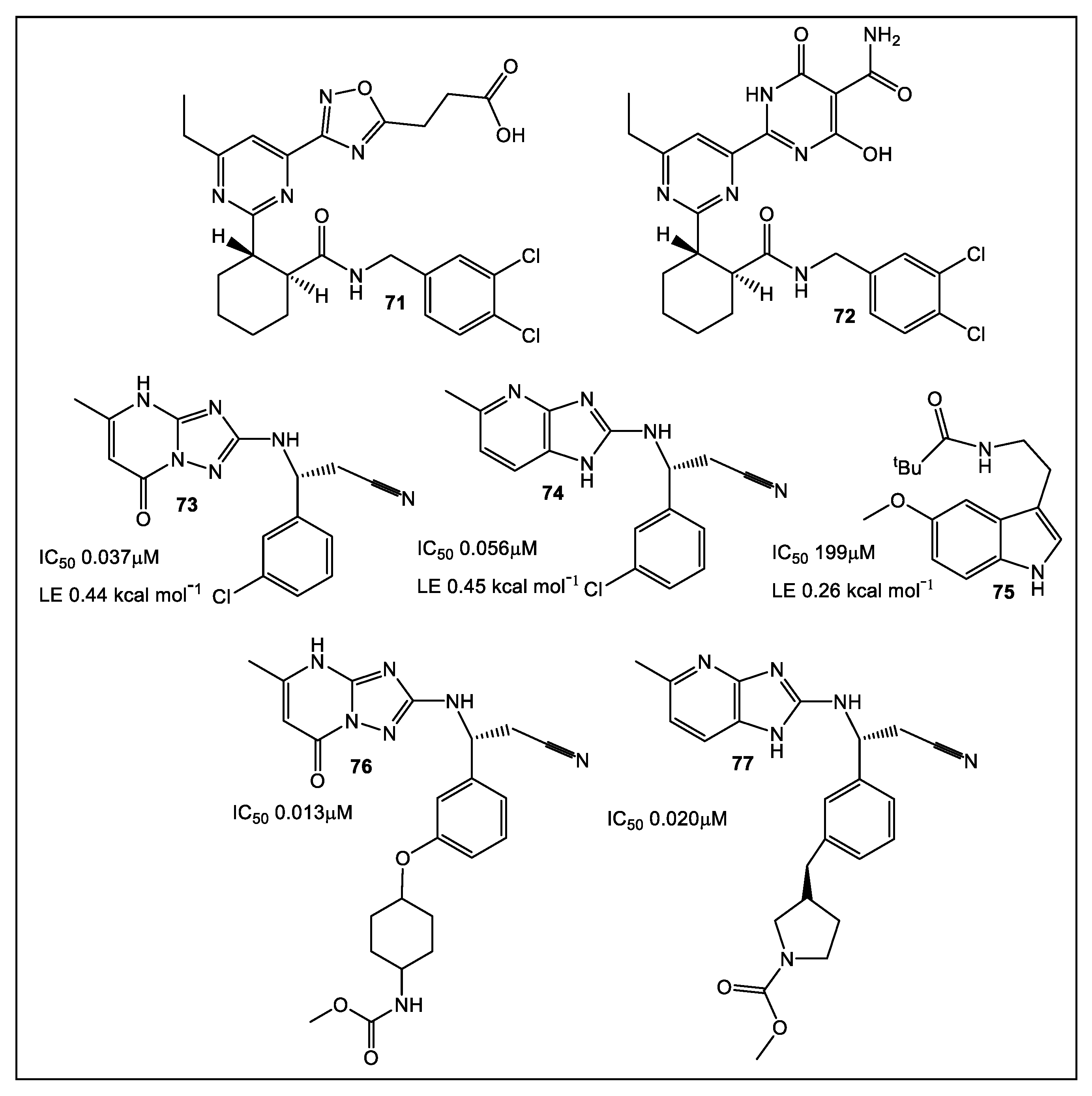
3.2. UDP-N-Acetylglucosamine Enolpyruvyl Transferase (MurA)
3.3. Phosphopantetheine Adenylyltransferase (PPAT, Also Known as CoaD)
4. Cell Wall Components: Lectins
5. Bacterial Virulence Factors: DsbA Enzymes of E. coli and V. cholerae
5.1. EcDsbA
5.2. VcDsbA
6. Quorum Sensing—QS
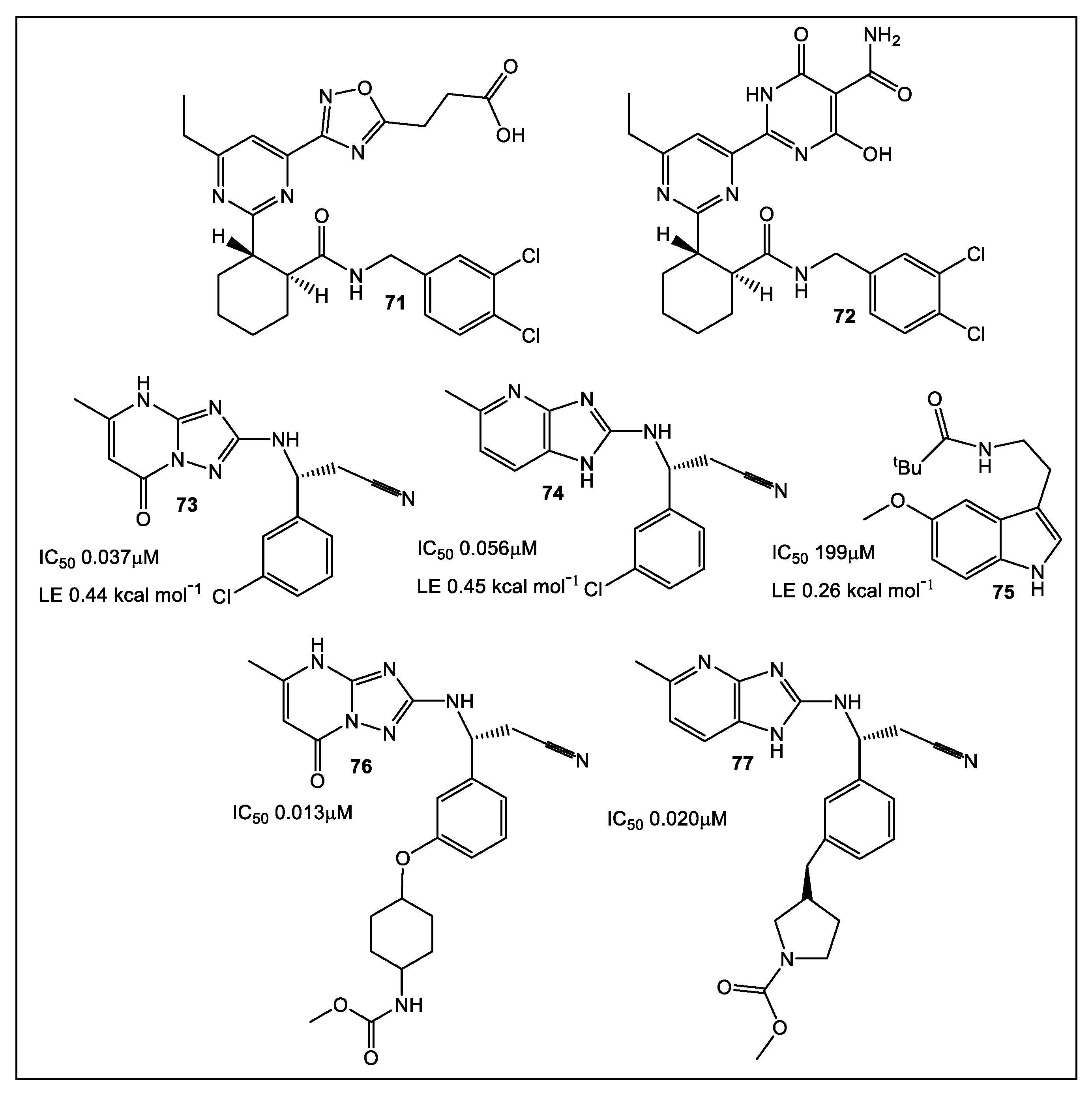
7. Bacterial Enzymes Catalyzing Purine Synthesis
7.1. Mycobacterium Abscessus Phosphoribosylaminoimidazole Succinocarboxamide Synthetase (PurC or SAICAR Synthetase)
7.2. tRNA (m1 G37) Methyltransferase (TrmD) from Mycobacterium Abscessus (MabTrmD)
7.3. Mycobacterium Thermoresistible (MthIMPDH) Inosine-5′-Monophosphate Dehydrogenase (IMPDH)
7.4. Threonyl-tRNA Synthetase from Salmonella Enterica
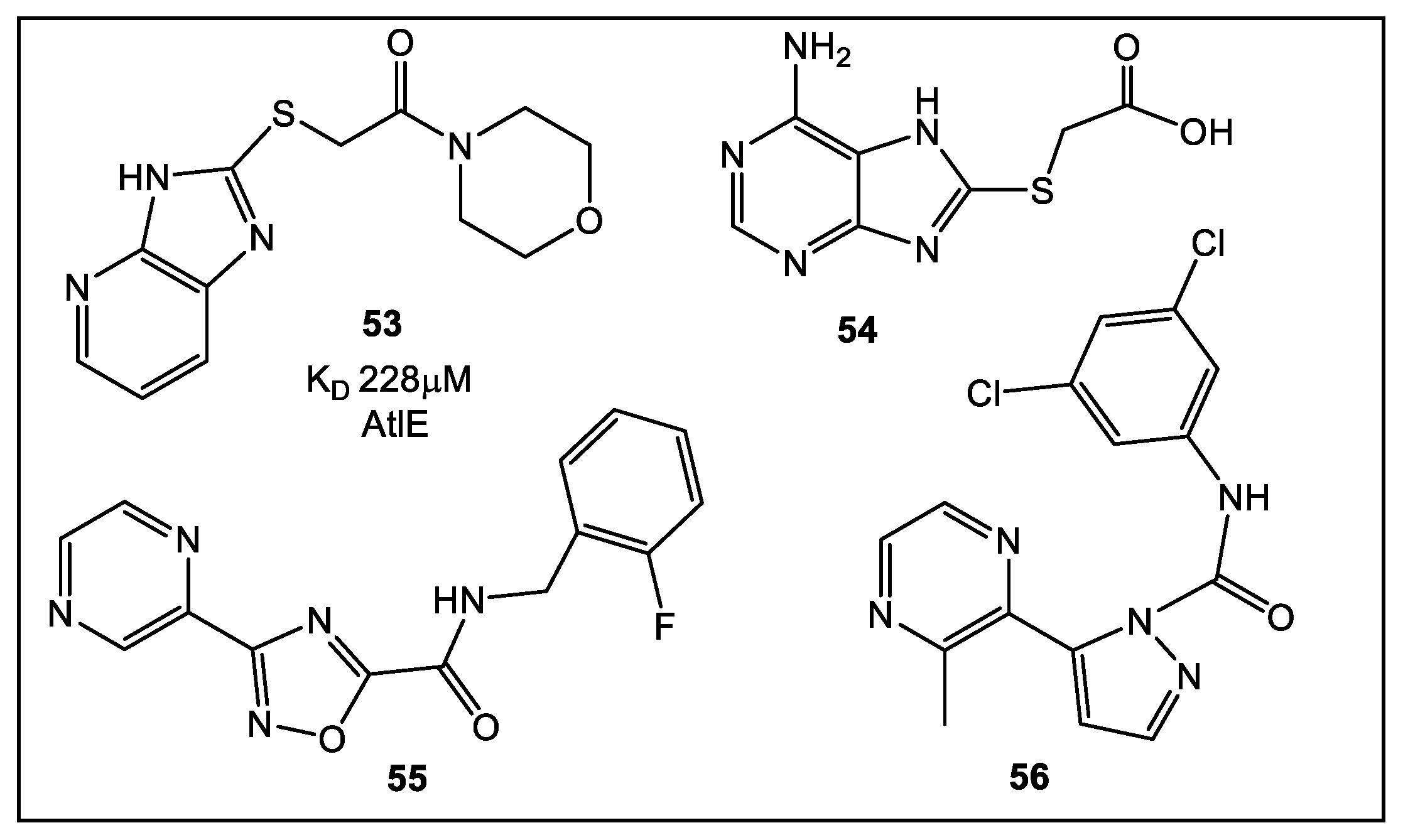
8. Primase/SSB-Ct Interaction
9. Fragment-Based Lead Discovery (FPLD); Cell-Based Screens for the Identification of Microbial Inhibitors of Leishmania, Plasmodium falciparum, Neisseria, Mycobacterium, and Flaviviruses
10. Conclusions
11. Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tenover, F.C. Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.; Kolter, R. Why are bacteria refractory to antimicrobials? Curr. Opin. Microbiol. 2002, 5, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Sefton, A.M. Mechanisms of Antimicrobial Resistance. Drugs 2002, 62, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.S.; Blomberg, N.; Breeze, A.L.; Brown, A.J.H.; Burrows, J.N.; Edwards, P.D.; Folmer, R.H.A.; Geschwindner, S.; Griffen, E.J.; Kenny, P.W.; et al. An Integrated Approach to Fragment-Based Lead Generation: Philosophy, Strategy and Case Studies from AstraZeneca’s Drug Discovery Programmes. Curr. Topics Med. Chem. 2007, 7, 1600–1629. [Google Scholar] [CrossRef]
- Jencks, W.P. On the Attribution and Additivity of Binding Energies. Proc. Natl Acad. Sci. USA 1981, 78, 4046–4050. [Google Scholar] [CrossRef]
- Bohm, H.J. The Computer Program LUDI: A New Method for the De Novo Design of Enzyme Inhibitors. J. Comp. Aided Mol. Design 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-Dimensional Relaxation- and Diffusion-Edited NMR Methods for Screening Compounds That Bind to Macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef]
- Canning, P.; Birchall, K.; Kettleborough, C.A.; Merritt, A.; Coombs, P.J. Fragment-based target screening as an empirical approach to prioritising targets: A case study on antibacterials. Drug Discov. Today 2020, 25, 2030–2037. [Google Scholar] [CrossRef]
- Lu, W.; Kostic, M.; Zhang, T.; Che, J.; Patricelli, M.P.; Jones, L.H.; Chouchani, E.T.; Gray, N.S. Fragment-based covalent ligand discovery. RSC Chem. Biol. 2021, 2, 354–367. [Google Scholar] [CrossRef]
- Denis, J.D.S.; Hall, R.J.; Murray, C.W.; Heightman, T.D.; Rees, D.C. Fragment-based drug discovery: Opportunities for organic synthesis. RSC Med. Chem. 2021, 12, 321–329. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Using Fragment-Based Approaches to Discover New Antibiotics. SLAS Discov. 2018, 23, 495–510. [Google Scholar] [CrossRef]
- Toleman, M.A.; Spencer, J.; Jones, L.; Walsh, T.R. BlaNDM-1 Is a Chimera, Likely Constructed in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 2773–2776. [Google Scholar] [CrossRef]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten years with New Delhi Metallo-β-lactamase-1 (NDM-1): From structural insights to inhibitor design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive rules for compound accumulation yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef]
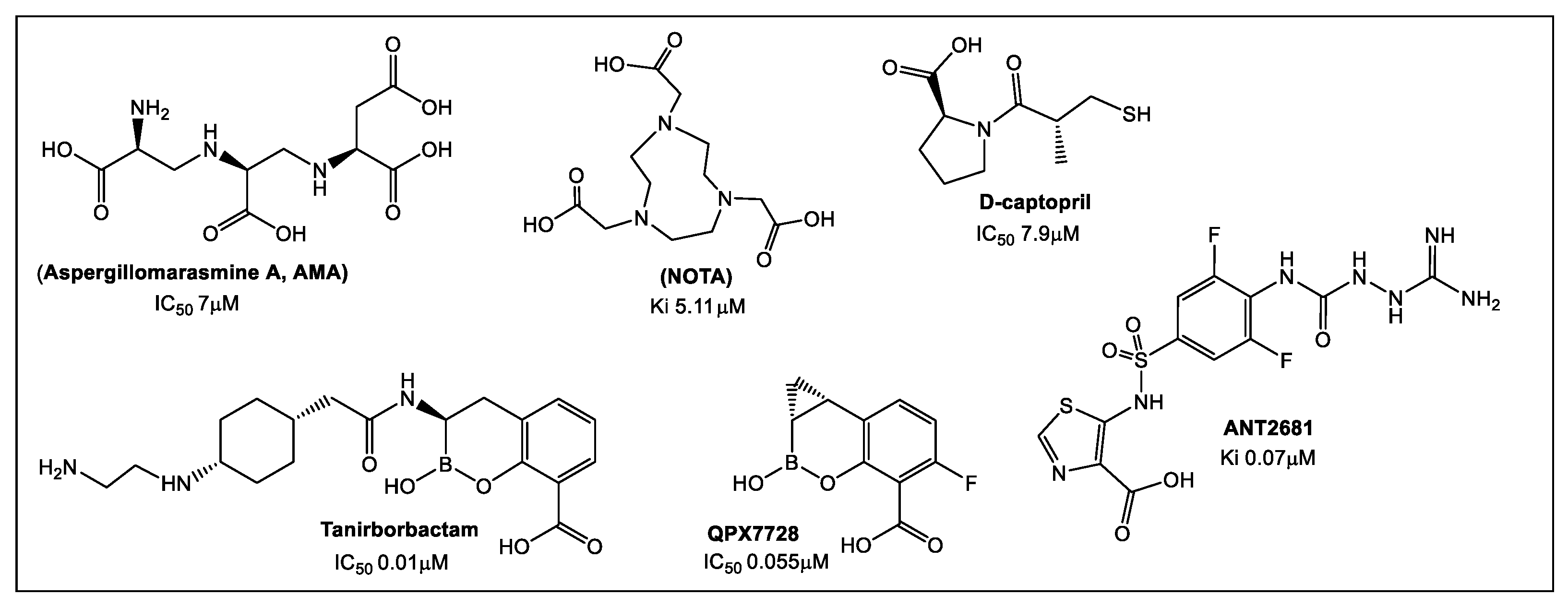
- Davies, D.T.; Leiris, S.; Sprynski, N.; Castandet, J.; Lozano, C.; Bousquet, J.; Zalacain, M.; Vasa, S.; Dasari, P.K.; Pattipati, R.; et al. ANT2681: SAR Studies Leading to the Identification of a Metallo-β-lactamase Inhibitor with Potential for Clinical Use in Combination with Meropenem for the Treatment of Infections Caused by NDM-Producing Enterobacteriaceae. ACS Infect. Dis. 2020, 6, 2419–2430. [Google Scholar] [CrossRef]
- Liu, B.; Trout, R.E.L.; Chu, G.-H.; McGarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; de Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2020, 63, 2789–2801. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-β-lactamases. J. Med. Chem. 2020, 63, 7491–7507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsivkovski, R.; Totrov, M.; Lomovskaya, O. Biochemical characterization of QPX7728, a new ultrabroad-spectrum beta-lactamase inhibitor of serine and metallo-betalactamases. Antimicrob. Agents Chemother. 2020, 64, e00130-20. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Tsivkovski, R.; Sun, D.; Reddy, R.; Totrov, M.; Hecker, S.; Griffith, D.; Loutit, J.; Dudley, M. QPX7728, An Ultra-Broad-Spectrum B-Lactamase Inhibitor for Intravenous and Oral Therapy: Overview of Biochemical and Microbiological Characteristics. Front. Microbiol. 2021, 12, 697180. [Google Scholar] [CrossRef] [PubMed]
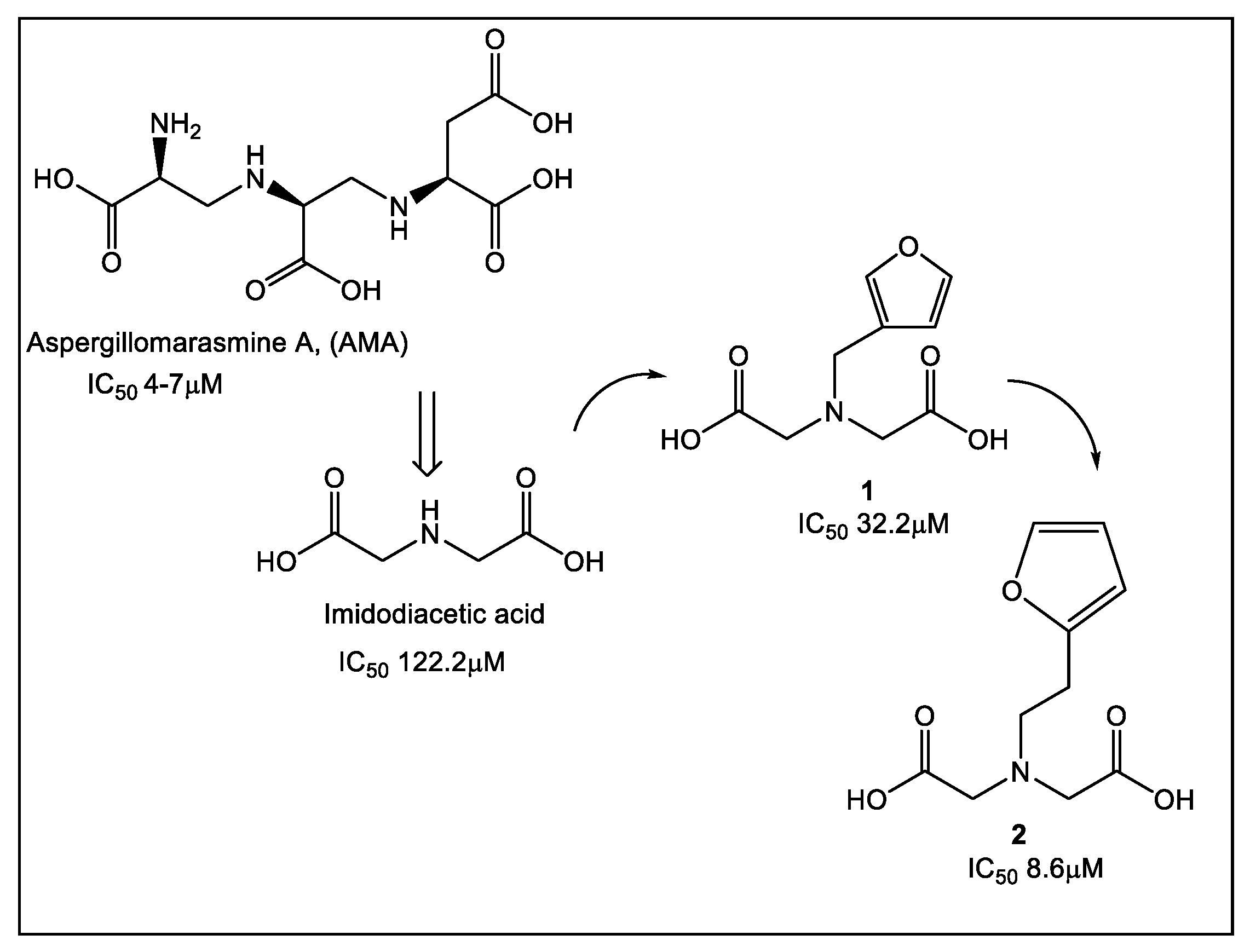
- Chen, A.Y.; Thomas, C.; Thomas, P.W.; Yangb, K.; Cheng, Z.; Fast, W.; Crowder, M.W.; Cohen, S.M. Iminodiacetic Acid as a Novel Metal-binding Pharmacophore for New Delhi Metallo-β-lactamase Inhibitor Development. ChemMedChem 2020, 15, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; de Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef]
- Bergstrom, A.; Katko, A.; Adkins, Z.; Hill, J.; Cheng, Z.; Burnett, M.; Yang, H.; Aitha, M.; Mehaffey, M.R.; Brodbelt, J.S.; et al. Probing the Interaction of Aspergillomarasmine A with Metallo-β-lactamases NDM-1, VIM-2, and IMP-7. ACS Infect. Dis. 2018, 4, 135–145. [Google Scholar] [CrossRef]
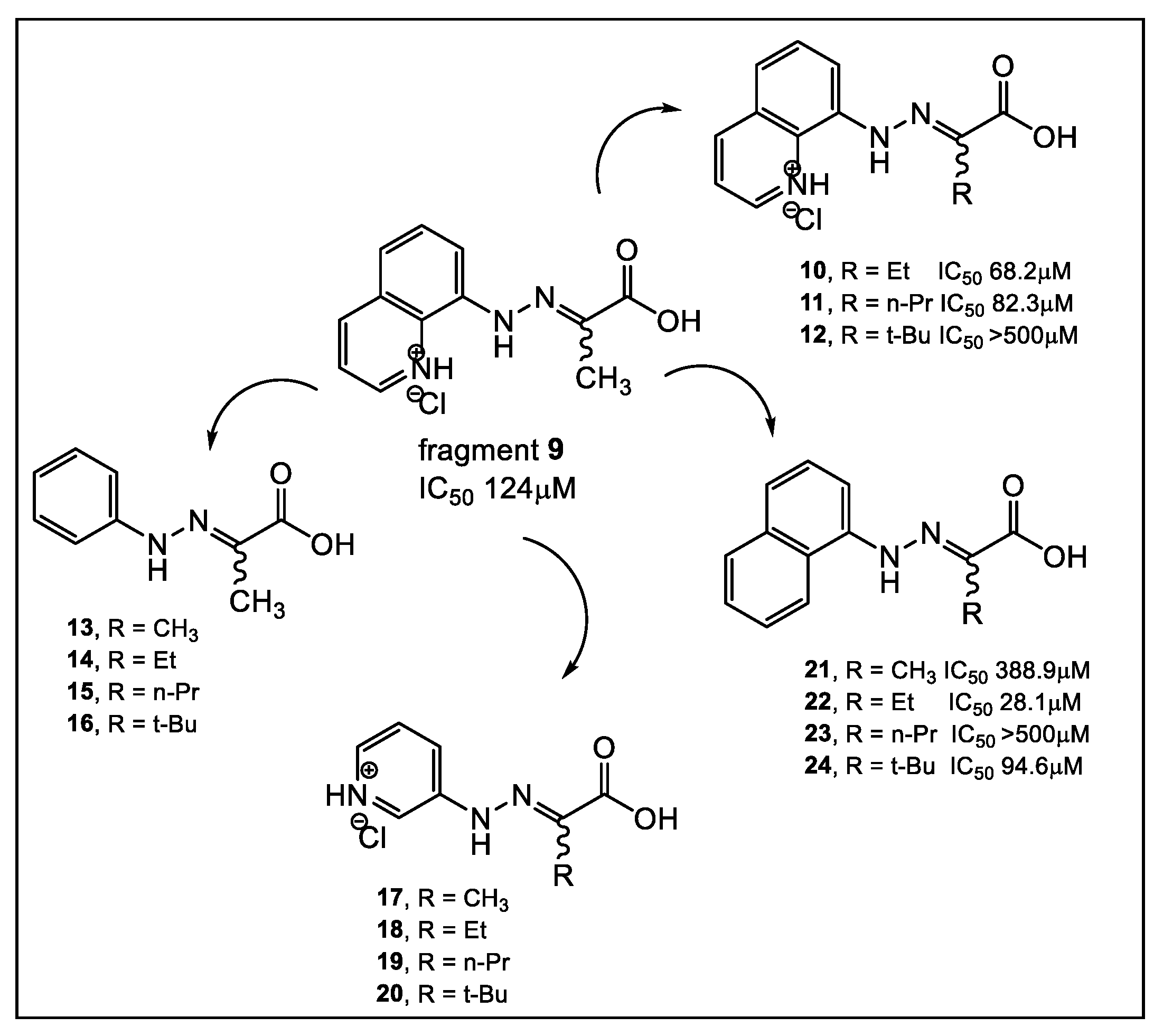
- Cain, R.; Brem, J.; Zollman, D.; McDonough, M.A.; Johnson, R.M.; Spencer, J.; Makena, A.; Abboud, M.I.; Cahill, S.; Lee, S.Y.; et al. In Silico Fragment-Based Design Identifies Subfamily B1 Metallo-β-lactamase Inhibitors. J. Med. Chem. 2018, 61, 1255–1260. [Google Scholar] [CrossRef]
- Zhang, H.; Hao, Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011, 25, 2574–2582. [Google Scholar] [CrossRef]
- Gillet, V.J.; Newell, W.; Mata, P.; Myatt, G.; Sike, S.; Zsoldos, Z.; Johnson, A.P. SPROUT: Recent developments in the de novo design of molecules. J. Chem. Inf. Model. 1994, 34, 207–217. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jimenez-Castellanos, J.C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef]
- Guo, H.; Cheng, K.; Gao, Y.; Bai, W.; Wu, C.; He, W.; Li, C.; Li, Z. A novel potent metal-binding NDM-1 inhibitor was identified by fragment virtual, SPR and NMR screening. Bioorg. Med. Chem. 2020, 28, 115437–115445. [Google Scholar] [CrossRef]
- Caburet, J.; Boucherle, B.; Bourdillon, S.; Simoncelli, G.; Verdirosa, F.; Docquier, J.-D.; Moreau, Y.; Krimm, I.; Crouzy, S.; Peuchmaur, M. A fragment-based drug discovery strategy applied to the identification of NDM-1 β-lactamase inhibitors. Eur. J. Med. Chem. 2022, 240, 114559–114614. [Google Scholar] [CrossRef]
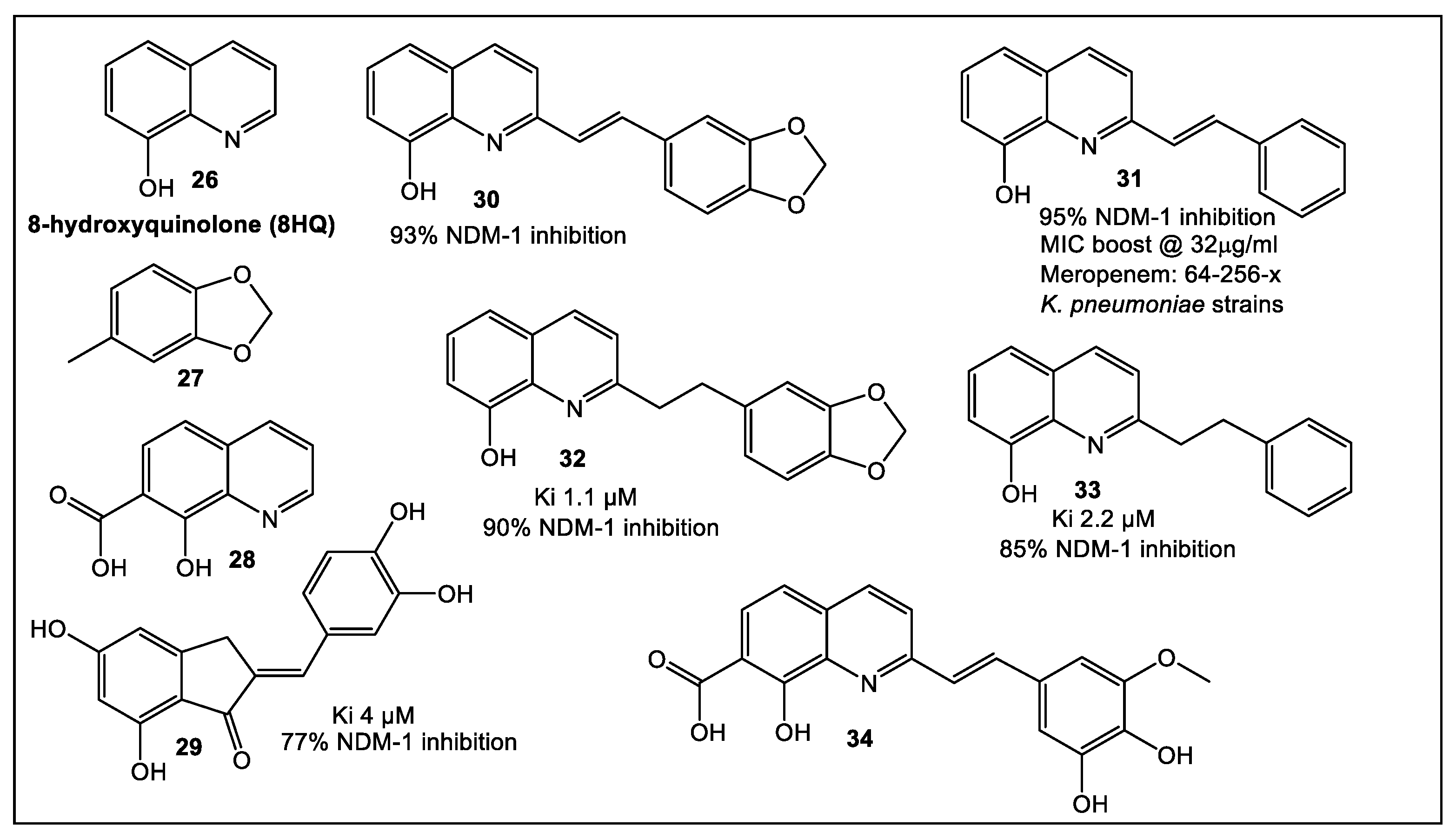
- Shin, W.S.; Nguyen, M.E.; Bergstrom, A.; Isabella, R.; Jennings, I.R.; Crowder, M.W.; Muthyala, R.; Sham, Y.Y. Fragment-based screening and hit-based substructure search: Rapid discovery of 8-hydroxyquinoline-7-carboxylic acid as a low-cytotoxic, nanomolar metallo β-lactamase inhibitor. Chem. Biol. Drug Des. 2021, 98, 481–492. [Google Scholar] [CrossRef]
- Zouhiri, F.; Mouscadet, J.-F.; Mekouar, K.; Desmaële, D.; Savouré, D.; Leh, H.; Subra, F.; le Bret, M.; Auclair, C.; d’Angelo, J. Structure-Activity relationships and binding mode of styrylquinolines as potent inhibitors of HIV-1 integrase and replication of HIV-1 in cell culture. J. Med. Chem. 2000, 43, 1533–1540. [Google Scholar] [CrossRef]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef]
- Erwin, A.L. Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme Lpxc. Cold Spring Harb. Perspect. Med. 2016, 6, a025304. [Google Scholar] [CrossRef]
- Kalinin, D.V.; Holl, R. Lpxc inhibitors: A patent review (2010–2016). Expert Opin. Ther. Patents 2017, 27, 1227–1250. [Google Scholar] [CrossRef]
- Cohen, F.; Aggen, J.B.; Andrews, L.D.; Assar, Z.; Boggs, J.; Choi, T.; Dozzo, P.; Easterday, A.N.; Haglund, C.M.; Hildebrandt, D.J.; et al. Optimization of LpxC inhibitors for antibacterial activity and cardiovascular safety. ChemMedChem 2019, 14, 1560–1572. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why hydroxamates may not be the best histone deacetylase inhibitors-what some may have forgotten or would rather forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Takashima, H.; Walmsley, D.L.; Ushiyama, F.; Matsuda, Y.; Kanazawa, H.; Yamaguchi-Sasaki, T.; Tanaka-Yamamoto, N.; Yamagishi, J.; Kurimoto-Tsuruta, R.; et al. Fragment-Based Discovery of Novel Non-Hydroxamate LpxC Inhibitors with Antibacterial Activity. J. Med. Chem. 2020, 63, 14805–14820. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Nicholson, J.D.; Hackley, B.E. Efficacy of a novel metalloprotease inhibitor on botulinum neurotoxin B activity. FEBS Lett. 1998, 429, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Agarwal, R.; Swaminathan, S. Small molecule non-peptide inhibitors of botulinum neurotoxin serotype E: Structure–activity relationship and a pharmacophore model. Bioorg. Med. Chem. 2016, 24, 3978–3985. [Google Scholar] [CrossRef]
- Zhou, Y.; McGillick, B.E.; Teng, Y.H.; Haranahalli, K.; Ojima, I.; Swaminathan, S.; Rizzo, R.C. Identification of small molecule inhibitors of botulinum neurotoxin serotype E via footprint similarity. Bioorg. Med. Chem. 2016, 24, 4875–4889. [Google Scholar] [CrossRef]
- Roxas-Duncan, V.; Enyedy, I.; Montgomery, V.A.; Eccard, V.S.; Carrington, M.A.; Lai, H.; Gul, N.; Yang, D.C.; Smith, L.A. Identification and biochemical characterization of small-molecule inhibitors of Clostridium botulinum neurotoxin serotype A. Antimicrob. Agents Chemother. 2009, 53, 3478–3486. [Google Scholar] [CrossRef]
- Lai, H.; Feng, M.; Roxas-Duncan, V.; Dakshanamurthy, S.; Smith, L.A.; Yang, D.C. Quinolinol and peptide inhibitors of zinc protease in botulinum neurotoxin A: Effects of zinc ion and peptides on inhibition. Arch. Biochem. Biophys. 2009, 491, 75–84. [Google Scholar] [CrossRef]
- Caglic, D.; Krutein, M.C.; Bompiani, K.M.; Barlow, D.J.; Benoni, G.; Pelletier, J.C.; Reitz, A.B.; Lairson, L.L.; Houseknecht, K.L.; Smith, G.R.; et al. Identification of clinically viable quinolinol inhibitors of botulinum neurotoxin A light chain. J Med. Chem. 2014, 57, 669–676. [Google Scholar] [CrossRef]
- Minnow, Y.V.; Goldberg, R.; Tummalapalli, S.R.; Rotella, D.P.; Goodey, N.M. Mechanism of inhibition of botulinum neurotoxin type A light chain by two quinolinol compounds. Arch. Biochem. Biophys. 2017, 618, 15–22. [Google Scholar] [CrossRef]
- Chauhan, R.; Vinita Chauhan, V.; Sonkara, P.; Vimal, M.; Dhaked, R.M. Targeted 8-hydroxyquinoline fragment based small molecule drug discovery against neglected botulinum neurotoxin type F. Bioorg. Chem. 2019, 92, 103297–103312. [Google Scholar] [CrossRef]
- Karamanos, Y. Endo-N-acetyl-β-D-glucosaminidases and their potential substrates: Structure/function relationships. Res. Microbiol. 1997, 148, 661–671. [Google Scholar] [CrossRef]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front. Microbiol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Tibaut, T.; Tomašič, T.; Hodnik, V.; Anderluh, M.; Pintar, S.; Novič, M.; Turk, D. Application of fragment based virtual screening towards inhibition of bacterial N-acetyglucosaminidase. SAR QSAR Environ. Res. 2018, 29, 647–660. [Google Scholar] [CrossRef]
- Keeley, A.; Ábrányi-Balogh, P.; Hrast, M.; Imre, T.; Ilaš, J.; Gobec, S.; Keserű, G.M. Heterocyclic electrophiles as new MurA inhibitors. Arch. Pharm. Chem. Life Sci. 2018, 351, e1800184. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Walsh, C.T. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: Enzymology, antibiotics, and antibiotic resistance. Nat. Prod. Rep. 1992, 9, 199–215. [Google Scholar] [CrossRef]
- Blake, K.L.; O’Neill, A.J.; Mengin-Lecreulx, D.; Henderson, P.J.F.; Bostock, J.M.; Dunsmore, C.J.; Simmons, K.J.; Fishwick, C.W.G.; Leeds, J.A.; Chopra, I. The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol. Microbiol. 2009, 72, 335–343. [Google Scholar] [CrossRef]
- Grabrijan, K.; Hrast, M.; Proj, M.; Dolšak, A.; Zdovc, I.; Imre, T.; Petri, L.; Abranyi-Balogh, P.; Keseru, G.M.; Gobec, S. Covalent inhibitors of bacterial peptidoglycan biosynthesis enzyme MurA with chloroacetamide warhead. Eur. J. Med. Chem. 2022, 243, 114752. [Google Scholar] [CrossRef]
- Allimuthu, D.; Adams, D.J. 2-Chloropropionamide as a low-reactivity electrophile for irreversible small-molecule probe identification. ACS Chem. Biol. 2017, 12, 2124–2131. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef]
- Masoudi, A.; Raetz, C.R.H.; Zhou, P.; Pemble, C.W. IV Chasing acyl carrier protein through a catalytic cycle of lipid A production. Nature 2014, 505, 422–426. [Google Scholar] [CrossRef]
- Begley, T.P.; Kinsland, C.; Strauss, E. The biosynthesis of coenzyme A in bacteria. Vitam. Horm. 2001, 61, 157–171. [Google Scholar] [PubMed]
- Genschel, U. Coenzyme A biosynthesis: Reconstruction of the pathway in archaea and an evolutionary scenario based on comparative genomics. Mol. Biol. Evol. 2004, 21, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.; Polanuyer, B.; Farrell, M.; Scholle, M.; Lykidis, A.; de Crecy-Lagard, V.; Osterman, A. Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics. J. Biol. Chem. 2002, 277, 21431–21439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Allanson, N.M.; Thomson, S.P.; Maclean, J.K.F.; Barker, J.J.; Primrose, W.U.; Tyler, P.D.; Lewendon, A. Inhibitors of phosphopantetheine adenylyltransferase. Eur. J. Med. Chem. 2003, 38, 345–349. [Google Scholar] [CrossRef]
- De Jonge, B.L.M.; Walkup, G.K.; Lahiri, S.D.; Huynh, H.; Neckermann, G.; Utley, L.; Nash, T.J.; Brock, J.; San Martin, M.; Kutschke, A.; et al. Discovery of inhibitors of 4′-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob. Agents Chemother. 2013, 57, 6005–6015. [Google Scholar] [CrossRef]
- Moreau, R.J.; Skepper, C.K.; Appleton, B.A.; Blechschmidt, A.; Balibar, C.J.; Benton, B.M.; Drumm, J.E.; Feng, B.Y.; Geng, M.; Li, C.; et al. Fragment-based drug discovery of inhibitors of phosphopantetheine adenylyltransferase from gram-negative bacteria. J. Med. Chem. 2018, 61, 3309–3324. [Google Scholar] [CrossRef]
- Skepper, C.K.; Moreau, R.J.; Appleton, B.A.; Benton, B.M.; Drumm, J.E., III; Feng, B.Y.; Geng, M.; Hu, C.; Li, C.; Lingel, A.; et al. Discovery and Optimization of Phosphopantetheine Adenylyltransferase Inhibitors with Gram-Negative Antibacterial Activity. J. Med. Chem. 2018, 61, 3325–3349. [Google Scholar] [CrossRef]
- Bishop, J.R.; Gagneux, P. Evolution of carbohydrate antigens—Microbial forces shaping host glycomes? Glycobiology 2007, 17, 23R–34R. [Google Scholar] [CrossRef]
- Meiers, J.; Siebs, E.; Zahorska, E.; Titz, A. Lectin antagonists in infection, immunity, and inflammation. Curr. Opin. Chem. Biol. 2019, 53, 51–67. [Google Scholar] [CrossRef]
- Shanina, E.; Kuhaudomlarp, S.; Lal, K.; Seeberger, P.H.; Imberty, A.; Rademacher, C. Druggable Allosteric Sites in b-Propeller Lectins. Angew. Chem. Int. Ed. 2022, 61, e202109339. [Google Scholar] [CrossRef]
- Audfray, A.; Claudinon, J.; Abounit, S.; Ruvoen-Clouet, N.; Larson, G.; Smith, D.F.; Wimmerov, M.; le Pendu, J.; Rçmer, W.; Varrot, A.; et al. Fucose-binding lectin from opportunistic pathogen Burkholderia ambifaria binds to both plant and human oligosaccharidic epitopes. J. Biol. Chem. 2012, 287, 4335–4347. [Google Scholar] [CrossRef] [Green Version]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef]
- Adams, L.A.; Sharma, P.; Mohanty, B.; Ilyichova, O.V.; Mulcair, M.D.; Williams, M.L.; Gleeson, E.C.; Totsika, M.; Doak, B.C.; Caria, S.; et al. Application of fragment-based screening to the design of inhibitors of Escherichia coli DsbA. Angew. Chem. Int. Ed. 2015, 54, 2179–2184. [Google Scholar] [CrossRef]
- Duncan, L.F.; Wang, G.; Ilyichova, O.V.; Scanlon, M.J.; Heras, B.; Abbott, B.M. The fragment-based development of a benzofuran hit as a new class of Escherichia coli DsbA inhibitors. Molecules 2019, 24, 3756. [Google Scholar] [CrossRef]
- Duncan, L.F.; Wang, G.; Ilyichova, O.V.; Dhouib, R.; Totsika, M.; Scanlon, M.J.; Heras, B.; Abbott, B.M. Elaboration of a benzofuran scaffold and evaluation of binding affinity and inhibition of Escherichia coli DsbA: A fragment-based drug design approach to novel antivirulence compounds. Bioorg. Med. Chem. 2021, 45, 116315–116327. [Google Scholar] [CrossRef]
- Wang, G.; Mohanty, B.; Williams, M.L.; Doak, B.C.; Dhouib, R.; Totsika, M.; McMahon, R.M.; Sharma, G.; Zheng, D.; Bentley, M.R.; et al. Selective Binding of Small Molecules to Vibrio cholerae DsbA Offers a Starting Point for the Design of Novel Antibacterials. ChemMedChem 2022, 17, e202100673. [Google Scholar] [CrossRef]
- Van Delden, C.; Iglewski, B.H. Cell-to-Cell Signaling and Pseudomonas aeruginosa Infections. Emerg. Infect. Dis. 1998, 4, 551–560. [Google Scholar] [CrossRef]
- Zender, M.; Witzgall, F.; Kiefer, A.; Kirsch, B.; Maurer, C.K.; Kany, A.M.; Xu, N.; Schmelz, S.; Börger, C.; Blankenfeldt, W.; et al. Flexible Fragment Growing Boosts Potency of Quorum Sensing Inhibitors against Pseudomonas aeruginosa Virulence. ChemMedChem 2020, 15, 188–194. [Google Scholar] [CrossRef]
- Déziel, E.; Gopalan, S.; Tampakaki, A.P.; Lépine, F.; Padfield, K.E.; Saucier, M.; Xiao, G.; Rahme, L.G. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: Multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol. Microbiol. 2005, 55, 998–1014. [Google Scholar] [CrossRef]
- Lu, C.; Kirsch, B.; Zimmer, C.; de Jong, J.C.; Henn, C.; Maurer, C.K.; Müsken, M.; Häussler, S.; Steinbach, A.; Hartmann, R.W. Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem. Biol. 2012, 19, 381–390. [Google Scholar] [CrossRef]
- Arif, S.M.; Floto, R.A.; Blundell, T.L. Using Structure-guided Fragment-Based Drug Discovery to Target Pseudomonas aeruginosa Infections in Cystic Fibrosis. Front. Mol. Biosci. 2022, 9, 857000. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book 2002, 1, e0018. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, K.; Jeyakanthan, J.; Sekar, K. Catalytic pathway, substrate binding and stability in SAICAR synthetase: A structure and molecular dynamics study. J. Struct Biol. 2015, 191, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Tuntland, M.L.; Fung, L.W. Substrate independent ATPase activity may complicate high throughput screening. Anal. Biochem. 2016, 510, 18–20. [Google Scholar] [CrossRef]
- Ginder, N.D.; Binkowski, D.J.; Fromm, H.J.; Honzatko, R.B. Nucleotide complexes of Escherichia coli phosphoribosylaminoimidazole succinocarboxamide synthetase. J. Biol. Chem. 2006, 281, 20680–20688. [Google Scholar] [CrossRef]
- Li, S.X.; Tong, Y.P.; Xie, X.C.; Wang, Q.H.; Zhou, H.N.; Han, Y.; Zhang, Z.Y.; Gao, W.; Li, S.G.; Zhang, X.C.; et al. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol. 2007, 366, 1603–1614. [Google Scholar] [CrossRef]
- Ducati, R.G.; Breda, A.; Basso, L.A.; Santos, D.S. Purine Salvage Pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1258–1275. [Google Scholar] [CrossRef]
- Wolf, N.M.; Abad-Zapatero, C.; Johnson, M.E.; Fung, L.W.M. Structures of SAICAR synthetase (PurC) from Streptococcus pneumoniae with ADP, Mg2+, AIR and Asp. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 841–850. [Google Scholar] [CrossRef]
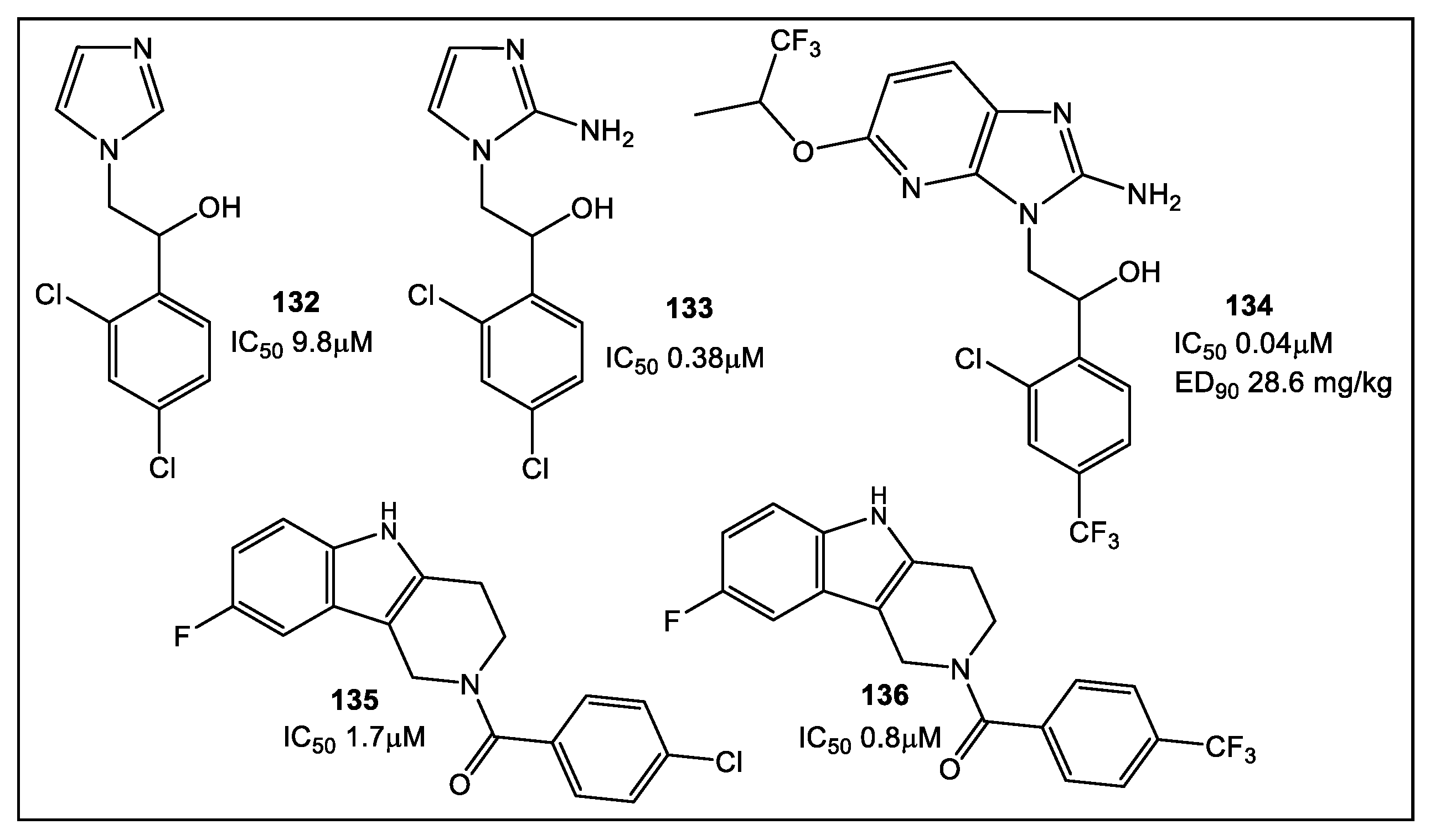
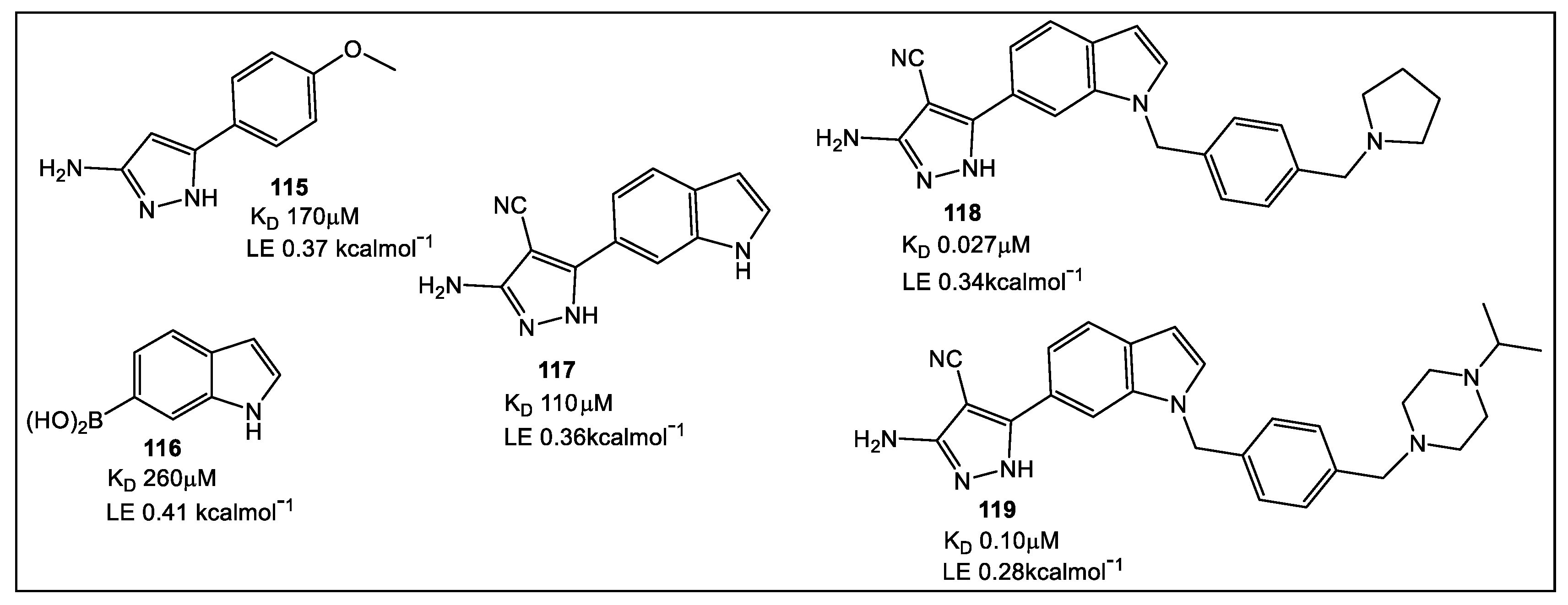
- Charoensutthivarakul, S.; Thomas, S.E.; Curran, A.; Brown, K.P.; Belardinelli, J.M.; Whitehouse, A.J.; Acebrón-García-de-Eulate, M.; Sangan, J.; Gramani, S.G.; Jackson, M.; et al. Development of Inhibitors of SAICAR Synthetase (PurC) from Mycobacterium abscessus Using a Fragment-Based Approach. ACS Infect. Dis. 2022, 8, 296–309. [Google Scholar] [CrossRef]
- Thomas, S.E.; Collins, P.; James, R.H.; Mendes, V.; Charoensutthivarakul, S.; Radoux, C.; Abell, C.; Coyne, A.G.; Floto, R.A.; von Delft, F.; et al. Structure-guided fragment-based drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philos. Trans A Math Phys. Eng. Sci. 2019, 377, 20180422. [Google Scholar] [CrossRef]
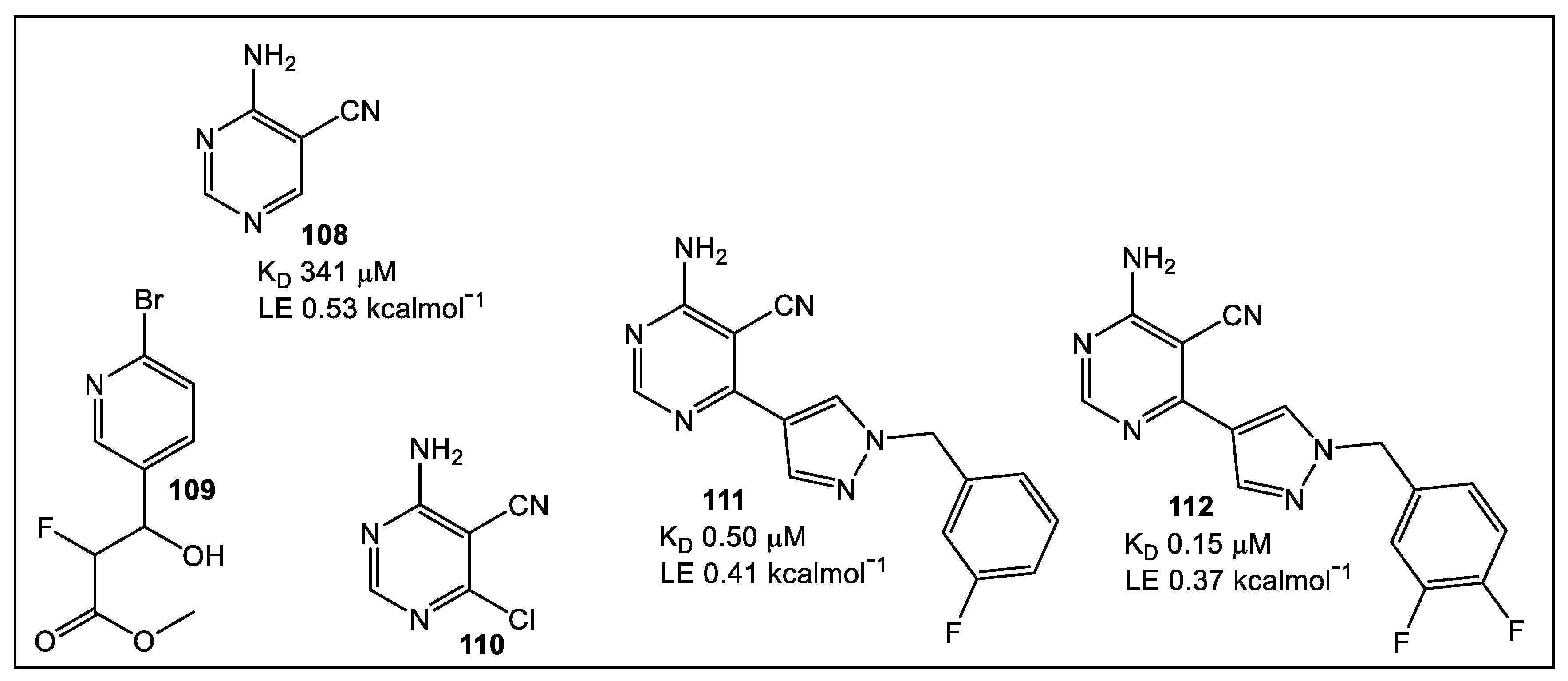
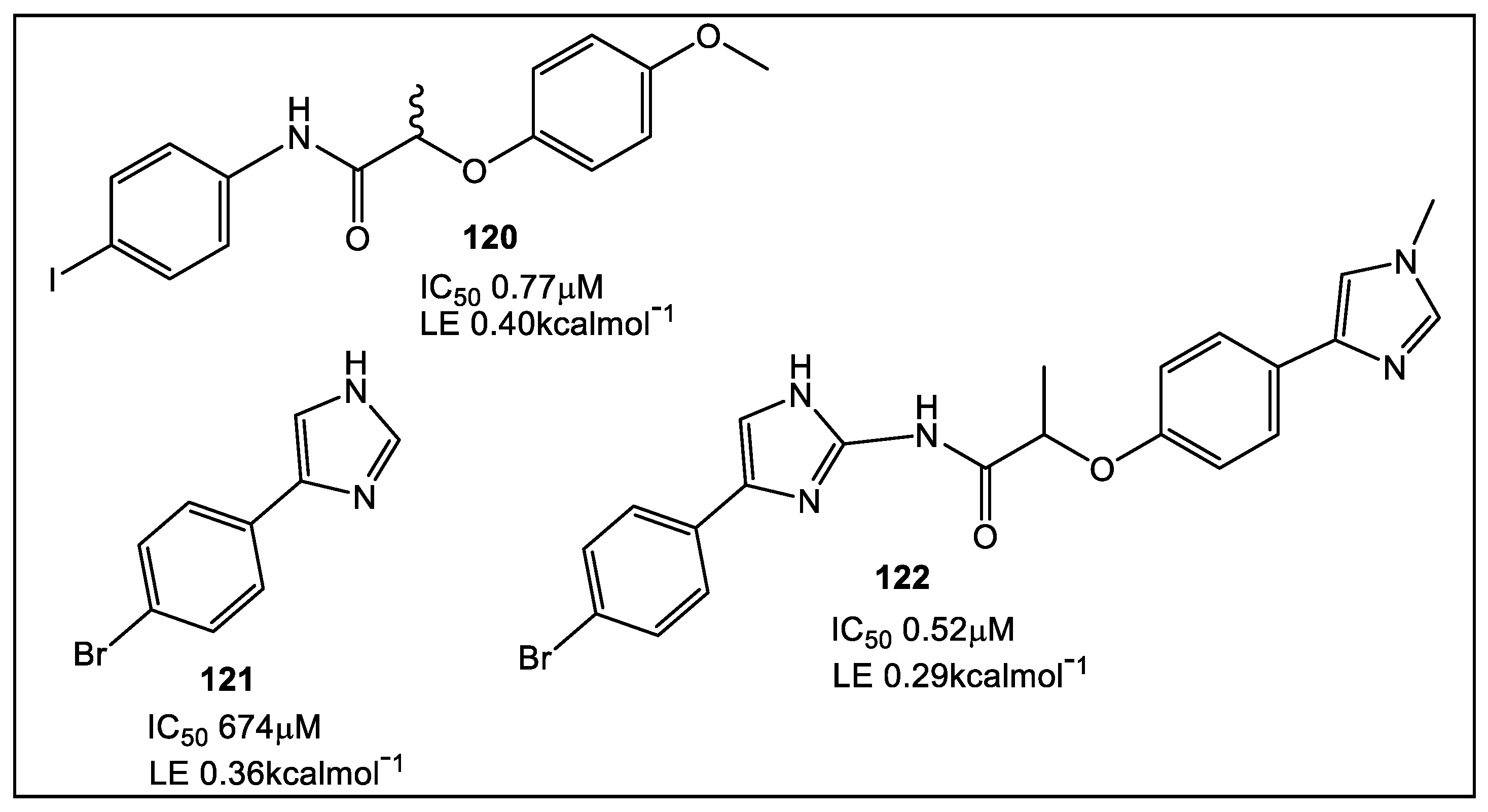
- Whitehouse, A.J.; Thomas, S.E.; Brown, K.P.; Fanourakis, A.; Chan, D.S.-H.; Libardo, M.D.J.; Mendes, V.; Boshoff, H.I.M.; Floto, R.A.; Abell, C.; et al. Development of Inhibitors against Mycobacterium abscessus tRNA (m1G37) Methyltransferase (TrmD) Using Fragment-Based Approaches. J. Med. Chem. 2019, 62, 7210–7232. [Google Scholar] [CrossRef] [Green Version]
- Goto-Ito, S.; Ito, T.; Yokoyama, S. Trm5 and TrmD: Two Enzymes from Distinct Origins Catalyze the Identical tRNA Modification, m1G37. Biomolecules 2017, 7, 32. [Google Scholar] [CrossRef]
- Forsyth, R.A.; Haselbeck, R.J.; Ohlsen, K.L.; Yamamoto, R.T.; Xu, H.; Trawick, J.D.; Wall, D.; Wang, L.; Brown-Driver, V.; Froelich, J.M.; et al. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 2002, 43, 1387–1400. [Google Scholar] [CrossRef]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 2015, 112, 4110–4115. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
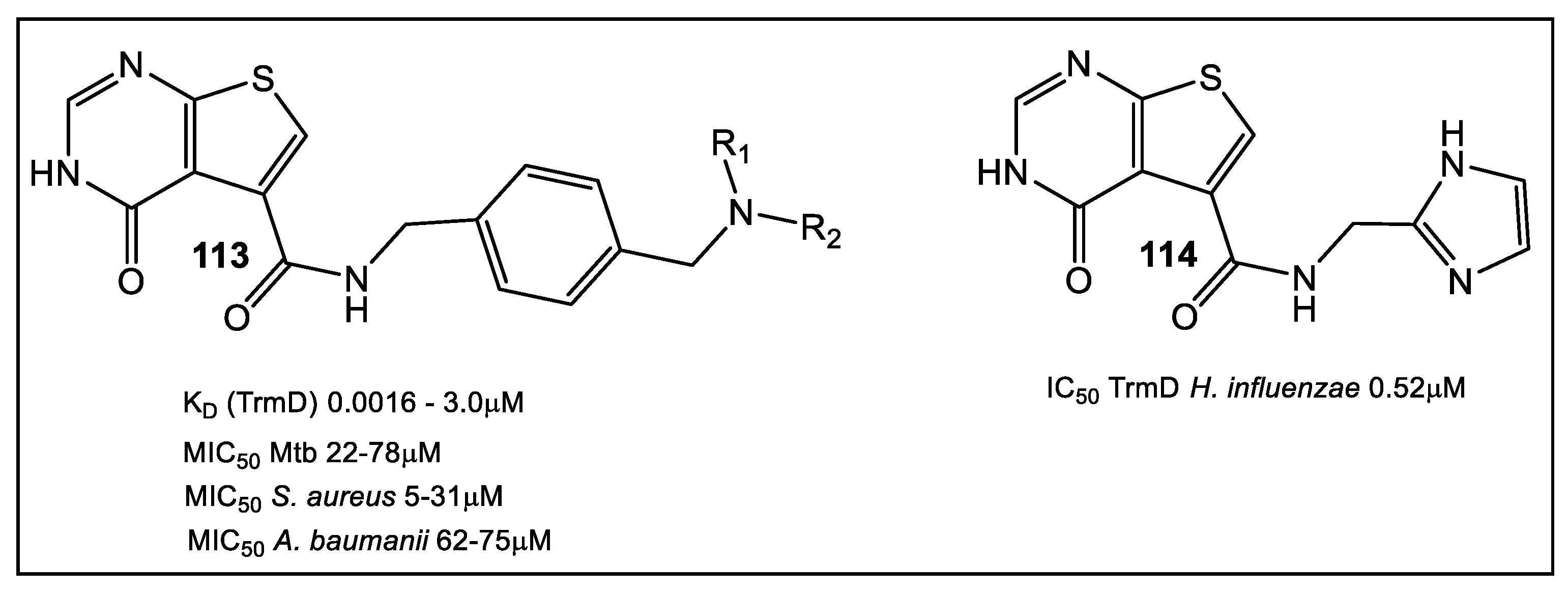
- Thomas, S.E.; Whitehouse, A.J.; Brown, K.; Belardinelli, J.M.; Lahiri, R.; Libardo, M.D.J.; Gupta, P.; Malhotra, S.; Boshoff, H.I.M.; Jackson, M.; et al. Fragment-based Discovery of a New Class of Inhibitors Targeting Mycobacterial tRNA Modification. Nucleic Acids Res. 2020, 48, 8099–8112. [Google Scholar] [CrossRef]
- Zhong, W.; Koay, A.; Ngo, A.; Li, Y.; Nah, Q.; Wong, Y.H.; Chionh, Y.H.; Ng, H.Q.; Koh-Stenta, X.; Poulsen, A.; et al. Targeting the bacterial epitranscriptome for antibiotic development: Discovery of novel tRNA-(N1G37) methyltransferase (TrmD) inhibitors. ACS Infect. Dis. 2019, 5, 326–335. [Google Scholar] [CrossRef]
- Zhong, W.; Pasunooti, K.K.; Balamkundu, S.; Wong, Y.H.; Nah, Q.; Gadi, V.; Gnanakalai, S.; Chionh, Y.H.; McBee, M.E.; Gopal, P.; et al. Thienopyrimidinone Derivatives That Inhibit Bacterial tRNA (Guanine37-N1)-Methyltransferase (TrmD) by Restructuring the Active Site with a Tyrosine-Flipping Mechanism. J. Med. Chem. 2019, 62, 7788–7805. [Google Scholar] [CrossRef]
- Hill, P.J.; Abibi, A.; Albert, R.; Andrews, B.; Gagnon, M.M.; Gao, N.; Grebe, T.; Hajec, L.I.; Huang, J.; Livchak, S.; et al. Selective Inhibitors of Bacterial t-RNA-(N1G37) Methyltransferase (TrmD) That Demonstrate Novel Ordering of the Lid Domain. J. Med. Chem. 2013, 56, 7278–7288. [Google Scholar] [CrossRef]
- Jackson, R.C.; Weber, G.; Morris, H.P. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature 1975, 256, 331–333. [Google Scholar] [CrossRef]
- Ratcliffe, A.J. Inosine 5′-monophosphate dehydrogenase inhibitors for the treatment of autoimmune diseases. Curr. Opin. Drug Discovery Dev. 2006, 9, 595–605. [Google Scholar]
- Chen, L.; Pankiewicz, K.W. Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr. Opin. Drug Discov. Dev. 2007, 10, 403–412. [Google Scholar]
- Olah, E.; Kokeny, S.; Papp, J.; Bozsik, A.; Keszei, M. Modulation of cancer pathways by inhibitors of guanylate metabolism. Adv. Enzyme Regul. 2006, 46, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Shu, Q. Inosine monophosphate dehydrogenase as a probe in antiviral drug discovery. Antiviral Chem. Chemother. 2007, 18, 245–258. [Google Scholar] [CrossRef]
- Shu, Q.; Nair, V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev. 2008, 28, 219–232. [Google Scholar] [CrossRef]
- Shah, C.P.; Kharkar, P.S. Inosine 5′-monophosphate dehydrogenase inhibitors as antimicrobial agents: Recent progress and future perspectives. Future Med. Chem. 2015, 7, 1415–1429. [Google Scholar] [CrossRef]
- Hedstrom, L.; Liechti, G.; Goldberg, J.B.; Gollapalli, D.R. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr. Med. Chem. 2011, 18, 1909–1918. [Google Scholar] [CrossRef]
- Hedstrom, L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef]
- Trapero, A.; Pacitto, A.; Singh, V.; Sabbah, M.; Coyne, A.G.; Mizrahi, V.; Blundell, T.L.; Ascher, D.B.; Abell, C. Fragment-Based Approach to Targeting Inosine-5′-monophosphate Dehydrogenase (IMPDH) from Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 2806–2822. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Gorla, S.K.; Wei, Y.; Mandapati, K.; Zhang, M.; Maltseva, N.; Modi, G.; Boshoff, H.I.; Gu, M.; et al. Mycobacterium tuberculosis IMPDH in complexes with substrates, products and antitubercular compounds. PLoS ONE 2015, 10, e0138976. [Google Scholar] [CrossRef]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Chin, J.E.; Liu, X.; Striepen, B.; Makowska-Grzyska, M.; Kim, Y.; Joachimiak, A.; Hedstrom, L.; et al. Optimization of benzoxazole-based inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2013, 56, 4028–4043. [Google Scholar] [CrossRef]
- Mendes, V.; Blundell, T.L. Targeting tuberculosis using structure-guided fragment based drug design. Drug Discov. Today 2017, 22, 546–554. [Google Scholar] [CrossRef]
- Togre, N.S.; Vargas, A.M.; Bhargavi, G.; Mallakuntla, M.K.; Tiwari, S. Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges. Int. J. Mol. Sci. 2022, 23, 10669. [Google Scholar] [CrossRef]
- Woese, C.R.; Olsen, G.J.; Ibba, M.; Soll, D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000, 64, 202–236. [Google Scholar] [CrossRef]
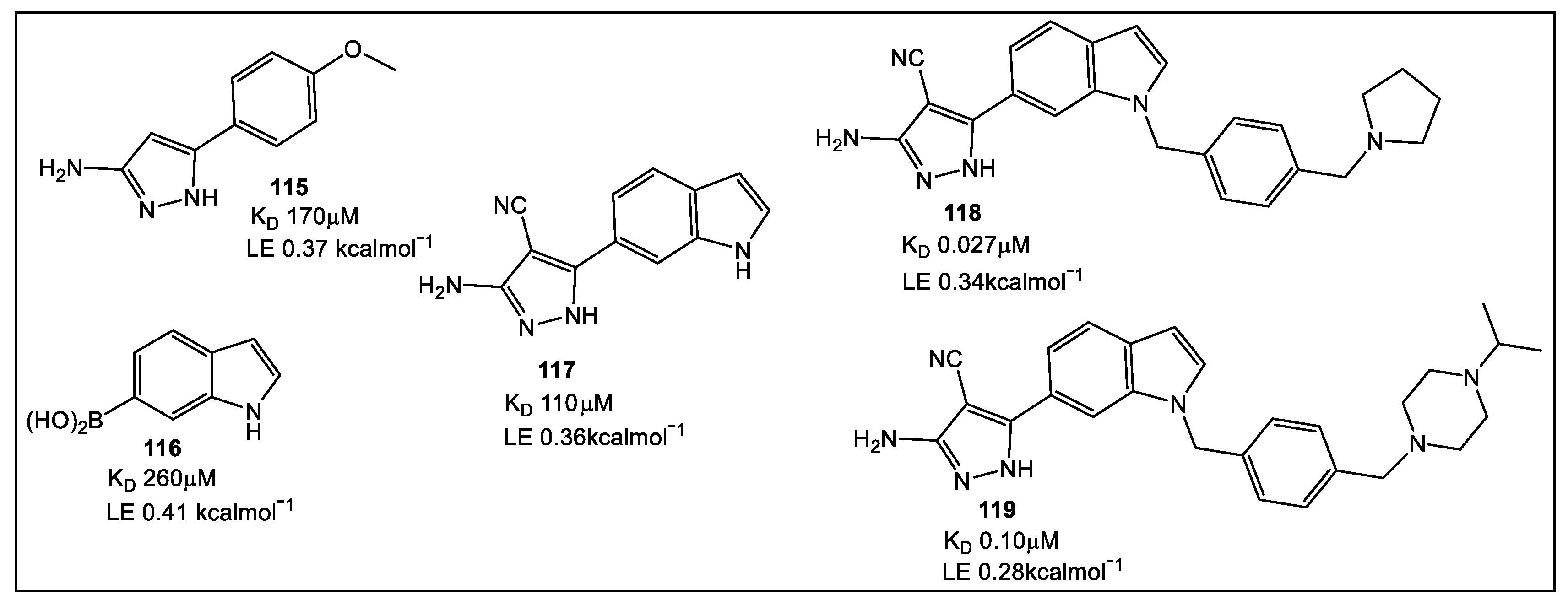
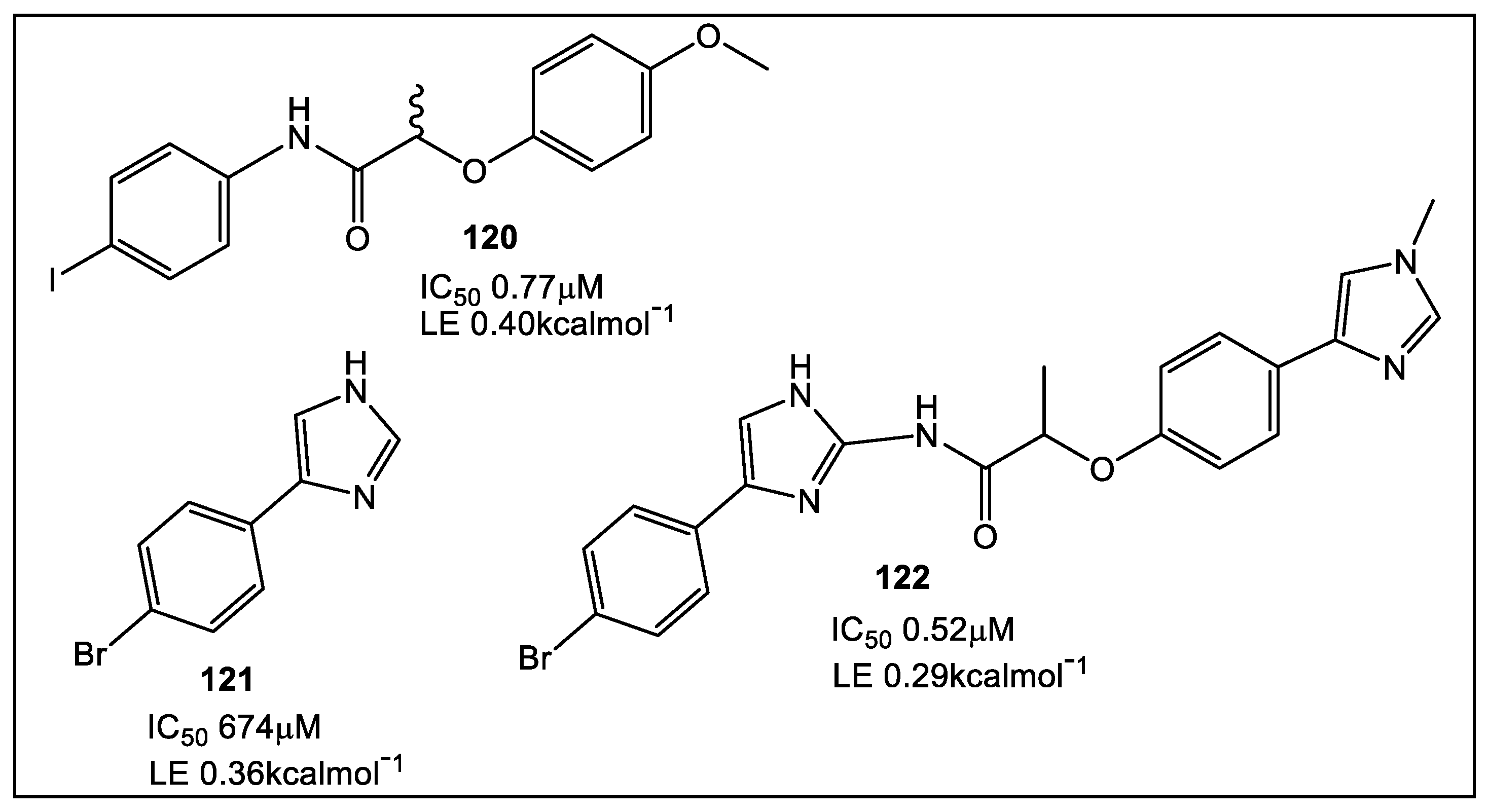
- Guo, J.; Chen, B.; Yu, Y.; Cheng, B.; Cheng, Y.; Ju, Y.; Gu, Q.; Xu, J.; Zhou, H. Discovery of novel tRNA-amino acid dual-site inhibitors against threonyl-tRNA synthetase by fragment-based target hopping. Euro. J. Med. Chem. 2020, 187, 111941. [Google Scholar] [CrossRef]
- Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; et al. Identification of bacteria-selective threonyl-tRNA synthetase substrate inhibitors by structure-based design. J. Med. Chem. 2013, 56, 1748–1760. [Google Scholar] [CrossRef]
- Bovee, M.L.; Pierce, M.A.; Francklyn, C.S. Induced fit and kinetic mechanism of adenylation catalyzed by Escherichia coli threonyl-tRNA synthetase. Biochemistry 2003, 42, 15102–15113. [Google Scholar] [CrossRef]
- Gadakh, B.; van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: A patent review from 2006 till present. Expert Opin. Ther. Pat. 2012, 22, 1453–1465. [Google Scholar] [CrossRef]
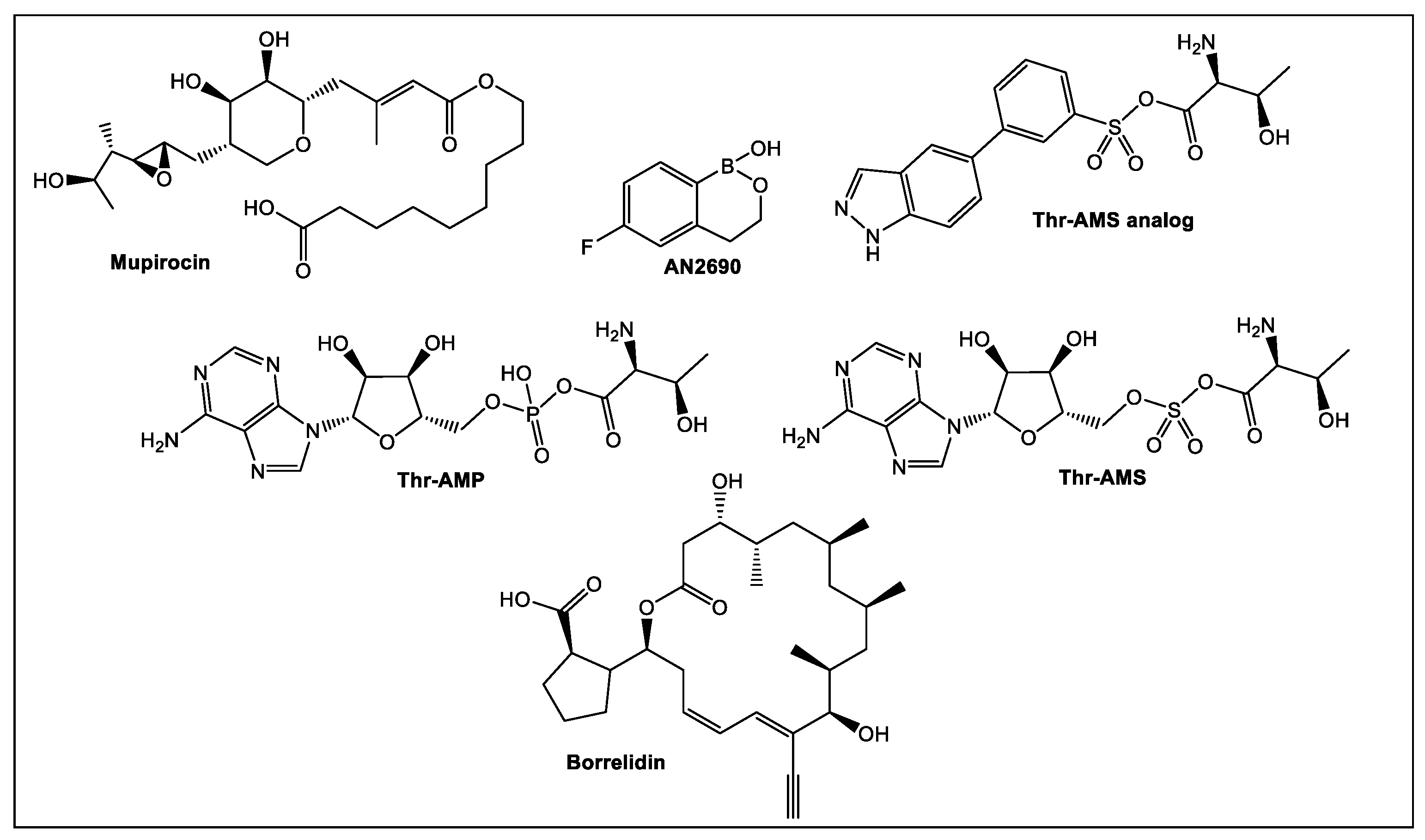
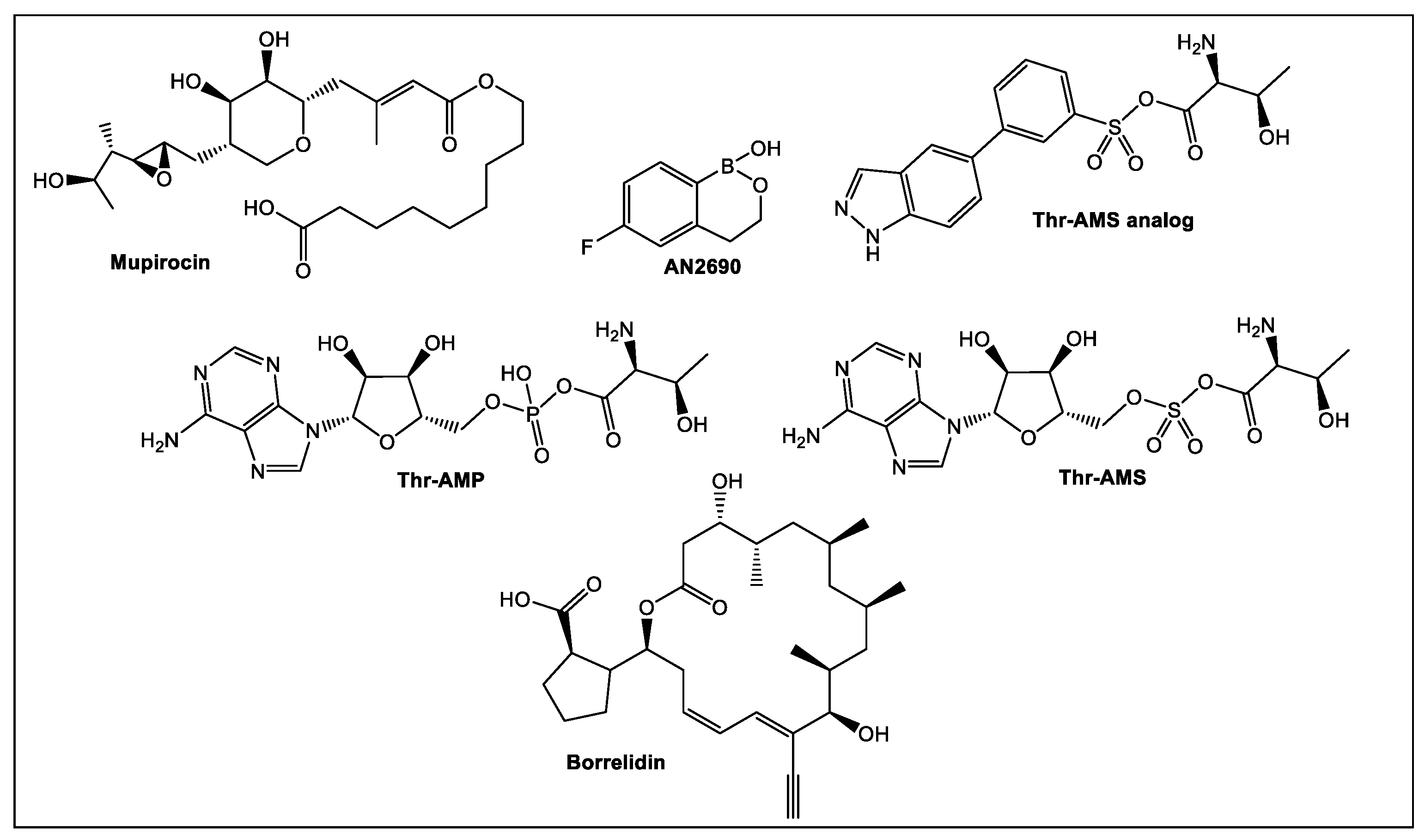
- Silvian, L.F.; Wang, J.; Steitz, T.A. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science 1999, 285, 1074–1077. [Google Scholar] [CrossRef]
- Ho, J.M.; Bakkalbasi, E.; Soll, D.; Miller, C.A. Drugging tRNA aminoacylation. RNA Biol. 2018, 15, 667–677. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyltRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Jampolsky, L.M.; Goldberg, M.W. Borrelidin, a new antibiotic with antiborrelia activity and penicillin enhancement properties. Arch. Biochem. 1949, 22, 476–478. [Google Scholar] [PubMed]
- Fang, P.; Yu, X.; Jeong, S.J.; Mirando, A.; Chen, K.; Chen, X.; Kim, S.; Francklyn, C.S.; Guo, M. Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase. Nat. Commun. 2015, 6, 6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Sun, L.; Yang, X.L.; Schimmel, P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature 2013, 494, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Yogavel, M.; Kikuchi, H.; Oshima, Y.; Hariguchi, N.; Matsumoto, M.; Goel, P.; Touquet, B.; Jumani, R.S.; Tacchini-Cottier, F.; et al. Targeting prolyl-tRNA synthetase to accelerate drug Discovery against malaria, Leishmaniasis, Toxoplasmosis, Cryptosporidiosis, and Coccidiosis. Structure 2017, 25, 1495–1505.e1496. [Google Scholar] [CrossRef]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014, 4, 6522. [Google Scholar] [CrossRef]
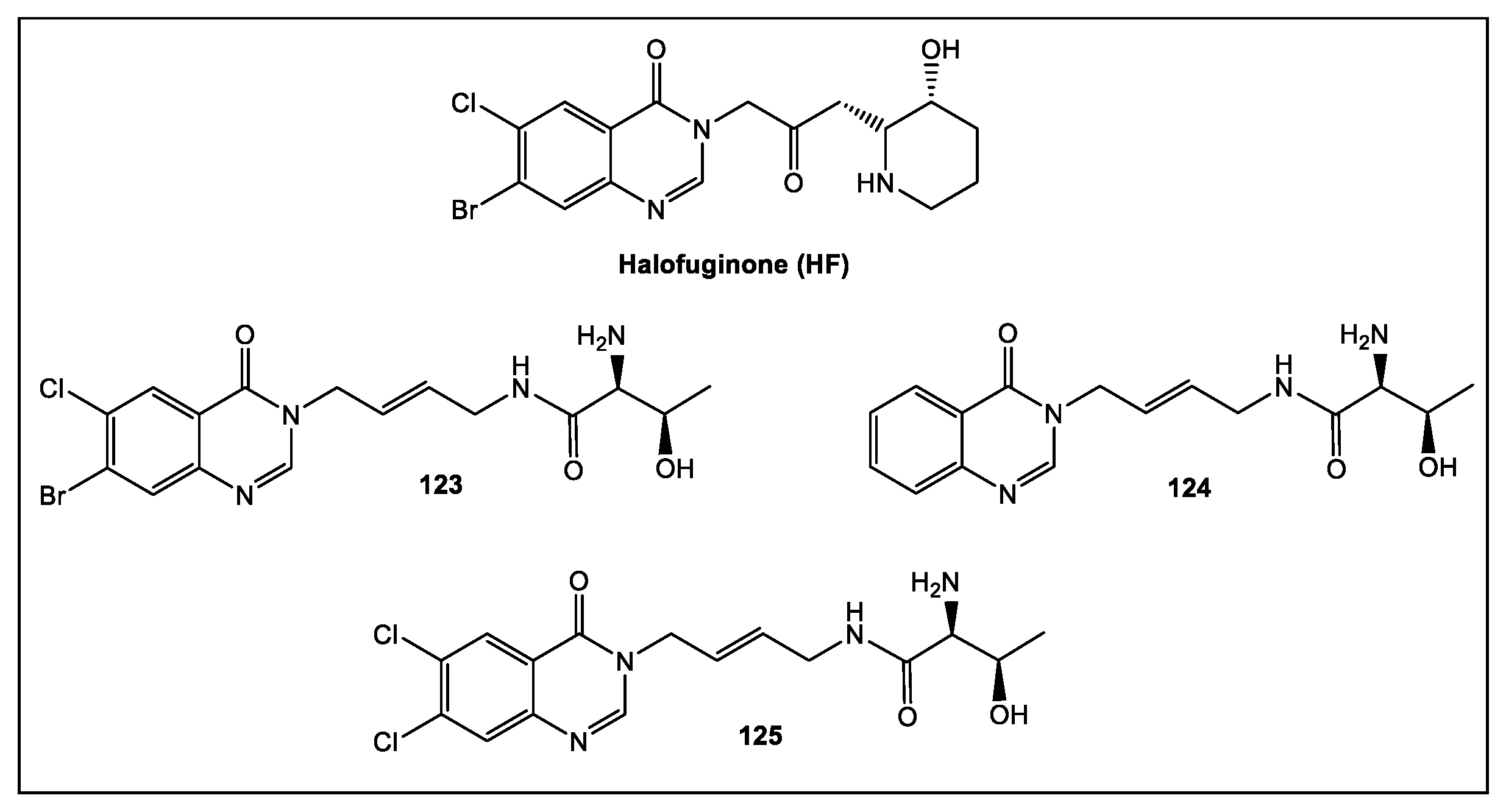
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyltRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef]
- Doak, B.C.; Morton, C.J.; Simpson, J.S.; Scanlon, M.J. Design and evaluation of the performance of an NMR screening fragment library. Aust. J. Chem. 2013, 66, 1465–1472. [Google Scholar] [CrossRef]
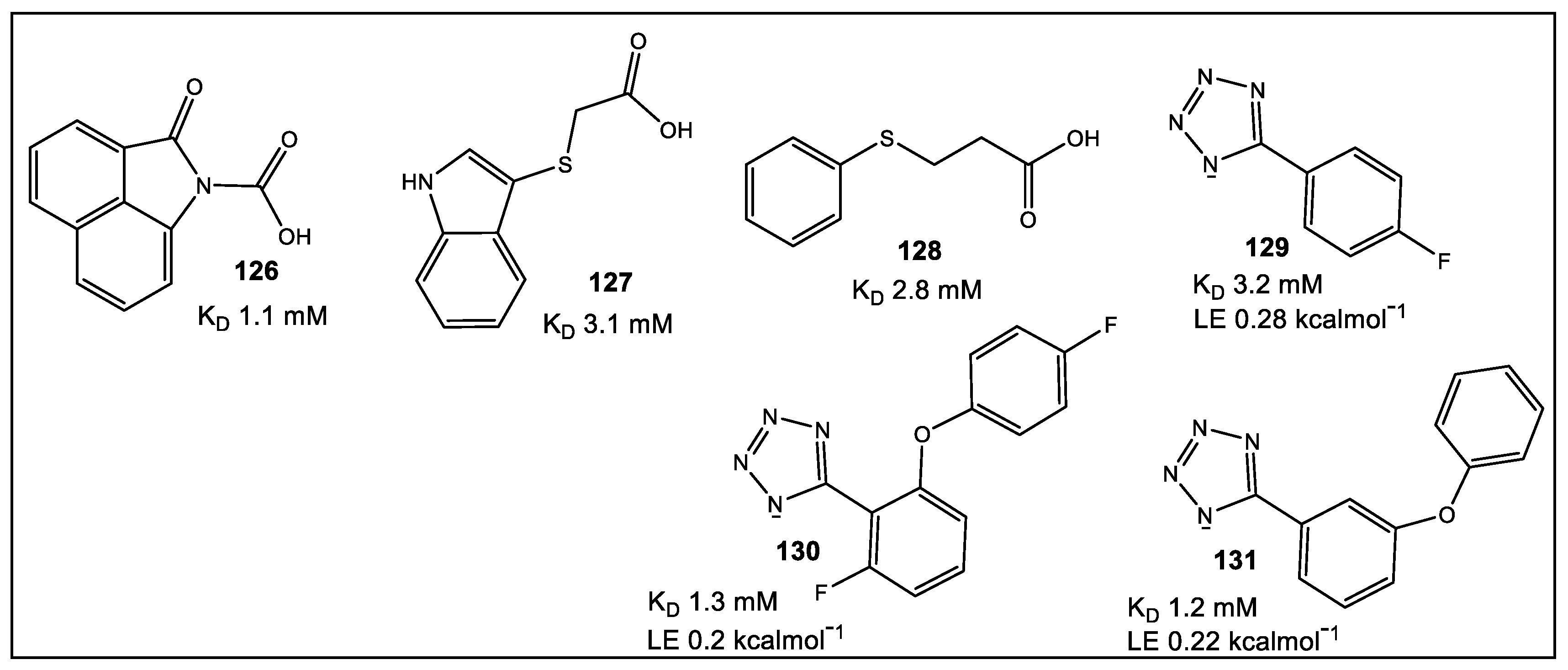
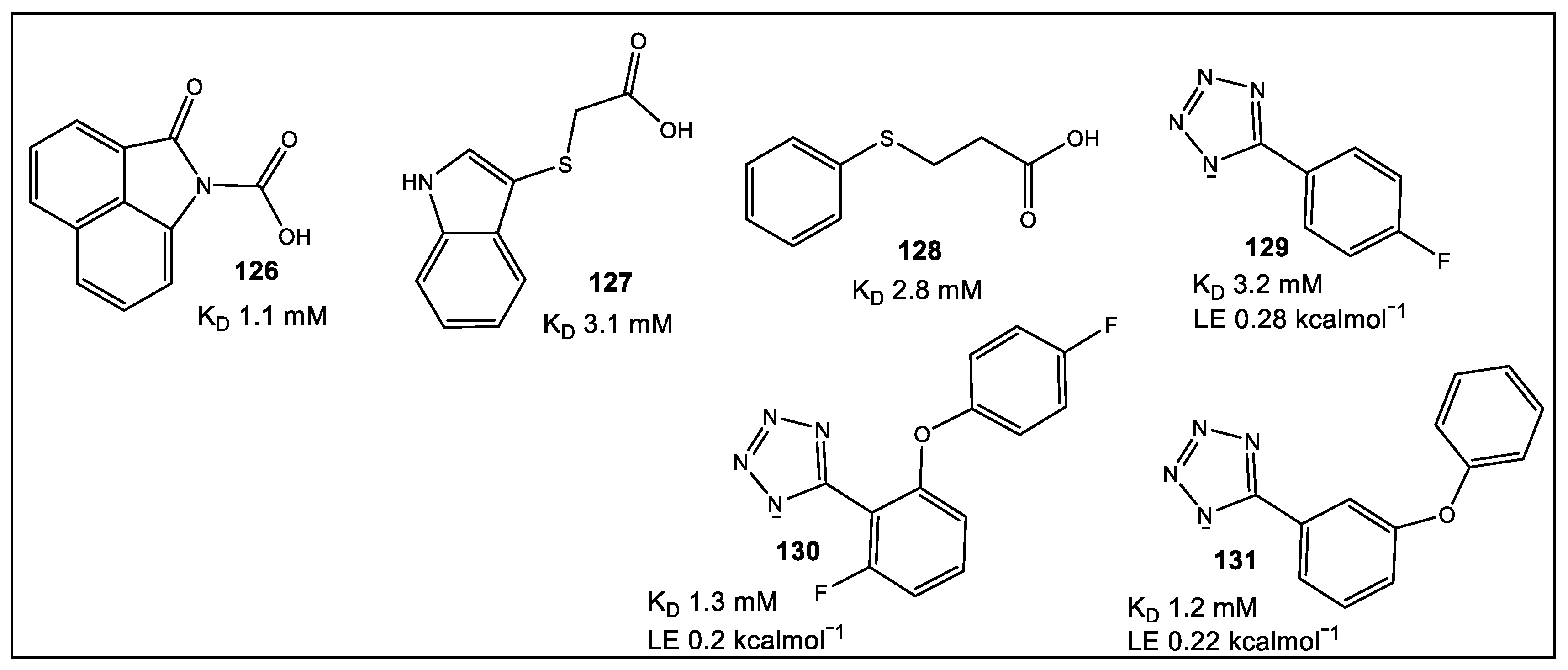
- Chilingaryan, Z.; Headey, S.J.; Lo, A.T.Y.; Xu, Z.-Q.; Otting, G.; Dixon, N.E.; Scanlon, M.J.; Oakley, A.J. Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction. Antibiotics 2018, 7, 14. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC−A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Moreira, W.; Lim, J.J.; Yeo, S.Y.; Ramanujulu, P.M.; Dymock, B.W.; Dick, T. Fragment-Based Whole Cell Screen Delivers Hits against M. tuberculosis and Non-tuberculous Mycobacteria. Front Microbiol. 2016, 7, 1392. [Google Scholar] [CrossRef] [Green Version]
- Negatu, D.A.; Liu, J.J.J.; Zimmerman, M.; Kaya, F.; Dartois, V.; Aldrich, C.C.; Gengenbacher, M.; Dick, T. Whole-Cell Screen of Fragment Library Identifies Gut Microbiota Metabolite Indole Propionic Acid as Antitubercular. Antimicrob. Agents Chemother. 2018, 62, e01571-17. [Google Scholar] [CrossRef]
- Ayotte, Y.; Bilodeau, F.; Descoteaux, A.; LaPlante, S.R. Fragment-Based Phenotypic Lead Discovery: Cell-Based Assay to Target Leishmaniasis. ChemMedChem 2018, 13, 1377–1386. [Google Scholar] [CrossRef]
- Basarab, G.S.; Hill, P.J.; Garner, C.E.; Hull, K.; Green, O.; Sherer, B.A.; Dangel, P.B.; Manchester, J.I.; Bist, S.; Hauck, S.; et al. Optimization of Pyrrolamide Topoisomerase II Inhibitors toward Identification of an Antibacterial Clinical Candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. [Google Scholar] [CrossRef]
- Ayotte, Y.; Bernet, E.; Bilodeau, F.; Cimino, M.; Gagnon, D.; Lebughe, M.; Mistretta, M.; Ogadinma, P.; Ouali, S.-L.; Sow, A.A.; et al. Fragment-Based Phenotypic Lead Discovery To Identify New Drug Seeds That Target Infectious Diseases. ACS Chem. Bio. 2021, 16, 2158–2163. [Google Scholar] [CrossRef]
- Parker, C.G.; Galmozzi, A.; Wang, Y.; Correia, B.E.; Sasaki, K.; Joslyn, C.M.; Kim, A.S.; Cavallaro, C.L.; Lawrence, R.M.; Johnson, S.R.; et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168, 527–541.e29. [Google Scholar] [CrossRef]
- Wang, Y.; Dix, M.M.; Bianco, G.; Remsberg, J.R.; Lee, H.-Y.; Kalocsay, M.; Gygi, S.P.; Forli, S.; Vite, G.; Lawrence, R.M.; et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 2019, 11, 1113–1123. [Google Scholar] [CrossRef]
- Gianni, D.; Farrow, S. Functional genomics for target identification. SLAS Discov. 2020, 25, 531–534. [Google Scholar] [CrossRef]
- Knight, S.; Gianni, D.; Hendricks, A. Fragment-based screening: A new paradigm for ligand and target discovery. SLAS Discov. 2022, 27, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Pasquer, Q.T.L.; Tsakoumagkos, I.A.; Hoogendoorn, S. From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems. Molecules 2020, 25, 5702. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konaklieva, M.I.; Plotkin, B.J. Fragment-Based Lead Discovery Strategies in Antimicrobial Drug Discovery. Antibiotics 2023, 12, 315. https://doi.org/10.3390/antibiotics12020315
Konaklieva MI, Plotkin BJ. Fragment-Based Lead Discovery Strategies in Antimicrobial Drug Discovery. Antibiotics. 2023; 12(2):315. https://doi.org/10.3390/antibiotics12020315
Chicago/Turabian StyleKonaklieva, Monika I., and Balbina J. Plotkin. 2023. "Fragment-Based Lead Discovery Strategies in Antimicrobial Drug Discovery" Antibiotics 12, no. 2: 315. https://doi.org/10.3390/antibiotics12020315
APA StyleKonaklieva, M. I., & Plotkin, B. J. (2023). Fragment-Based Lead Discovery Strategies in Antimicrobial Drug Discovery. Antibiotics, 12(2), 315. https://doi.org/10.3390/antibiotics12020315