
Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus aureus Clinical Isolates


Abstract
:1. Introduction
2. Results
2.1. Isolation and Identification of Isolates
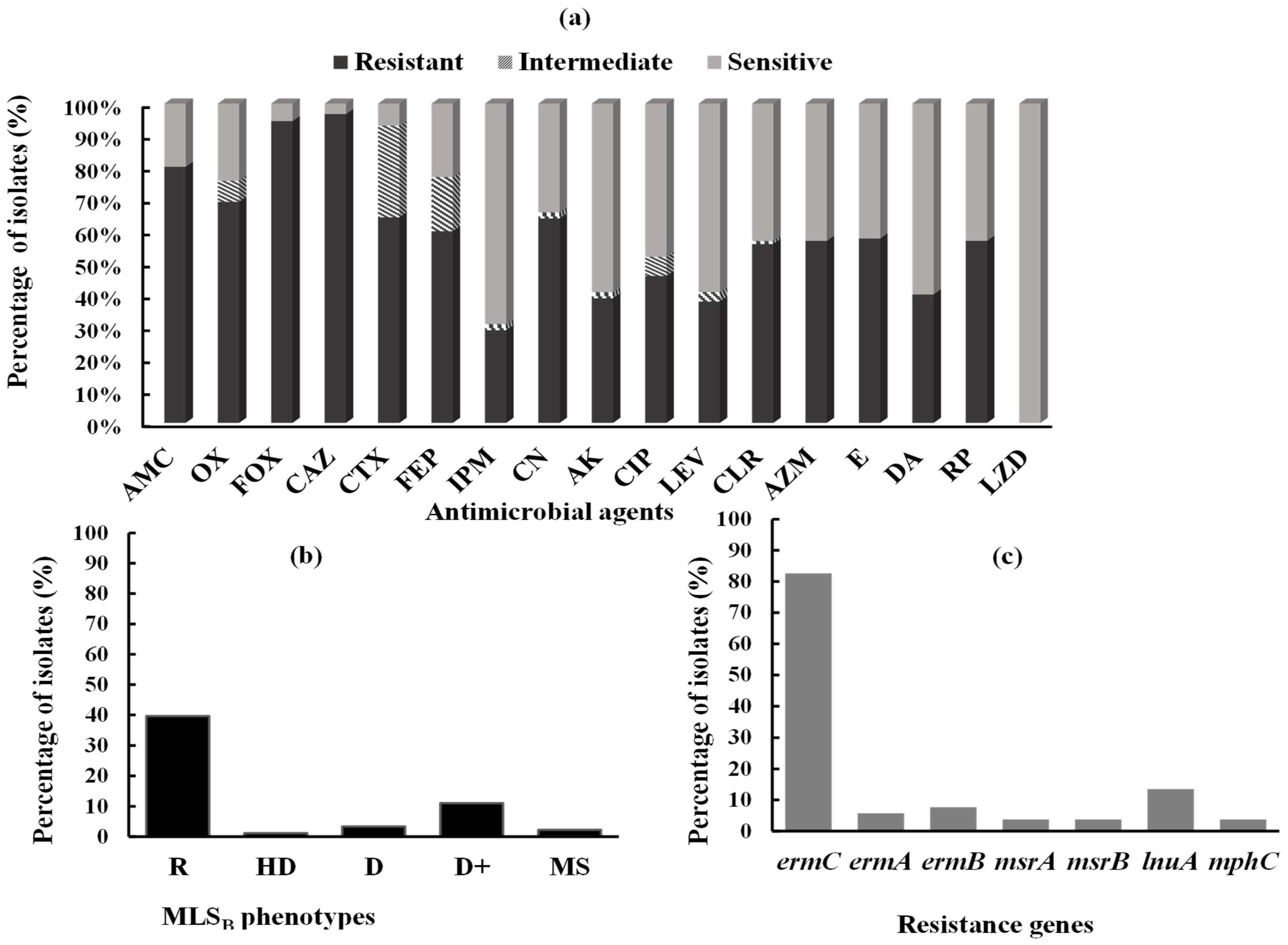
2.2. Antimicrobial Susceptibility Testing
2.3. Phenotypic Detection of MLSB Phenotypes
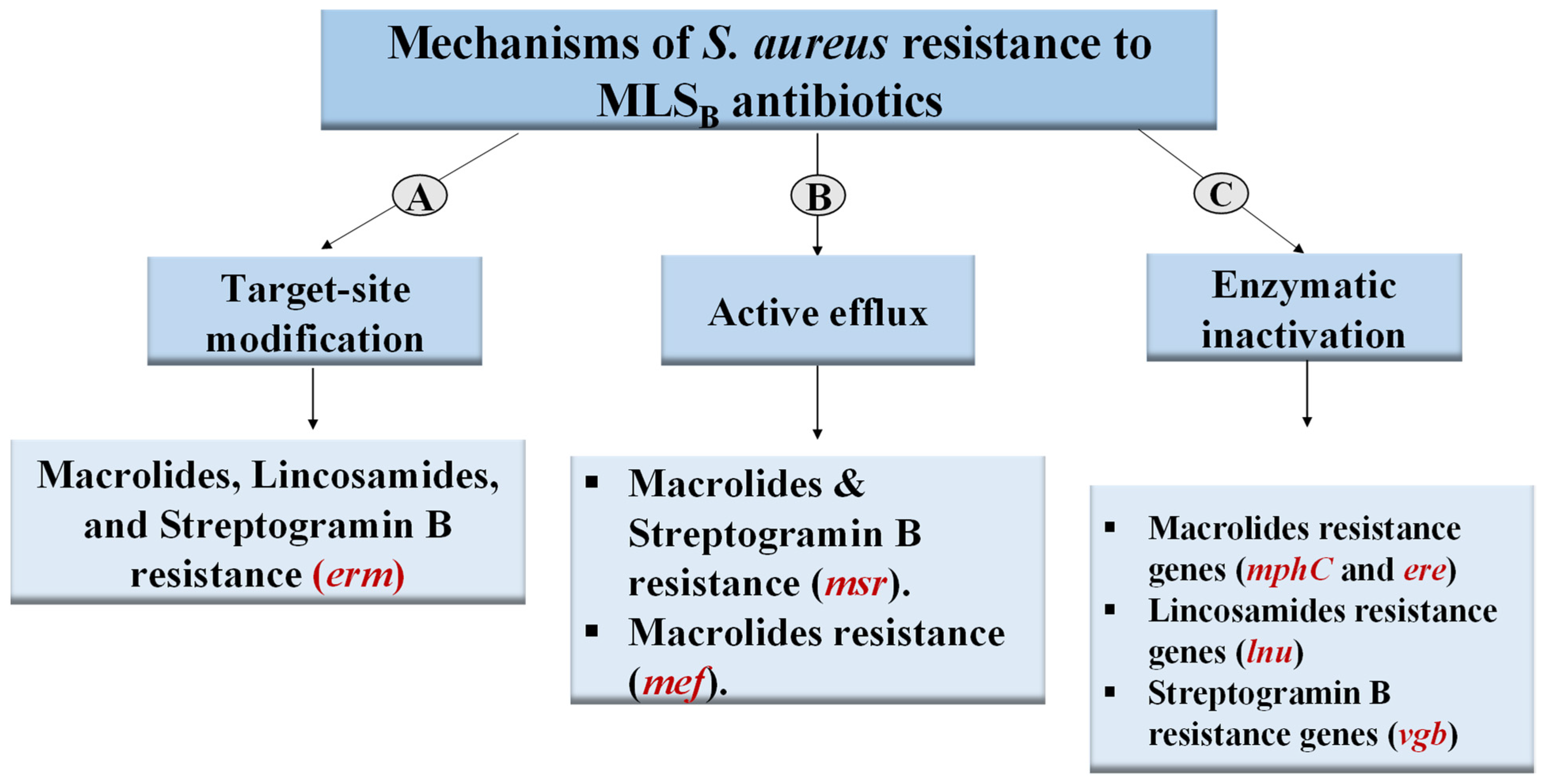
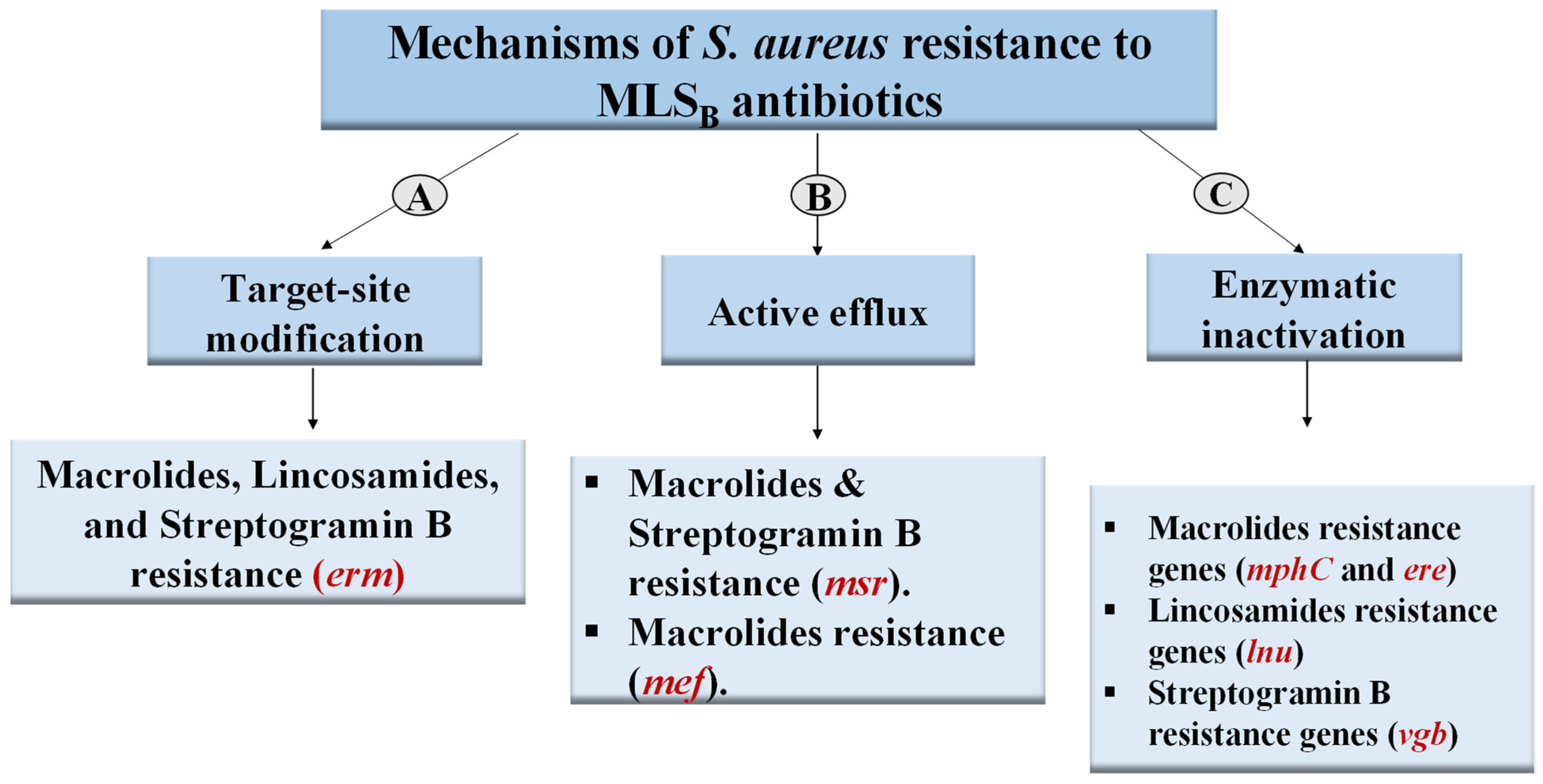
2.4. Prevalence of MLSB-Resistance Genes
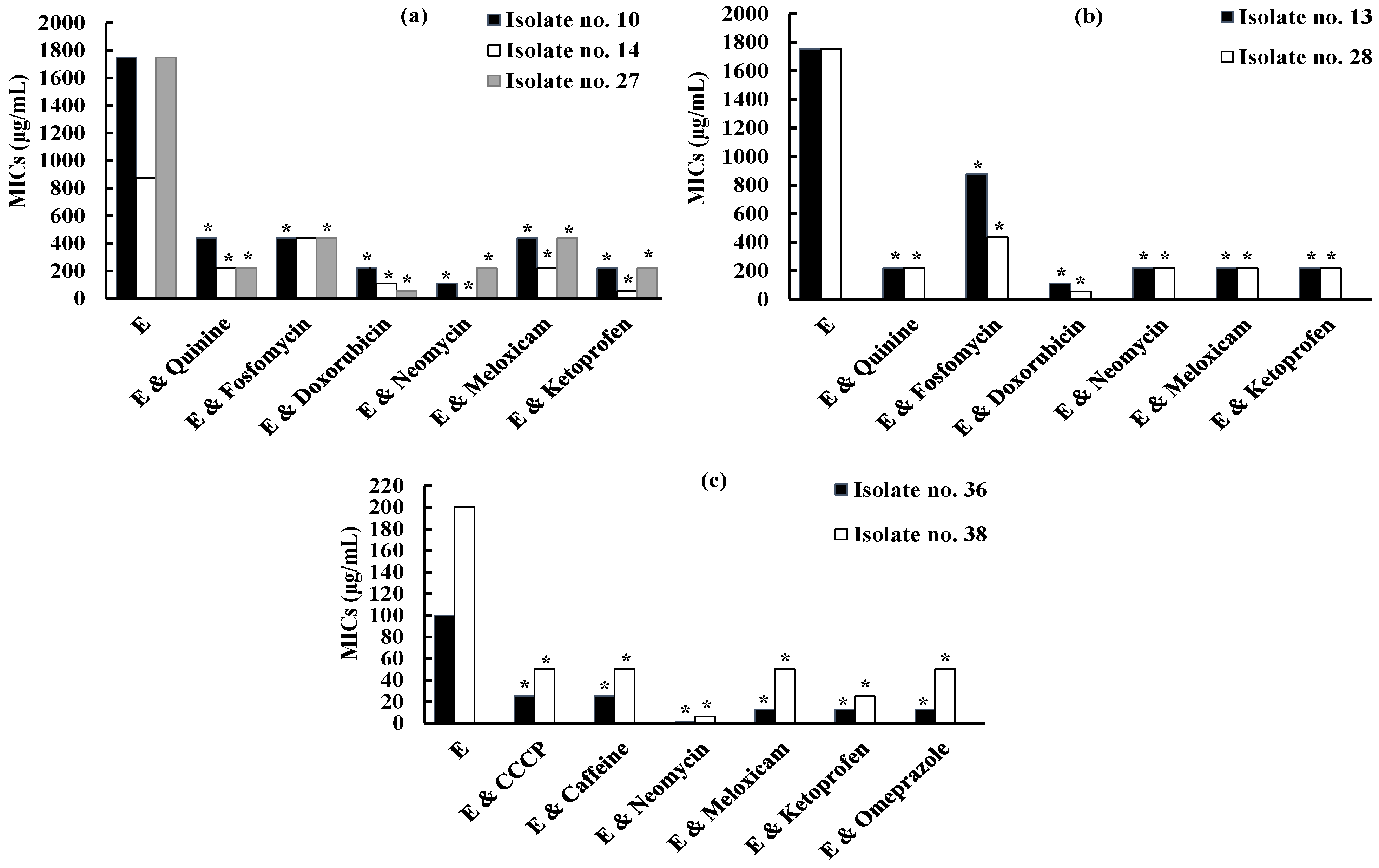
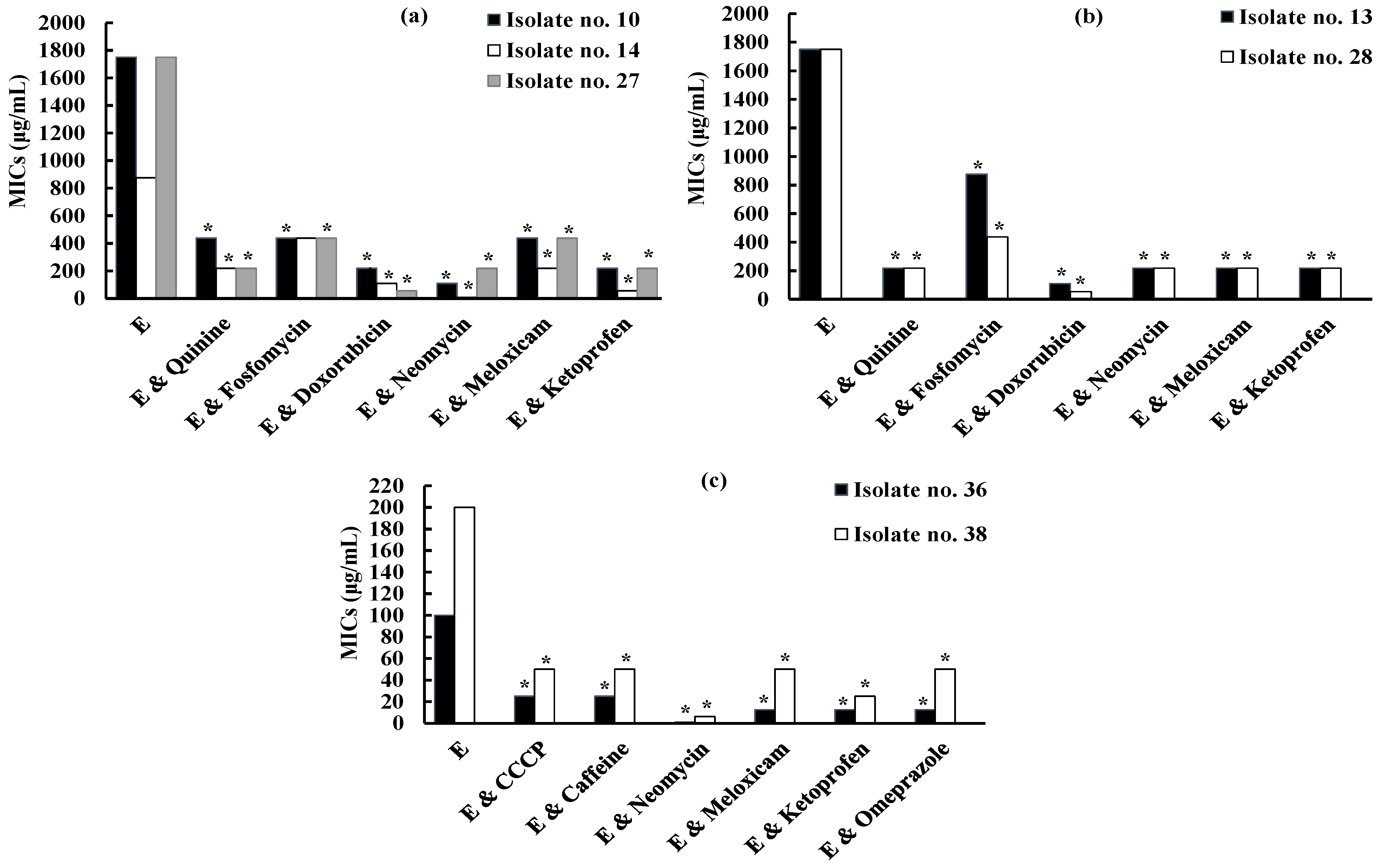
2.5. Screening for the Effect of Different Compounds on Erythromycin Resistance
2.5.1. Screening for the Effect of Different Compounds on Erythromycin Resistance against iMLSB S. aureus Isolates
2.5.2. Screening for the Effect of Different Compounds on Erythromycin Resistance against MS S. aureus Isolates
2.6. Screening for the Effect of Different Compounds on Inducible Clindamycin Resistance against iMLSB S. aureus Isolates
2.7. Checkerboard Microdilution Assay
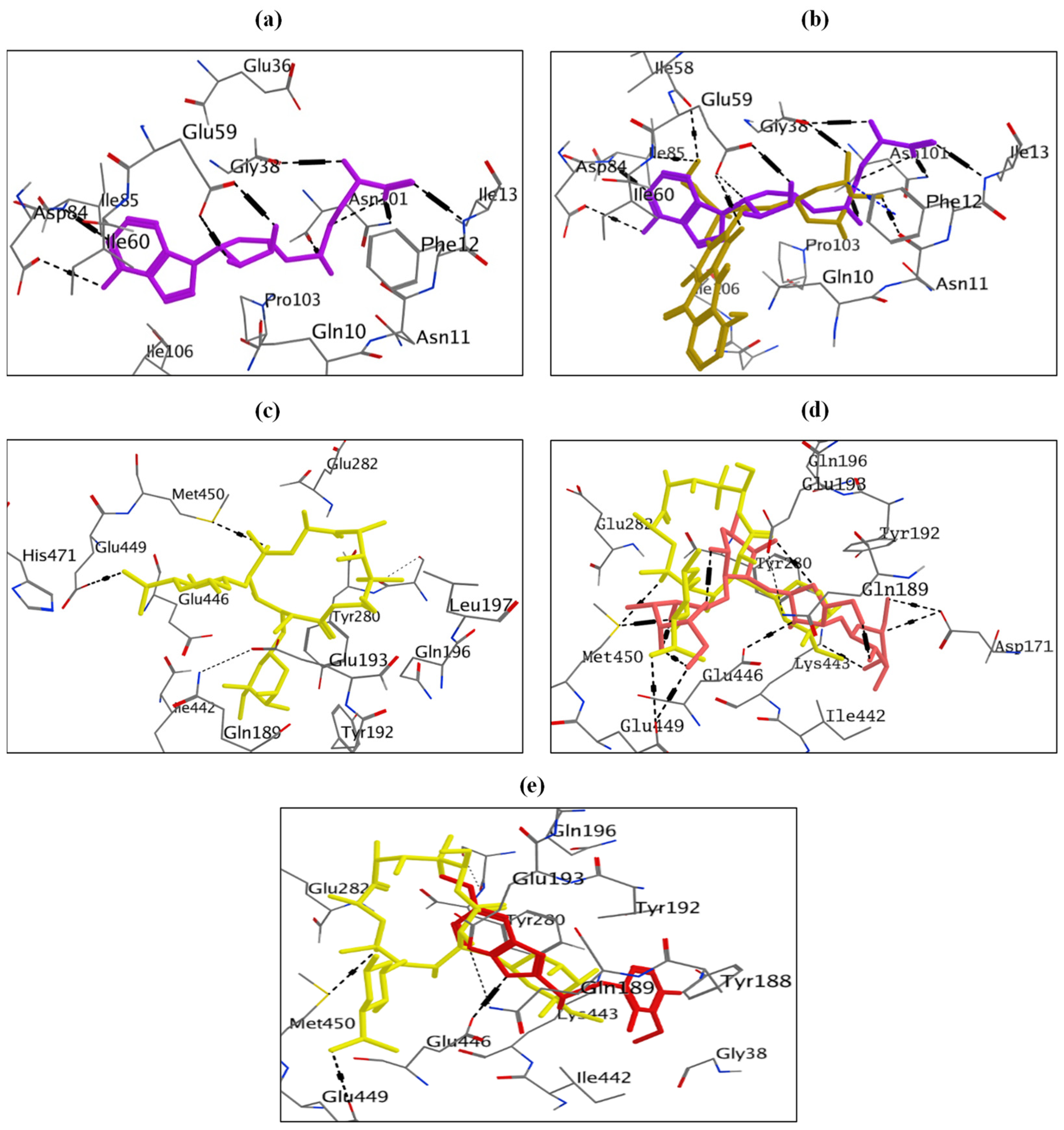
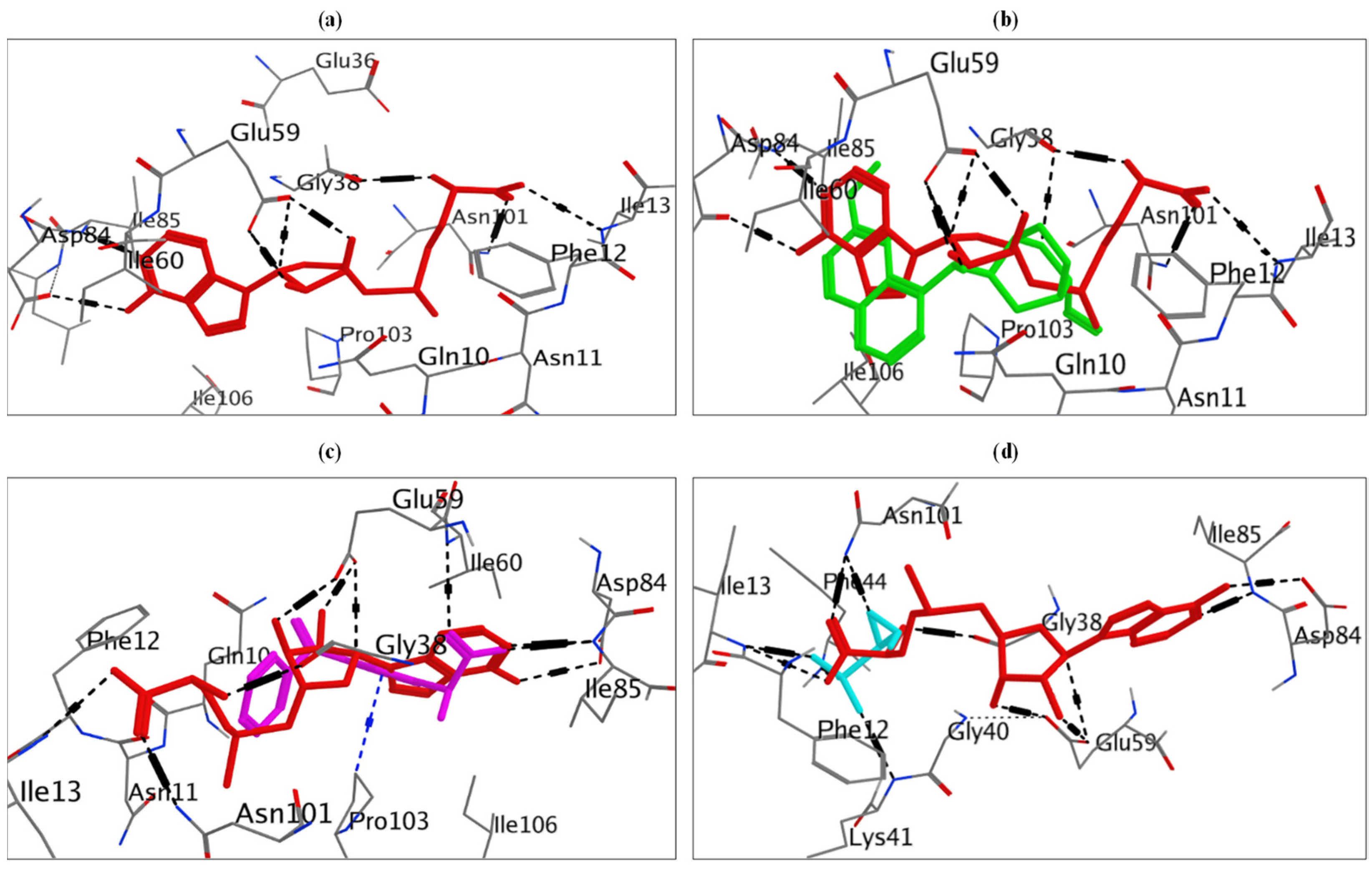
2.8. Molecular Docking Study for Potential Inhibitors of Erythromycin Resistance
2.8.1. Molecular Docking Study of Doxorubicin at S-Adenosyl-L-Methionine (SAM)-Binding Site of ErmC’ Protein
2.8.2. Molecular Docking Study of Neomycin and Omeprazole at Binding Site of MsrA Protein
2.9. Molecular Docking Study of Quinine, Fosfomycin and Ketoprofen at SAM-Binding Site of ErmC’ Protein
3. Discussion
4. Materials and Methods
4.1. Isolation and Identification of S. aureus Clinical Isolates
4.2. Antimicrobial Susceptibility Testing
4.3. Phenotypic Detection of MLSB Phenotypes
4.4. Detection of MLSB Determinants by PCR
4.5. Screening for the Effect of Different Compounds on Erythromycin Resistance
4.6. Screening for the Effect of Different Compounds on Inducible Clindamycin Resistance
4.6.1. Induction of Clindamycin Resistance by Erythromycin
4.6.2. The Effect of Tested Compounds on Erythromycin-Induced Resistance to Clindamycin
4.7. Checkerboard Microdilution Method
4.8. Docking Studies
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bitrus, A.; Peter, O.; Abbas, M.; Goni, M. Staphylococcus aureus: A review of antimicrobial resistance mechanisms. Vet. Sci. Res. J. 2018, 4, 43–54. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, K. Pathogenesis of Staphylococcus aureus. In Staphylococcus Aureus, 1st ed.; Fetsch, A., Ed.; Elsevier: Berlin, Germany, 2018; pp. 13–38. [Google Scholar]
- Che Hamzah, A.M.; Yeo, C.C.; Puah, S.M.; Chua, K.H.; Chew, C.H. Staphylococcus aureus infections in Malaysia: A review of antimicrobial resistance and characteristics of the clinical isolates, 1990–2017. Antibiotics 2019, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, J.; Boldock, E.; Prince, L.R.; Renshaw, S.A.; Whyte, M.K.; Foster, S.J. Identification of Staphylococcus aureus factors required for pathogenicity and growth in human blood. Infect. Immun. 2017, 85, e00337-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, W.; Kirst, H.A. Macrolide Antibiotics; Springer Science & Business Media: Cham, Switzerland, 2002. [Google Scholar]
- Roberts, M.C.; Sutcliffe, J.; Courvalin, P.; Jensen, L.B.; Rood, J.; Seppala, H. Nomenclature for macrolide and macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob. Agents Chemother. 1999, 43, 2823–2830. [Google Scholar] [CrossRef] [Green Version]
- Aktas, Z.; Aridogan, A.; Kayacan, C.B.; Aydin, D. Resistance to macrolide, lincosamide and streptogramin antibiotics in staphylococci isolated in Istanbul, Turkey. J. Microbiol. 2007, 45, 286–290. [Google Scholar]
- Reynolds, E.; Ross, J.I.; Cove, J.H. Msr (A) and related macrolide/streptogramin resistance determinants: Incomplete transporters? Int. J. Antimicrob. Agents 2003, 22, 228–236. [Google Scholar] [CrossRef]
- Luna, V.A.; Coates, P.; Eady, E.A.; Cove, J.H.; Nguyen, T.T.; Roberts, M.C. A variety of gram-positive bacteria carry mobile mef genes. J. Antimicrob. Chemother. 1999, 44, 19–25. [Google Scholar] [CrossRef]
- Matsuoka, M.; Inoue, M.; Endo, Y.; Nakajima, Y. Characteristic expression of three genes, msr (A), mph (C) and erm (Y), that confer resistance to macrolide antibiotics on Staphylococcus aureus. FEMS Microbiol. Lett. 2003, 220, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Zelazny, A.M.; Ferraro, M.J.; Glennen, A.; Hindler, J.F.; Mann, L.M.; Munro, S.; Murray, P.R.; Reller, L.B.; Tenover, F.C.; Jorgensen, J.H. Selection of strains for quality assessment of the disk induction method for detection of inducible clindamycin resistance in staphylococci: A CLSI collaborative study. J. Clin. Microbiol. 2005, 43, 2613–2615. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saribas, Z.; Tunckanat, F.; Pinar, A. Prevalence of erm genes encoding macrolide-lincosamide-streptogramin (MLS) resistance among clinical isolates of Staphylococcus aureus in a Turkish university hospital. Clin. Microbiol. Infect. 2006, 12, 797–799. [Google Scholar] [CrossRef] [Green Version]
- Lina, G.; Quaglia, A.; Reverdy, M.-E.; Leclercq, R.; Vandenesch, F.; Etienne, J. Distribution of genes encoding resistance to macrolides, lincosamides, and streptogramins among staphylococci. Antimicrob. Agents Chemother. 1999, 43, 1062–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ounissi, H.; Courvalin, P. Nucleotide sequence of the gene ereA encoding the erythromycin esterase in Escherichia coli. Gene 1985, 35, 271–278. [Google Scholar] [CrossRef]
- Chesneau, O.; Tsvetkova, K.; Courvalin, P. Resistance phenotypes conferred by macrolide phosphotransferases. FEMS Microbiol. Lett. 2007, 269, 317–322. [Google Scholar] [CrossRef]
- Petinaki, E.; Papagiannitsis, C. Resistance of Staphylococci to Macrolides-Lincosamides-Streptogramins B (MLS): Epidemiology and Mechanisms of Resistance. In Staphylococcus Aureus; InTech Open: Rijeka, Croatia, 2019; Volume 117. [Google Scholar] [CrossRef] [Green Version]
- Brisson-Noël, A.; Delrieu, P.; Samain, D.; Courvalin, P. Inactivation of lincosaminide antibiotics in Staphylococcus. Identification of lincosaminide O-nucleotidyltransferases and comparison of the corresponding resistance genes. J. Biol. Chem. 1988, 263, 15880–15887. [Google Scholar] [CrossRef]
- Allignet, J.; El Solh, N. Comparative analysis of staphylococcal plasmids carrying three streptogramin-resistance genes: vat–vgb–vga. Plasmid 1999, 42, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Daurel, C.; Huet, C.; Dhalluin, A.; Bes, M.; Etienne, J.; Leclercq, R. Differences in potential for selection of clindamycin-resistant mutants between inducible erm (A) and erm (C) Staphylococcus aureus genes. J. Clin. Microbiol. 2008, 46, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Drinkovic, D.; Fuller, E.R.; Shore, K.P.; Holland, D.J.; Ellis-Pegler, R. Clindamycin treatment of Staphylococcus aureus expressing inducible clindamycin resistance. J. Antimicrob. Chemother. 2001, 48, 315–316. [Google Scholar] [CrossRef] [Green Version]
- Siberry, G.K.; Tekle, T.; Carroll, K.; Dick, J. Failure of clindamycin treatment of methicillin-resistant Staphylococcus aureus expressing inducible clindamycin resistance in vitro. Clin. Infect. Dis. 2003, 37, 1257–1260. [Google Scholar] [CrossRef]
- Gould, I. Antibiotic resistance: The perfect storm. Int. J. Antimicrob. Agents 2009, 34, S2–S5. [Google Scholar] [CrossRef]
- Clancy, J.; Schmieder, B.J.; Petitpas, J.W.; Manousos, M.; Williams, J.A.; Faiella, J.A.; Girard, A.E.; Mcguirk, P.R. Assays to detect and characterize synthetic agents that inhibit the ErmC methyltransferase. J. Antibiot. 1995, 48, 1273–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahirrao, P.; Tambat, R.; Chandal, N.; Mahey, N.; Kamboj, A.; Jain, U.K.; Singh, I.P.; Jachak, S.M.; Nandanwar, H.S. MsrA efflux pump inhibitory activity of Piper cubeba lf and its phytoconstituents against Staphylococcus aureus RN4220. Chem. Biodivers. 2020, 17, e2000144. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, F.G.; Parente, R.E.L.; Cavalcante-Figueredo, M.R.; Figueiredo, J.G.; da Silva, R.L.P.; Ferreira Matias, E.F.; Silva, T.M.S.; Camara, C.A.; de Morais Oliveira-Tintino, C.D.; Tintino, S.R. Inhibition of Staphylococcus aureus TetK and MsrA efflux pumps by hydroxyamines derived from lapachol and norlachol. J. Bioenerg. Biomembr. 2021, 53, 149–156. [Google Scholar] [CrossRef]
- Smith, E.C.; Kaatz, G.W.; Seo, S.M.; Wareham, N.; Williamson, E.M.; Gibbons, S. The phenolic diterpene totarol inhibits multidrug efflux pump activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 4480–4483. [Google Scholar] [CrossRef] [Green Version]
- Fung, K.; Han, Q.; Ip, M.; Yang, X.; Lau, C.B.; Chan, B.C. Synergists from Portulaca oleracea with macrolides against methicillin-resistant Staphylococcus aureus and related mechanism. Hong Kong Med. J. 2017, 23, 38–42. [Google Scholar] [PubMed]
- Kaatz, G.W.; Seo, S.M.; Ruble, C.A. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1993, 37, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Saber, N.; Kandala, N.J. The inhibitory effect of fluphenazinedecanoate and caffeine on Staphylococcus aureus efflux pumps. Curr. Res. Microbiol. Biotechnol. 2018, 6, 1530–1535. [Google Scholar]
- Kumar, A.; Khan, I.A.; Koul, S.; Koul, J.L.; Taneja, S.C.; Ali, I.; Ali, F.; Sharma, S.; Mirza, Z.M.; Kumar, M. Novel structural analogues of piperine as inhibitors of the NorA efflux pump of Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 61, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Vidaillac, C.; Guillon, J.; Arpin, C.; Forfar-Bares, I.; Ba, B.B.; Grellet, J.; Moreau, S.; Caignard, D.-H.; Jarry, C.; Quentin, C. Synthesis of omeprazole analogues and evaluation of these as potential inhibitors of the multidrug efflux pump NorA of Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintino, S.R.; Morais-Tintino, C.D.; Campina, F.F.; Pereira, R.L.; Costa, M.d.S.; Braga, M.F.; Limaverde, P.W.; Andrade, J.C.; Siqueira-Junior, J.P.; Coutinho, H.D.M. Action of cholecalciferol and alpha-tocopherol on Staphylococcus aureus efflux pumps. EXCLI J. 2016, 15, 315–322. [Google Scholar] [PubMed]
- Tintino, S.R.; Oliveira-Tintino, C.D.; Campina, F.F.; Weslley Limaverde, P.; Pereira, P.S.; Siqueira-Junior, J.P.; Coutinho, H.D.; Quintans-Júnior, L.J.; da Silva, T.G.; Leal-Balbino, T.C. Vitamin K enhances the effect of antibiotics inhibiting the efflux pumps of Staphylococcus aureus strains. Med. Chem. Res. 2018, 27, 261–267. [Google Scholar] [CrossRef]
- Aeschlimann, J.R.; Dresser, L.D.; Kaatz, G.W.; Rybak, M.J. Effects of NorA inhibitors on in vitro antibacterial activities and postantibiotic effects of levofloxacin, ciprofloxacin, and norfloxacin in genetically related strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 1999, 43, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharate, J.B.; Singh, S.; Wani, A.; Sharma, S.; Joshi, P.; Khan, I.A.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B. Discovery of 4-acetyl-3-(4-fluorophenyl)-1-(p-tolyl)-5-methylpyrrole as a dual inhibitor of human P-glycoprotein and Staphylococcus aureus Nor A efflux pump. Org. Biomol. Chem. 2015, 13, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Brooks, L.; Beckley, A.; Colquhoun, J.; Dewhurst, S.; Dunman, P.M. Neomycin sulfate improves the antimicrobial activity of mupirocin-based antibacterial ointments. Antimicrob. Agents Chemother. 2016, 60, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Rumynska, T.; Hural, A.; Konechnyi, Y.; Vynnytska, R.; Lozynskyi, A.; Salyha, Y.; Korniychuk, O.; Lesyk, R. Microbial biofilms and some aspects of anti-inflammatory drug use. Biopolym. Cell 2021, 37, 247–258. [Google Scholar] [CrossRef]
- Khalaf, A.; Kamal, M.; Mokhtar, S.; Mohamed, H.; Salah, I.; Abbas, R.; Ali, S.; Abd El-Baky, R.M. Antibacterial, anti-biofilm activity of some non-steroidal anti-inflammatory drugs and N-acetyl cysteine against some biofilm producing uropathogens. Am. J. Epidemiol. 2015, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Oselin, K.; Anier, K. Inhibition of human thiopurine S-methyltransferase by various nonsteroidal anti-inflammatory drugs in vitro: A mechanism for possible drug interactions. Drug Metab. Dispos. 2007, 35, 1452–1454. [Google Scholar] [CrossRef] [Green Version]
- Abreu, A.C.; Saavedra, M.J.; Simões, L.C.; Simões, M. Combinatorial approaches with selected phytochemicals to increase antibiotic efficacy against Staphylococcus aureus biofilms. Biofouling 2016, 32, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Olateju, O.A.; Babalola, C.P.; Olubiyi, O.O.; Kotila, O.A.; Kwasi, D.A.; Oaikhena, A.O.; Okeke, I.N.J. Quinoline antimalarials increase the antibacterial activity of ampicillin. Front. Microbiol. 2021, 12, 556550. [Google Scholar] [CrossRef] [PubMed]
- Bodet, C., 3rd; Jorgensen, J.; Drutz, D. Antibacterial activities of antineoplastic agents. Antimicrob. Agents Chemother. 1985, 28, 437–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, V.; Oppenheim, B. Antimicrobial activity of cytotoxic drugs may influence isolation of bacteria and fungi from blood cultures. J. Clin. Pathol. 1993, 46, 1124–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieringer, J.H.; Wenz, A.F.; Just, H.-M.; Daschner, F.D. Effect of 5-fluorouracil, mitoxantrone, methotrexate, and vincristine on the antibacterial activity of ceftriaxone, ceftazidime, cefotiam, piperacillin, and netilmicin. Chemotherapy 1986, 32, 418–424. [Google Scholar] [CrossRef]
- Yokochi, T.; Robertson, K.D. Doxorubicin inhibits DNMT1, resulting in conditional apoptosis. Mol. Pharmacol. 2004, 66, 1415–1420. [Google Scholar] [CrossRef]
- Chuang-Smith, O.N.; Schlievert, P.M. Staphylococcal Enterotoxin C Subtypes Are Differentially Associated with Human Infections and Immunobiological Activities. mSphere 2021, 6, e01153-20. [Google Scholar] [CrossRef]
- Mehraj, J.; Witte, W.; Akmatov, M.K.; Layer, F.; Werner, G.; Krause, G. Epidemiology of Staphylococcus aureus nasal carriage patterns in the community. Curr. Top. Microbiol. Immunol. 2016, 398, 55–87. [Google Scholar] [CrossRef]
- Kuehl, R.; Morata, L.; Meylan, S.; Mensa, J.; Soriano, A. When antibiotics fail: A clinical and microbiological perspective on antibiotic tolerance and persistence of Staphylococcus aureus. J. Antimicrob. Chemother. 2020, 75, 1071–1086. [Google Scholar] [CrossRef]
- Razeghi, M.; Saffarian, P.; Goudarzi, M. Incidence of inducible clindamycin resistance and antibacterial resistance genes variability in clinical Staphylococcus aureus strains: A two-year multicenter study in Tehran, Iran. Gene Rep. 2019, 16, 100411. [Google Scholar] [CrossRef]
- Ahmed, S.; Ahmed, S.; Mohamed, W.; Feky, M.; Daef, E.; Badary, M.; Hetta, H. Nosocomial vancomycin and methicillin resistant staphylococcal infections in intensive care units in Assiut University Hospitals. Egypt. J. Med. Microbiol. 2011, 20, 127–140. [Google Scholar]
- Guzmán-Blanco, M.; Mejía, C.; Isturiz, R.; Alvarez, C.; Bavestrello, L.; Gotuzzo, E.; Labarca, J.; Luna, C.M.; Rodríguez-Noriega, E.; Salles, M.J. Epidemiology of meticillin-resistant Staphylococcus aureus (MRSA) in Latin America. Int. J. Antimicrob. Agents 2009, 34, 304–308. [Google Scholar] [CrossRef]
- Jiménez Quiceno, J.N.; Ocampo Ríos, A.M.; Vanegas Múnera, J.M.; Rodríguez Tamayo, E.A.; Mediavilla, J.; Chen, L.; Muskus, C.; Vélez, L.; Rojas, C.; Restrepo Gouzy, A. CC8 MRSA strains harboring SCCmec Type IVc are predominant in Colombian hospitals. PLoS ONE 2012, 7, e38576. [Google Scholar] [CrossRef]
- Youssef, C.R.; Kadry, A.A.; Shaker, G.H.; El-Ganiny, A.M. The alarming association between antibiotic resistance and reduced susceptibility to biocides in nosocomial MRSA isolates from two regional hospitals in Egypt. Arch. Microbiol. 2021, 203, 3295–3303. [Google Scholar] [CrossRef] [PubMed]
- Kishk, R.M.; Anani, M.M.; Nemr, N.A.; Soliman, N.M.; Fouad, M.M. Inducible clindamycin resistance in clinical isolates of staphylococcus aureus in Suez Canal University Hospital, Ismailia, Egypt. J. Infect. Dev. Ctries. 2020, 14, 1281–1287. [Google Scholar] [CrossRef]
- Adhikari, R.; Shrestha, S.; Barakoti, A.; Amatya, R. Inducible clindamycin and methicillin resistant Staphylococcus aureus in a tertiary care hospital, Kathmandu, Nepal. BMC Infect. Dis. 2017, 17, 483. [Google Scholar] [CrossRef] [PubMed]
- Majhi, S.; Dash, M.; Mohapatra, D.; Mohapatra, A.; Chayani, N. Detection of inducible and constitutive clindamycin resistance among Staphylococcus aureus isolates in a tertiary care hospital, Eastern India. Avicenna J. Med. 2016, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, A.M.C.; Yeo, C.C.; Puah, S.M.; Chua, K.H.; Rahman, N.I.A.; Abdullah, F.H.; Othman, N.; Chew, C.H. Tigecycline and inducible clindamycin resistance in clinical isolates of methicillin-resistant Staphylococcus aureus from Terengganu, Malaysia. J. Med. Microbiol. 2019, 68, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Jarajreh, D.A.; Aqel, A.; Alzoubi, H.; Al-Zereini, W. Prevalence of inducible clindamycin resistance in methicillin-resistant Staphylococcus aureus: The first study in Jordan. J. Infect. Dev. Ctries. 2017, 11, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Sarrou, S.; Malli, E.; Tsilipounidaki, K.; Florou, Z.; Medvecky, M.; Skoulakis, A.; Hrabak, J.; Papagiannitsis, C.C.; Petinaki, E. MLSB-resistant Staphylococcus aureus in central Greece: Rate of resistance and molecular characterization. Microb. Drug Resist. 2019, 25, 543–550. [Google Scholar] [CrossRef]
- Mama, M.; Aklilu, A.; Misgna, K.; Tadesse, M.; Alemayehu, E. Methicillin-and inducible clindamycin-resistant Staphylococcus aureus among patients with wound infection attending Arba Minch Hospital, South Ethiopia. Int. J. Microbiol. 2019, 2019, 2965490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maravic, G. Macrolide resistance based on the Erm-mediated rRNA methylation. Curr. Drug Targets Infect. Disord. 2004, 4, 193–202. [Google Scholar] [CrossRef]
- Elkammoshi, A.M.; Ghasemzadeh-Moghaddam, H.; Nordin, S.A.; Taib, N.M.; Subbiah, S.K.; Neela, V.; Hamat, R.A. A low prevalence of inducible macrolide, lincosamide, and streptogramin b resistance phenotype among methicillin-susceptible Staphylococcus aureus isolated from malaysian patients and healthy individuals. Jundishapur J. Microbiol. 2016, 9, e37148. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.N.d.P.; Rabelo, M.A.; Lima, J.L.d.C.; Neto, A.M.B.; Lopes, A.C.d.S.; Maciel, M.A.V. Phenotypic and molecular characterization of resistance to macrolides, lincosamides and type B streptogramin of clinical isolates of Staphylococcus spp. of a university hospital in Recife, Pernambuco, Brazil. Braz. J. Infect. Dis. 2016, 20, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, R.; Fernández-Barat, L.; Motos, A.; López-Aladid, R.; Vázquez, N.; Panigada, M.; Álvarez-Lerma, F.; López, Y.; Muñoz, L.; Castro, P. Molecular characterization of methicillin-resistant Staphylococcus aureus clinical strains from the endotracheal tubes of patients with nosocomial pneumonia. Antimicrob. Resist. Infect. Control 2020, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Lüthje, P.; Schwarz, S. Antimicrobial resistance of coagulase-negative staphylococci from bovine subclinical mastitis with particular reference to macrolide–lincosamide resistance phenotypes and genotypes. J. Antimicrob. Chemother. 2006, 57, 966–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaku, P.; Lebughe, M.; Strauß, L.; Peters, G.; Herrmann, M.; Mumba, D.; Mellmann, A.; Muyembe-Tamfum, J.-J.; Schaumburg, F. Unveiling the molecular basis of antimicrobial resistance in Staphylococcus aureus from the Democratic Republic of the Congo using whole genome sequencing. Clin. Microbiol. Infect. 2016, 22, 644.e1–644.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akpaka, P.E.; Roberts, R.; Monecke, S. Molecular characterization of antimicrobial resistance genes against Staphylococcus aureus isolates from Trinidad and Tobago. J. Infect. Public Health 2017, 10, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, M.; Al Nasbeh, A.; Rafei, R.; Mallat, H.; Achkar, M.; Dabboussi, F.; Hamze, M. Characterization of resistance genes to macrolides, lincosamides and streptogramins (MLS) among clinical isolates of Staphylococcus aureus in North Lebanon. Int. Arab. J. Antimicrob. Agents 2015, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Lüthje, P.; Schwarz, S. Molecular basis of resistance to macrolides and lincosamides among staphylococci and streptococci from various animal sources collected in the resistance monitoring program BfT-GermVet. Int. J. Antimicrob. Agents 2007, 29, 528–535. [Google Scholar] [CrossRef]
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Michael, G.B.; Feßler, A.T.; Vester, B. Lincosamides, streptogramins, phenicols, and pleuromutilins: Mode of action and mechanisms of resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a027037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Tu, C.; Tan, C.; El-Sayed, M.A.E.-G.; Dai, M.; Xia, Y.; Liu, Y.; Zhong, L.-L.; Shen, C.; Chen, G. Antimicrobial resistance, virulence genes profiling and molecular relatedness of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in Guangdong Province, China. Infect. Drug Resist. 2019, 12, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, S.; Lozano, C.; Aspiroz, C.; Ruiz-Ripa, L.; Eguizábal, P.; Campaña-Burguet, A.; Cercenado, E.; López-Calleja, A.I.; Castillo, J.; Azcona-Gutiérrez, J.M. Beyond CC398: Characterisation of Other Tetracycline and Methicillin-Resistant Staphylococcus aureus Genetic Lineages Circulating in Spanish Hospitals. Pathogens 2022, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Yebra, M.J.; Sanchez, J.; Martin, C.G.; Hardisson, C.; Barbes, C. The effect of sinefungin and synthetic analogues on RNA and DNA methyltransferases from Streptomyces. J. Antibiot. 1991, 44, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Barbes, C.; Sanchez, J.; Yebra, M.; Robert-Gero, M.; Hardisson, C. Effects of sinefungin and S-adenosylhomocysteine on DNA and protein methyltransferases from Streptomyces and other bacteria. FEMS Microbiol. Lett. 1990, 69, 239–243. [Google Scholar] [CrossRef]
- Schluckebier, G.; Zhong, P.; Stewart, K.D.; Kavanaugh, T.J.; Abad-Zapatero, C. The 2.2 Å structure of the rRNA methyltransferase ErmC′ and its complexes with cofactor and cofactor analogs: Implications for the reaction mechanism. J. Mol. Biol. 1999, 289, 277–291. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Syroegin, E.A.; Aleksandrova, E.V.; Atkinson, G.C.; Gregory, S.T.; Mankin, A.S.; Polikanov, Y.S. Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance. Nat. Chem. Biol. 2021, 17, 412–420. [Google Scholar] [CrossRef]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Cousin, M.A.; Ojasoo, T.; Raynaud, J.P. Paromomycin and dihydrostreptomycin binding to Escherichia coli ribosomes. Eur. J. Biochem. 1976, 66, 597–606. [Google Scholar] [CrossRef]
- Vázquez, D. Protein synthesis and translation inhibitors. In Inhibitors of Protein Biosynthesis, 30th ed.; Kleinzeller, A., Springer, G.F., Wittmann, H.G., Eds.; Springer: New York, NY, USA, 1979; pp. 1–14. [Google Scholar]
- Dahlberg, A.E.; Horodyski, F.; Keller, P. Interaction of neomycin with ribosomes and ribosomal ribonucleic acid. Antimicrob. Agents Chemother. 1978, 13, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, L.K.; Edwards, T.A.; O’Neill, A.J. ABC-F proteins mediate antibiotic resistance through ribosomal protection. MBio 2016, 7, e01975–e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, S.; Oluwatuyi, M.; Kaatz, G.W. A novel inhibitor of multidrug efflux pumps in Staphylococcus aureus. J. Antimicrob. Chemother. 2003, 51, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, S. Anti-staphylococcal plant natural products. Nat. Prod. Rep. 2004, 21, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Maton, P.N. Omeprazole. N. Engl. J. Med. 1991, 324, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Spížek, J.; Řezanka, T. Lincomycin, clindamycin and their applications. Appl. Microbiol. Biotechnol. 2004, 64, 455–464. [Google Scholar] [CrossRef]
- Lee, H.J.; Jhang, S.T.; Jin, H.J. Potential Target Site for Inhibitors in MLSB Antibiotic Resistance. Antibiotics 2021, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Vandepitte, J.; Verhaegen, J.; Engbaek, K.; Piot, P.; Heuck, C.C.; Rohner, P.; Heuck, C. Basic Laboratory Procedures in Clinical Bacteriology; World Health Organization: Geneva, Switzerland, 2003.
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2021. [Google Scholar]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.; Giske, C.; Harbarth, S.; Hindler, J.; Kahlmeter, G.; Olsson-Liljequist, B. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Steward, C.D.; Raney, P.M.; Morrell, A.K.; Williams, P.P.; McDougal, L.K.; Jevitt, L.; McGowan Jr, J.E.; Tenover, F.C. Testing for induction of clindamycin resistance in erythromycin-resistant isolates of Staphylococcus aureus. J. Clin. Microbiol. 2005, 43, 1716–1721. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [Green Version]
- Tintino, S.R.; Morais-Tintino, C.D.; Campina, F.F.; Costa, M.d.S.; Menezes, I.R.; de Matos, Y.M.L.; Calixto-Júnior, J.T.; Pereira, P.S.; Siqueira-Junior, J.P.; Leal-Balbino, T.C. Tannic acid affects the phenotype of Staphylococcus aureus resistant to tetracycline and erythromycin by inhibition of efflux pumps. Bioorg. Chem. 2017, 74, 197–200. [Google Scholar] [CrossRef]
- Cetin, E.S.; Tekeli, A.; Ozseven, A.G.; Us, E.; Aridogan, B.C. Determination of in vitro activities of polymyxin B and rifampin in combination with ampicillin/sulbactam or cefoperazone/sulbactam against multidrug-resistant Acinetobacter baumannii by the E-test and checkerboard methods. Jpn. J. Infect. Dis. 2013, 66, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.H.; Chen, M.Y.; Victor, L.Y.; Chow, J.W. Synergy assessed by checkerboard a critical analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–349. [Google Scholar] [CrossRef] [PubMed]
- EUCAST. Terminology relating to methods for the determination of susceptibility of bacteria to antimicrobial agents. Clin. Microbiol. Infect. 2000, 6, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2019.01, Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite#910, Montreal, QC, Canada, H3A 2R7. 2019. Available online: https://www.chemcomp.com/Products.htm (accessed on 20 January 2023).
- Matsuoka, M.; Endou, K.; Kobayashi, H.; Inoue, M.; Nakajima, Y. A plasmid that encodes three genes for resistance to macrolide antibiotics in Staphylococcus aureus. FEMS Microbiol. Lett. 1998, 167, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Strommenger, B.; Kettlitz, C.; Werner, G.; Witte, W. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resistance genes in Staphylococcus aureus. J. Clin. Microbiol. 2003, 41, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, R.; Bauduret, F.; Soussy, C. Selection of constitutive mutants of gram-positive cocci inducible resistant to macrolides, lincosamides and streptogramins (MLS): Comparison of the selective effects of the MLS. Pathol. Biol. 1989, 37, 568–572. [Google Scholar]
- Ross, J.; Eady, E.; Cove, J.; Cunliffe, W.; Baumberg, S.; Wootton, J. Inducible erythromycin resistance in staphlyococci is encoded by a member of the ATP-binding transport super-gene family. Mol. Microbiol. 1990, 4, 1207–1214. [Google Scholar] [CrossRef]
- Bozdogan, B.L.; Berrezouga, L.; Kuo, M.-S.; Yurek, D.A.; Farley, K.A.; Stockman, B.J.; Leclercq, R. A new resistance gene, linB, conferring resistance to lincosamides by nucleotidylation in Enterococcus faecium HM1025. Antimicrob. Agents Chemother. 1999, 43, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, J.; Grebe, T.; Tait-Kamradt, A.; Wondrack, L. Detection of erythromycin-resistant determinants by PCR. Antimicrob. Agents Chemother. 1996, 40, 2562–2566. [Google Scholar] [CrossRef] [Green Version]
- Allignet, J.; Loncle, V.; El Solh, N. Sequence of a staphylococcal plasmid gene, vga, encoding a putative ATP-binding protein involved in resistance to virginiamycin A-like antibiotics. Gene 1992, 117, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Loncle, V.; Casetta, A.; Buu-Hoi, A.; El Solh, N. Analysis of pristinamycin-resistant Staphylococcus epidermidis isolates responsible for an outbreak in a Parisian hospital. Antimicrob. Agents Chemother. 1993, 37, 2159–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerum, A.M.; Jensen, L.B.; Aarestrup, F.M.l. Detection of the satA gene and transferability of virginiamycin resistance in Enterococcus faecium from food-animals. FEMS Microbiol. Lett. 1998, 168, 145–151. [Google Scholar] [CrossRef]
- Allignet, J.; Liassine, N.; El Solh, N. Characterization of a staphylococcal plasmid related to pUB110 and carrying two novel genes, vatC and vgbB, encoding resistance to streptogramins A and B and similar antibiotics. Antimicrob. Agents Chemother. 1998, 42, 1794–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Number | Genes Present | Result of D Test |
|---|---|---|
| 1, 3, 4, 5, 6, 7, 8, 9, 21, 22, 23, 24, 25, 26, 29, 30, 31, 33, 34, 35, 39, 40, 41, 42, 43, 44, 45, 46, 48, 50. | ermC | R |
| 2, 37, 49 | ermB | R |
| 10 | ermA | D |
| 11, 15 | ermC, lnuA | D+ |
| 12 | ermC, lnuA | HD |
| 13, 18, 28, 52, 16, 17, 19 | ermC | D+ |
| 14 | ermC | D |
| 20, 32 | ermC, lnuA | R |
| 27 | ermA, lnuA | D |
| 36 | msrA, msrB, mphC, lnuA | MS |
| 38 | msrA, msrB, mphC | MS |
| 47 | ermA | R |
| 51 | ermB | D+ |
| Isolate No. | MIC of E (μg/mL) | The Effect of Potential Inhibitors on Erythromycin Resistance | |||||
|---|---|---|---|---|---|---|---|
| Potential Inhibitor | MICs (μg/mL) | Conc. Of the Inhibitor (μg/mL) | MICs of E in Combination with Inhibitor (μg/mL) | Fold Change | p-Value | ||
| 10 (D) | 1750 | Quinine | 1250 | 625 | 437.5 | 4-fold (-) | 0.003 * |
| 14 (D) | 875 | 1250 | 625 | 218.75 | 4-fold (-) | 0.02 * | |
| 27 (D) | 1750 | 1250 | 625 | 218.75 | 8-fold (-) | 0.003 * | |
| 13 (D+) | 1750 | 1250 | 625 | 218.75 | 8-fold (-) | 0.003 * | |
| 28 (D+) | 1750 | 1250 | 625 | 218.75 | 8-fold (-) | 0.003 * | |
| 10 (D) | 1750 | Fosfomycin | 2 | 1 | 437.5 | 4-fold (-) | 0.02 * |
| 14 (D) | 875 | 2 | 1 | 437.5 | 2-fold (-) | 0.2 | |
| 27 (D) | 1750 | 2 | 1 | 437.5 | 4-fold (-) | 0.02 * | |
| 13 (D+) | 1750 | 2 | 1 | 875 | 2-fold (-) | 0.02 * | |
| 28 (D+) | 1750 | 1 | 0.5 | 437.5 | 4-fold (-) | 0.02 * | |
| 10 (D) | 1750 | Doxorubicin | 8 | 4 | 218.75 | 8-fold (-) | 0.0005 * |
| 14 (D) | 875 | 8 | 4 | 109.375 | 8-fold (-) | 0.0005 * | |
| 27 (D) | 1750 | 8 | 4 | 54.688 | 32-fold (-) | 0.0001 * | |
| 13 (D+) | 1750 | 8 | 4 | 109.375 | 16-fold (-) | 0.0001 * | |
| 28 (D+) | 1750 | 8 | 4 | 54.688 | 32-fold (-) | 0.0001 * | |
| 10 (D) | 1750 | Neomycin | 1 | 0.5 | 109.375 | 16-fold (-) | 0.0005 * |
| 14 (D) | 875 | 2 | 1 | 6.836 | 128-fold (-) | <0.0001 * | |
| 27 (D) | 1750 | 1 | 0.5 | 218.75 | 8-fold (-) | 0.003 * | |
| 13 (D+) | 1750 | 16 | 8 | 218.75 | 8-fold (-) | 0.02 * | |
| 28 (D+) | 1750 | 2 | 1 | 218.75 | 8-fold (-) | 0.004 * | |
| 36 (MS) | 100 | 64 | 32 | 0.781 | 128-fold (-) | <0.0001 * | |
| 38 (MS) | 200 | 0.0625 | 0.3125 | 6.25 | 32-fold (-) | 0.0001 * | |
| 10 (D) | 1750 | Meloxicam | 2048 | 1024 | 437.5 | 4-fold (-) | 0.003 * |
| 14 (D) | 875 | 1024 | 512 | 218.75 | 4-fold (-) | 0.02 * | |
| 27 (D) | 1750 | 2048 | 1024 | 437.5 | 4-fold (-) | 0.003 * | |
| 13 (D+) | 1750 | 2048 | 1024 | 218.75 | 8-fold (-) | 0.003 * | |
| 28 (D+) | 1750 | 2048 | 1024 | 218.75 | 8-fold (-) | 0.003 * | |
| 36 (MS) | 100 | 1024 | 512 | 12.5 | 8-fold (-) | 0.003 * | |
| 38 (MS) | 200 | 128 | 64 | 50 | 4-fold (-) | 0.003 * | |
| 10 (D) | 1750 | Ketoprofen | 3125 | 1562.5 | 218.75 | 8-fold (-) | 0.003 * |
| 14 (D) | 875 | 3125 | 1562.5 | 54.688 | 16-fold (-) | 0.0005 * | |
| 27 (D) | 1750 | 3125 | 1562.5 | 218.75 | 8-fold (-) | 0.003 * | |
| 13 (D+) | 1750 | 3125 | 1562.5 | 218.75 | 8-fold (-) | 0.003 * | |
| 28 (D+) | 1750 | 3125 | 1562.5 | 218.75 | 8-fold (-) | 0.003 * | |
| 36 (MS) | 100 | 3125 | 1562.5 | 12.5 | 8-fold (-) | 0.003 * | |
| 38 (MS) | 200 | 1562.5 | 781.25 | 25 | 8-fold (-) | 0.003 * | |
| 36 (MS) | 100 | CCCP | 2 | 1 | 25 | 4-fold (-) | 0.02 * |
| 38 (MS) | 200 | 2 | 1 | 50 | 4-fold (-) | 0.02 * | |
| 36 (MS) | 100 | Caffeine | 22,000 | 2750 | 25 | 4-fold (-) | 0.02 * |
| 38 (MS) | 200 | 5500 | 2750 | 50 | 4-fold (-) | 0.02 * | |
| 36 (MS) | 100 | Omeprazole | 5000 | 2500 | 12.5 | 8-fold (-) | 0.0005 * |
| 38 (MS) | 200 | 625 | 312.5 | 50 | 4-fold (-) | 0.003 * | |
| Isolate No. | ICR | Fold Change | Inhibition of ICR | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DA (μg/mL) | DA/E (μg/mL) | Potential Inhibitor | MICs (μg/mL) | Conc. of Inhibitor (μg/mL) | MICs of DA/E in Combination with Inhibitor (μg/mL) | Fold Change | p-Value | ||
| 10 (D) | 0.0625 | 1 | 16-fold (+) | Quinine | 1250 | 625 | 0.25 | 4-fold (-) | 0.02 * |
| 14 (D) | 0.0625 | 1 | 1250 | 625 | 0.25 | 4-fold (-) | 0.02 * | ||
| 27 (D) | 0.0625 | 1 | 1250 | 625 | 0.25 | 4-fold (-) | 0.02 * | ||
| 13 (D+) | 0.0625 | 16 | 256-fold (+) | 1250 | 625 | 2 | 8-fold (-) | 0.003 * | |
| 28 (D+) | 0.0625 | 16 | 1250 | 625 | 4 | 4-fold (-) | 0.003 * | ||
| 10 (D) | 0.0625 | 1 | 16-fold (+) | Fosfomycin | 2 | 1 | 0.25 | 4-fold (-) | 0.02 * |
| 14 (D) | 0.0625 | 1 | 2 | 1 | 0.25 | 4-fold (-) | 0.02 * | ||
| 27 (D) | 0.0625 | 1 | 2 | 1 | 0.5 | 2-fold (-) | 0.02 * | ||
| 13 (D+) | 0.0625 | 16 | 256-fold (+) | 2 | 1 | 4 | 4-fold (-) | 0.003 * | |
| 28 (D+) | 0.0625 | 16 | 1 | 0.5 | 4 | 4-fold (-) | 0.003 * | ||
| 10 (D) | 0.0625 | 1 | 16-fold (+) | Ketoprofen | 3125 | 1562.5 | 0.25 | 4-fold (-) | 0.02 * |
| 14 (D) | 0.0625 | 1 | 3125 | 1562.5 | 0.25 | 4-fold (-) | 0.02 * | ||
| 27 (D) | 0.0625 | 1 | 3125 | 1562.5 | 0.25 | 4-fold (-) | 0.02 * | ||
| 13 (D+) | 0.0625 | 16 | 256-fold (+) | 3125 | 1562.5 | 0.5 | 32-fold (-) | 0.0001 * | |
| 28 (D+) | 0.0625 | 16 | 3125 | 1562.5 | 0.5 | 32-fold (-) | 0.0001 * | ||
| Isolate No. | Potential Inhibitor | FICI | Combined Effect | |
|---|---|---|---|---|
| 10 (D) | Quinine | E/Q | 0.625 | Additive |
| Fosfomycin | E/F | 0.75 | Additive | |
| Doxorubicin | E/D | 0.5 | Synergism | |
| Neomycin | E/N | 0.5 | Synergism | |
| Meloxicam | E/M | 0.75 | Additive | |
| Ketoprofen | E/K | 0.625 | Additive | |
| 36 (MS) | CCCP | E/CCCP | 0.625 | Additive |
| Caffeine | E/C | 0.531 | Additive | |
| Omeprazole | E/O | 0.5 | Synergism | |
| Neomycin | E/N | 0.313 | Synergism | |
| Meloxicam | E/M | 0.625 | Additive | |
| Ketoprofen | E/K | 0.625 | Additive | |
| Isolate No. | FICI and Combined Effect of DA/E | Potential Inhibitor | FICI | Combined Effect | |
|---|---|---|---|---|---|
| Isolate no. 10 | 16.668 (antagonism) | Quinine | DA/Q | 1 | Additive |
| DA&E/Q | 0.56 | Additive | |||
| Fosfomycin | DA/F | 0.375 | Synergism | ||
| DA&E/F | 0.56 | Additive | |||
| Ketoprofen | DA/K | 0.5 | Synergism | ||
| DA&E/K | 0.75 | Additive | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahfouz, A.A.; Said, H.S.; Elfeky, S.M.; Shaaban, M.I. Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus aureus Clinical Isolates. Antibiotics 2023, 12, 503. https://doi.org/10.3390/antibiotics12030503
Mahfouz AA, Said HS, Elfeky SM, Shaaban MI. Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus aureus Clinical Isolates. Antibiotics. 2023; 12(3):503. https://doi.org/10.3390/antibiotics12030503
Chicago/Turabian StyleMahfouz, Aya A., Heba S. Said, Sherin M. Elfeky, and Mona I. Shaaban. 2023. "Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus aureus Clinical Isolates" Antibiotics 12, no. 3: 503. https://doi.org/10.3390/antibiotics12030503
APA StyleMahfouz, A. A., Said, H. S., Elfeky, S. M., & Shaaban, M. I. (2023). Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus aureus Clinical Isolates. Antibiotics, 12(3), 503. https://doi.org/10.3390/antibiotics12030503