1. Introduction
Biofilm formation in
Pseudomonas aeruginosa is believed to be responsible for 65% of
P. aeruginosa-related deaths and poses a significant threat to individuals with cystic fibrosis (CF) due to antibiotic resistance [
1]. Therefore, blocking biofilm formation in
P. aeruginosa could help reduce mortality and curb antibiotic resistance. A biofilm consists of bacterial cells embedded in a matrix of extracellular polymeric substances (EPS) composed of lipids, macromolecules, DNA, exopolysaccharides, and proteins [
2], rendering the organisms therein impermeable to antibiotics. Accordingly, the planktonic
P. aeruginosa is 1000 times less resistant to antibiotics than biofilm-embedded
P. aeruginosa [
3,
4,
5]. Biofilm formation also allows
P. aeruginosa to evade recognition by the host immune system [
3], and this is coordinated by its quorum sensing (QS) system, which plays a crucial role in the formation, secretion, processing, and recognition of autoinducer (AL), a cell-to-cell communication molecule implicated in biofilm formation [
6,
7,
8]. At specific density, as measured by the ambient AL concentration, the transcription of bacterial genes in the community becomes synchronized, allowing the organisms to act cooperatively. Thus, virulence factor secretion, swimming motility, secondary metabolite synthesis, biofilm development, and antibiotic resistance in the organism community are all controlled by ALs [
9]. Consequently, disruption of
P. aeruginosa QS-mediated signaling disrupts communication in the organism community, reducing pathogenicity and ultimately eradicating the organism from the host [
10,
11,
12].
Pseudomonas aeruginosa has three main QS systems: LasI-LasR, RhlI-RhlR, and PQS-MvfR, all of which contribute to the formation of mature and differentiated biofilms [
13,
14,
15]. However, the Las system regulates the expression of the other two systems with its autoinducer 3O-C12-HSL; thus, targeting the Las QS system could help prevent the expression of other QS systems [
14,
15,
16]. In this regard, the molecular docking approach could help in the in silico screening of metabolites against the Las QS system due to its ability to identify lead metabolites from a library of compounds. Interestingly, the technique has been employed in the recent screening of metabolites against a bacterial QS system [
17] and, thus, could serve as a less expensive approach to understanding compounds’ interactions with the QS system. However, due to the limitations of virtual screening relating to the lack of subsequent refinement (rescoring) and assessment of binding pose stability [
18,
19], further refinement and assessment of thermodynamic information are often encouraged using molecular mechanics/GB surface approach and molecular dynamics (MD) simulations [
18,
19], and in this study, these approaches were employed.
Several synthetic compounds such as azithromycin, erythromycin, levamisole, propranolol, and chloroquine have been shown to have anti-QS activities in the search for QS inhibitors via drug repurposing [
20]. However, the side effects of these drugs and the increase in the frequency of
P. aeruginosa strains with increased virulence following their use [
11,
21] have prompted researchers to investigate alternative QS inhibitors, such as plant-derived compounds. Many plants, such as
Pisum sativum seedlings,
Citrus reticulata, and
Syzygium aromaticum, have been recently studied as potent QS modulators [
11,
14]. Interestingly,
Helianthus annuus (sunflower) and its oil have been found to have nutritional and therapeutic relevance as anti-inflammatory, antimalarial, antiasthmatic, antioxidant, antitumor, and antibacterial agents [
22]. However, despite the antibacterial significance of
H. annuus and its derived products, there is a paucity of information on its antipathogenic activity through QS inhibition to date. Hence, this study evaluated the anti-QS activity of two sunflower seed oils using both in vitro and in silico techniques. In the in vitro assay, a biomonitor strain,
Chromobacterium violaceum, was employed to understand and easily detect QS inhibition in
P. aeruginosa due to
C. violaceum’s ability to produce the purple pigment violacein [
23]. Thus, in addition to identifying active Las modulators of
P. aeruginosa, metabolites of sunflower seed oil were screened in silico against the main QS regulator in
C. violaceum, the CviR QS system [
23]. Identifying lead compounds from plant-derived products, such as oils, against important QS regulators of
P. aeruginosa and
C. violaceum could help to identify active anti-QS modulators that could fast-track novel drug discovery and development and contribute toward the increased commercial importance of plants.
4. Discussion
There have been numerous studies conducted on sunflower seeds to determine their antibacterial, anticancer, antioxidant, and other health benefits [
38,
39,
40]. However, no information exists on their anti-QS potential to date, hence the motivation for this study. Solvent extraction is the traditional method for obtaining oil from oilseeds, and in this study, n-hexane was employed as a solvent for extraction because of its benefits, including easy recovery, low latent heat of vaporization (330 kJ/kg), non-polarity, and good selectivity to solvents [
41].
The MIC of an antibiotic is the lowest concentration at which bacterial growth is prevented [
42]. In this study, the two oils (AGSUN 5102 CLP and AGSUN 5106 CLP) had remarkable MIC, which was higher than those obtained for the reference standards (azithromycin and cinnamaldehyde), and the one reported by Liu et al. [
38] but were significantly lower than the values reported by Benites et al. [
43]. Interestingly, despite the fact that the oils were not refined as the standards used in this study, the values obtained with the two cultivars fall within the limit reported by Ács et al. [
44], which is >43.4 mg/mL. Thus, it can be inferred that
P. aeruginosa showed considerable susceptibility to the active constituents of the oils from the two investigated cultivars of sunflower seeds. Hence, both oils could be further explored as possible antibacterial agents against the test pathogen in this study.
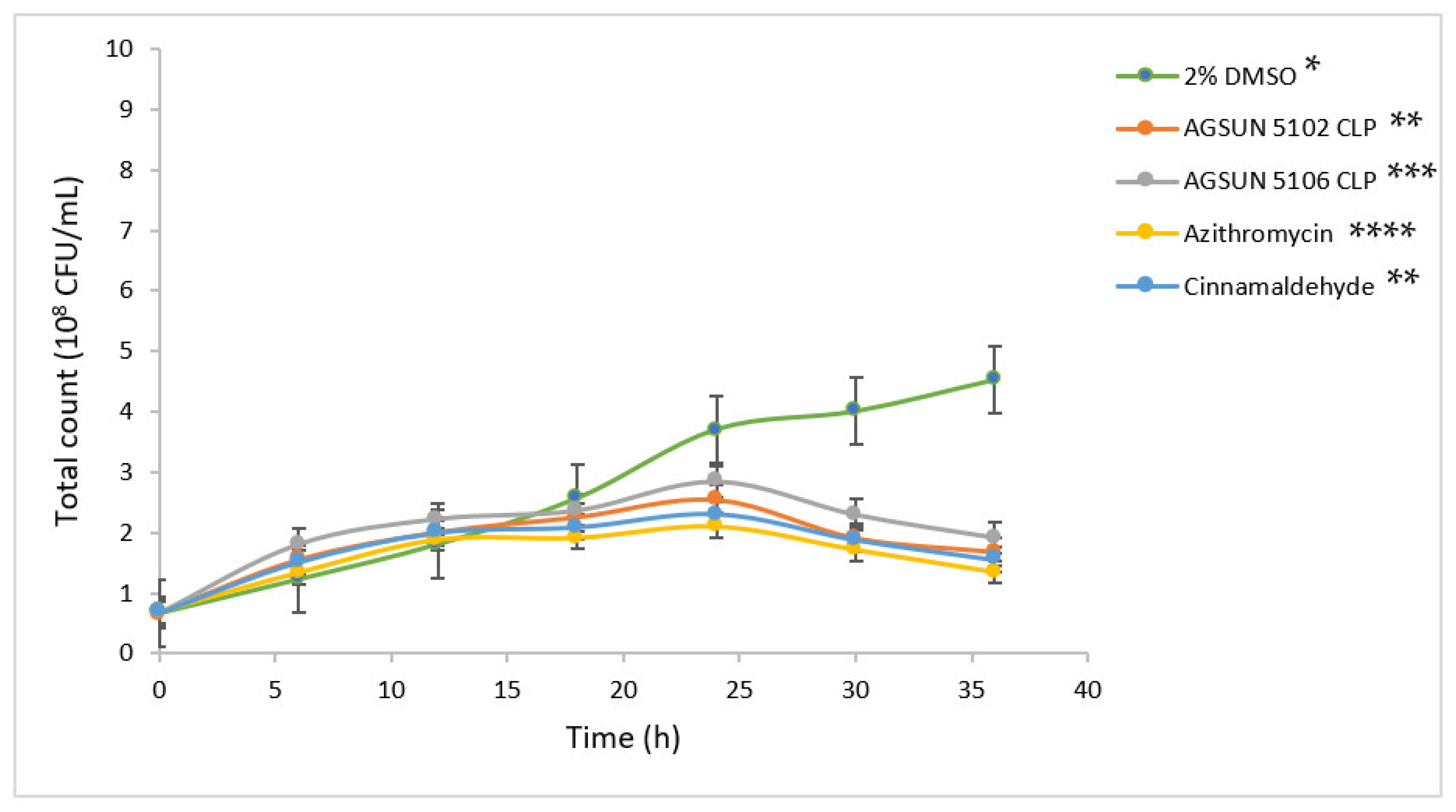
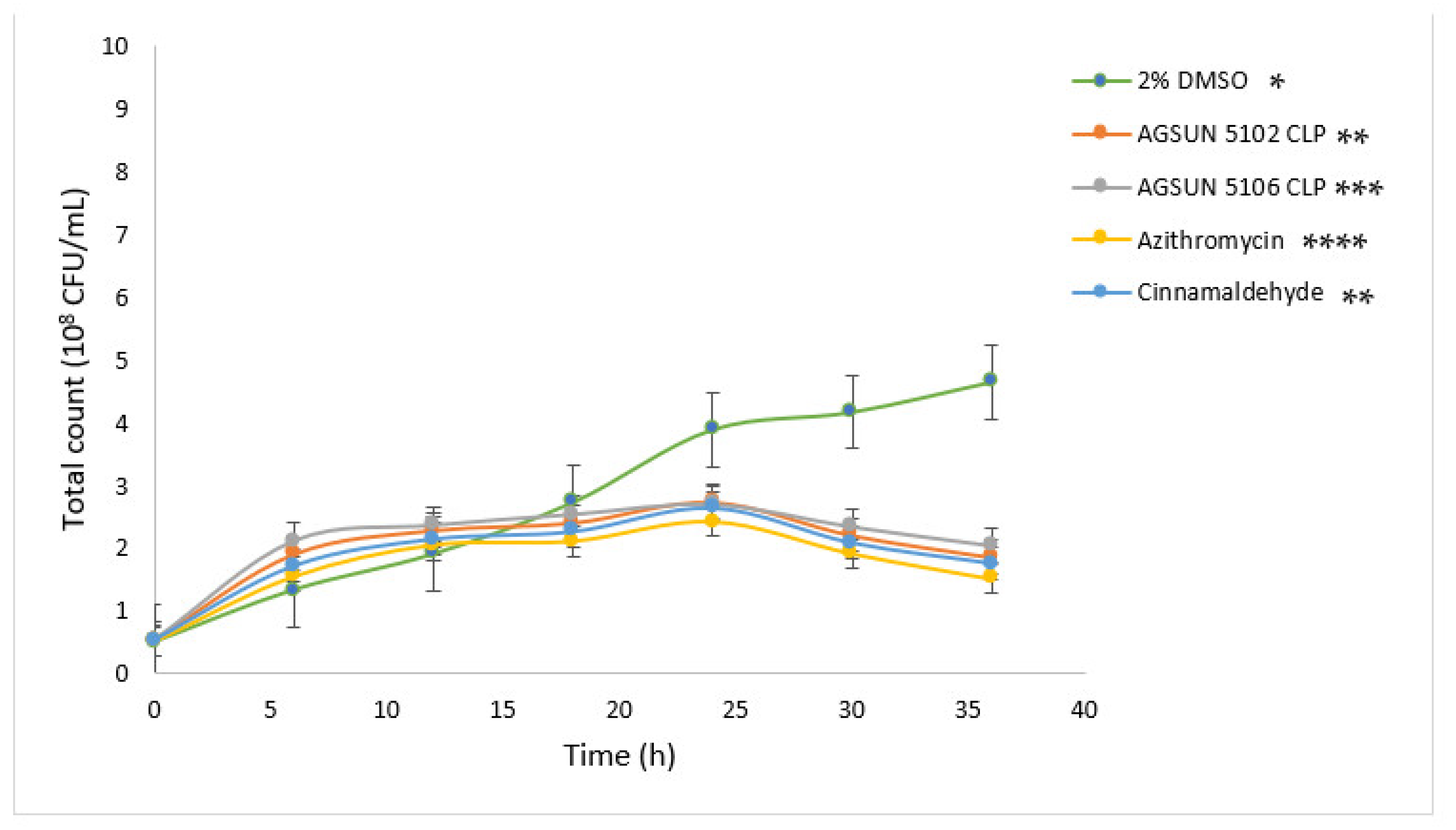
The pathogens were treated with the oils and reference standards for 36 h to understand the growth pattern of
P. aeruginosa and
C. violaceum. The initial steady growth observed before 15 h for the oils and reference standard-treated cells could be due to the low permeability of the outer membrane of Gram-negative bacteria, which enables them to express or exhibit innate resistance [
45]. However, the observed decrease in bacterial growth beyond 15 h in cells treated with the oils and the reference standards could be attributed to the minute penetration of the outer membrane by the administered treatments, which could have initiated a slow release of bacterial cell components that eventually led to cumulative cell death. Ideally, for the oils to act as AQS or potential antipathogenic agents, they only need to show less hindrance to bacterial development, as the speculation of antipathogenic drugs ought not to be bactericidal but to hinder or disrupt the virulence factors [
45].
Chromobacterium violaceum ATCC 12472, a well–recognized QSI biomonitor strain that generates the purple pigment known as violacein, was chosen as the QSI biomonitor strain as it is an efficient bacterium to visually detect and quantify pigment suppression by metabolites of the investigated oils in this study. Generally, violacein pigmentation in
C. violaceum ATCC 12472, regulated by QS chemical communications, produces a naturally occurring and easily observable phenotype without the need for additional substrates, making it simple to assess how well a substance inhibits QS [
46]. However, it is worth noting that this screening technique does not provide information on the precise kinds and quantities of active chemical compounds that are present [
47]. In this study, treatment with the oils might have impacted violacein pigment, which could indicate that the pathogen’s QS system had been tampered with, as seen by the sizes of the opaque zones surrounding the wells administered with the oils. However, the small size of the opaque zones following treatments with the oils could indicate that the biomonitor strain exhibited resistant characteristics to the oils, which according to Kowalska–Krochmal and Dudek–Wicher [
42], suggest a substantial possibility of therapeutic failure even with increasing doses of the oils. On the other hand, azithromycin, which is a refined drug, demonstrated an intermediate characteristic at MIC towards the biomonitor strain, which was largely anticipated, suggesting a likelihood of therapeutic success by increasing the dosage of the medication. Generally, judging by the results obtained, it could be logically inferred that qualitative violacein inhibitory assay might not be the best method to explore AQS potentials as a therapeutic agent, and this was consistent with the submission of Cosa et al. [
28]. Hence, the need for quantitative studies to complement qualitative assays in evaluating the AQS potential of an agent.
In this study, to further validate the interference of the QSS of
C. violaceum by the oils through inhibition of the purple pigment as suggested by Cosa et al. [
28], a quantitative AQS was performed. The two oils exhibited varying levels of AQS activity against
C. violaceum, as confirmed by the significantly reduced production of violacein following treatment by AGSUN 5102 CLP and AGSUN 5106 CLP at both MIC and sub-MIC doses. This observation agrees with a previous study that demonstrated that antipathogenic medications should show their AQS potential with their effectiveness shown at sub-MIC concentrations and upward [
45]. In a related investigation, Khan et al. [
48] noted a remarkable halt in the formation of the violacein pigment in
C. violaceum when essential oils of cinnamon, peppermint, and lavender were present. Similarly, according to Noumi et al. [
49], tea tree oil demonstrated significant inhibition of violacein in
C. violaceum ATCC 12472 at MIC and sub-MIC. In this study, like the essential oils, both cinnamaldehyde and azithromycin also had significant AQS activity at all doses. These findings are significant, as they demonstrate that some metabolites of the oils had a structural resemblance to the analogs of signaling molecules (AHL), allowing competitive structural binding between AHL and the oils’ metabolites with the appropriate receptor protein in the organism, thus, affecting the transmission of signal molecules and QS due to essential oils’ metabolites interfering with the
C. violaceum CviR-QSS. The results of this study showed that the oils could prevent the development of QS.
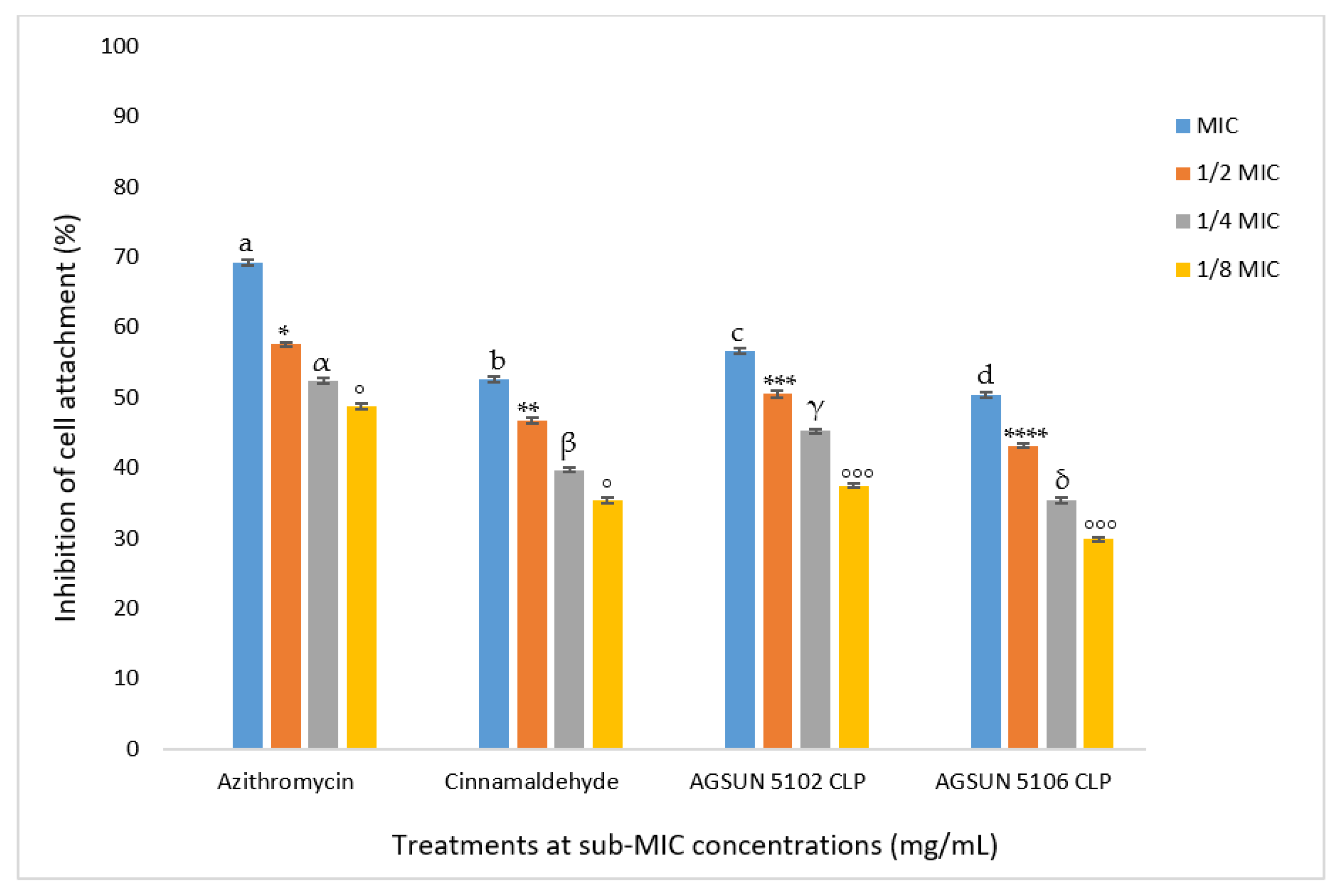
The current study also aimed to investigate the ability of the oils to get rid of biofilms of
P. aeruginosa both in early and mature stages. The oils significantly inhibited cell attachment in a concentration-dependent manner, coherent with what was seen in the quantitative AQS. Both oils favorably competed with the standards in reducing cell attachment. The anti-adhesion activity observed with the oils in this study agrees with the findings of Pejčić et al. [
50], who observed that the presence of sage and basil essential oils significantly inhibited cell attachment. Since cell attachment is significant in the advancement of infection caused by
P. aeruginosa [
17], their inhibition during the early stages of biofilm formation by the essential oil suggested their importance as an AQS antimicrobial. As in the inhibition of cell attachment, a similar trend in the inhibition of biofilm development was observed in this study, where both oils competed relatively well with the controls in reducing biofilm formation. Remarkably, the reduction of biofilm formation by both oils was observed at all concentrations except at
MIC of AGSUN 5106 CLP, where the biofilm formation was enhanced. This observation points to the AQS potency of the essential oils. This phenomenon has also been reported by other researchers [
17,
51]. However, one explanation for biofilm formation enhancement at
MIC of AGSUN 5106 CLP could be due to the efflux pump resistance mechanism during the cell attachment stage, where AGSUN 5106 CLP oil might have been expelled from the cells [
17]. Another possible reason for biofilm formation enhancement at
MIC could be because some of the metabolites of the oil were utilized by the bacteria as a source of nourishment and aided in the growth of the bacteria.
Examining biofilm structure in relation to the geographic localization of significant biofilm matrix components is possible using microscopy techniques. Confocal laser scanning microscopy (CLSM) is an effective tool for studying biofilms and may be used to quantitatively examine the biofilm matrix and the amount of adhering biomass [
52]. Additionally, the spatiotemporal impacts of various nutritional conditions or antibiotic treatments can be observed [
53]. In this study, CLSM was used to study the three-dimensional architectural complexity of biofilms in the presence of the oils. The marked reduction in biofilm thickness and biomass following treatment with the oils compared to the 2% DMSO-treated cells could be indicative of the anti-biofilm activity of the oils. Compared to the reference standards, a complete inhibition of biofilm thickness and biomass was observed in the azithromycin-treated cells. This finding corroborates the cell attachment and biofilm formation inhibition assay, as azithromycin was the most effective anti-biofilm agent. However, treatment with the oils, especially AGSUN 5102 CLP, showed activities comparable to cinnamaldehyde-treated cells, where the biofilm thickness and biomass were significantly reduced, which also corroborates the findings of the cell attachment and biofilm inhibition assay. This is a significant finding as it shows that the oils had the ability to turn off the expression of genes responsible for the biofilm matrix formation, thus, reducing the chances of virulence/resistance-forming genes in
P. aeruginosa forming.
One of the primary factors determining the virulence of
P. aeruginosa is pyocyanin, a blue redox-active secondary metabolite that can generate free radicals. Patients with cystic fibrosis frequently experience this substance’s effects, which involve interfering with ion transport and mucus secretion in respiratory epithelial cells [
54]. Reactive oxygen species (ROS) produced by pyocyanin significantly impact the development of both acute and chronic respiratory infections and play a crucial role in changing the host immune system [
55]. In this study, the observation that the oils significantly reduced the production of pyocyanin in a concentration-dependent manner could be suggestive of their probable anti-pyocyanin effect. At MIC, the fact that both oils reduced pyocyanin in a manner comparable to the reference standards points to their beneficial potential as AQS antimicrobials. This is a significant discovery since pyocyanin synthesis is crucial to the virulence of
P. aeruginosa, and the observation noted in this study agrees with the report of Pejčić et al. [
50], where basil and sage oils significantly decreased pyocyanin production.
The potential of
P. aeruginosa to colonize various settings through motility influence is another factor supporting its classification as a life-threatening opportunistic pathogen [
54]. Motility in
P. aeruginosa is regulated by QS, where swimming on a soft surface and swarming on a semisolid surface are made possible by flagella and pili IV [
32]. A distinct reduction in the diameter zone following the treatment of
P. aeruginosa cells with the oils revealed their remarkable anti-swarming and swimming motility effect relative to the untreated bacterial cells. The anti-swarming and swimming motility effects demonstrated by the oils were comparable to those displayed by the reference standards. Interestingly, this observation is consistent with the report of Pejčić et al. [
50], who found that the swimming and swarming motility were reduced by sage and basil oils. As the swarming and swimming motility of
P. aeruginosa plays a pivotal role in the emergence, development, and upkeep of the biofilm’s structural framework [
56], limiting this mobility may lessen the organism’s virulence ability. This is significant as the oils limited the degree of motility in
P. aeruginosa in a way that suggested possible interference in the production of virulence factors.
Since the mechanism of action of the oils was unknown, they were profiled using chromatographic techniques to analyze the constituents that might be responsible for the AQS potential observed against
P. aeruginosa in the in vitro evaluation. The 15 metabolites identified in the oils were essentially fatty acids and vitamins, and the oils could be said to be high linoleic edible oils, as they are rich in polyunsaturated fatty acids (linoleic acids), monounsaturated fatty acids (oleic acid), and others, thus, they are also cholesterol and nutritionally friendly [
57].
Emran et al. [
58] demonstrated in silico docking as a promising method to support findings from in vitro analysis. Upon identifying the constituents, a molecular docking analysis was performed with all the identified constituents to evaluate which of the compounds are active and bind at the catalytic regions of the Las proteins system of
P. aeruginosa and the CviR system of
C. violaceum. Following molecular docking with all the identified constituents from the oils against all the proteins involved in the Las and CviR systems, the compound(s) with the highest negative binding score and interactions were considered to have the greatest affinity for the respective investigated protein [
59]. Additionally, the proteins in each case of the Las and CviR systems that were most receptive to the constituents were taken further for analysis. Specifically, the binding affinities from LasR (Las system) and CviR (CviR) proteins were considered as they had the best affinity for the test metabolites. Phylloquinone having the highest negative score against LasR was an indication of its significant affinity for the protein relative to the other test metabolites. This also holds for linoleic acid and oleic acid as the next-ranked metabolites relative to the reference standards and is suggestive of their higher affinity and interactions against LasR. This observation could be due to the higher number of interactions formed between the metabolites and, most especially, phylloquinone with LasR, which was higher than those formed between the reference standards and LasR. This opinion is consistent with the report of Chen et al. [
60], where a higher number of interactions and hydrogen bonds of anthocyanins against α–Glucosidase enhanced the affinity of the compound against α–Glucosidase [
60]. Furthermore, the top three metabolites interacted with key amino acid residues such as Tyr56, Trp60, Asp73, Ser129 Leu36, Leu40, Tyr47, Val76, and Cys79 in LasR, which is consistent with those reported by Bottomley et al. [
61] between LasR’s autoinducer (3–oxo–C12–HSL) and LasR at the catalytic site, with phylloquinone interacting more with these amino acids towards LasR. This is indicative that the top three compounds are binding at the catalytic site of LasR and exhibiting similar interactions and traits as LasR’s autoinducer (3–oxo–C12–HSL). This was similarly observed against CviR, with the top-ranked compounds, including phylloquinone, linoleic acid, myristic acid, and lauric acid, having competitive docking scores and interactions relative to the reference standards, with phylloquinone having the best affinity for the receptor, and suggesting phylloquinone as the best inhibitor of the protein. Measuring the ligand’s RMSD from its reference point in the resulting complex following optimum superimposition is one of the most popular methods for assessing the accuracy of docking geometry [
62]. Remarkably, confirmatory superimposition analysis of the docking protocols in this study indicates the same binding position with the native inhibitors of LasR and CviR, hence eliminating the selection of any pseudo-positive binding conformations as the greatest energy-minimized posture. However, due to the limitations of molecular docking, which can only be used as a preliminary investigation of a ligand’s affinity for a protein’s binding pocket, an MD simulation over 100 ns was conducted to gain further insight into LasR and CviR protein’s behavior upon the binding of the top-ranked compounds at the catalytic region. This is important to evaluate the residing time of the metabolites and reference standards at the catalytic region by measuring the binding free energy as well as important conformational information regarding the thermodynamic structural stability, flexibility, and compactness of the complexes taken as post-MD simulation indices [
63].
The top three compounds from molecular docking were then further taken to MD simulation. The binding free energy estimates the distinction in energy between a complex and its unbound receptor component, and the higher the negative value, the better the affinity of the ligand toward the enzyme [
64]. Phylloquinone, linoleic acid, and oleic acid, when bound with LasR at the catalytic region, had higher negative binding free energy values than the reference standards. This finding suggests the top three compounds as promising and better potential inhibitors of LasR, especially phylloquinone which had the highest negative binding free energy value. Moreover, this observation is suggestive of phylloquinone having a higher residence time at the catalytic region of LasR, thus, resulting in an enhancement of activity towards LasR; it also correlates with the results obtained in the molecular docking studies, where phylloquinone was identified as the most promising metabolite. Similarly, against the CviR of
C. violaceum, the higher binding free energy values observed with the top four metabolites relative to the cinnamaldehyde point to their advantage as better inhibitors of CviR. While compared to azithromycin, the observation that only phylloquinone and linoleic acid had higher binding free energy values, with phylloquinone having the highest value, demonstrated the benefit of phylloquinone and linoleic acid as an anti-CviR agent, with phylloquinone again having the best inhibitory effect.
The RMSD trajectory was analyzed to evaluate the structural stability of the resulting complexes at the catalytic site of LasR over 100 ns and, thus, the stability of a complex structure is shown by its proximity to the unbound structure, which implies a lower RMSD value [
64]. As reported by Ramírez and Caballero [
65], a desirable and generally acceptable RMSD number should be less than 3 Å. In this study, the complexes converged at around 15 ns and 45 ns; however, after 60 ns, they were all at equilibration and were relatively stable and compact. The LasR deviated from its native conformation upon the binding of ligand compounds accounting for a maximum fluctuation of less than 2.5 Å average RMSD value, suggesting that all the complexes were within the acceptable RMSD limit of less than 3 Å. This observation points to the thermodynamic structural stability of the top-ranked metabolites towards LasR and further enhances their benefit as AQS agents. Interestingly, the phylloquinone–LasR complex, with the highest negative binding free energy, had the lowest mean RMSD value comparable to the unbound LasR, thus, further indicating the advantage of the compound as a potential LasR inhibitor. Similarly, the observation that only linoleic acid among the top-ranked metabolites against CviR caused increased RMSD relative to unbound CviR suggests the thermodynamic structural stability of top-ranked metabolites with CviR. Moreover, all the RMSD values of the top-ranked metabolites and reference standards were less than 3 Å, with myristic acid having the lowest value.
The RMSF assesses the fluctuation of the amino residues of LasR and CviR protein and can be related to the stability of the intra- and inter-molecular bonds within the complex, and the lower the fluctuation at the catalytic location, the stronger the binding and affinity of the ligand to the protein [
66]. The binding of the investigated metabolites to LasR and CviR led to random fluctuations that arose from possible structural conformation changes. For all the metabolites investigated, the lowest fluctuations at the catalytic region of LasR were generally observed between residues 30 and 70. This indicates that metabolite binding at these residues was stable due to fewer fluctuations. This observation is favorable for the top-ranked metabolites and reference standards as inhibitors of LasR and is consistent with the report of Husain et al. [
67], where similarly reduced fluctuations in the catalytic region of the protein were observed. This result shows that the three key metabolites from the oils are promising inhibitors of LasR, especially phylloquinone, which is the metabolite that leads to the lowest volatility and flexibility of LasR after binding to the protein, reflecting its increased attractiveness and ability to improve the stability of LasR–amino acid residues. Interestingly, this finding is also coherent with those of the binding free energy. A similar observation was noted for the metabolites and reference standard binding of CviR where, except for linoleic acid and azithromycin, CviR ligand binding caused decreased RMSF. This observation points to the increased attractiveness and ability of the ligands to enhance the stability of CviR amino acid residues. However, in contrast to LasR, where phylloquinone showed the least fluctuation of LasR amino acid residues after its binding, the myristic acid–CviR complex had the least fluctuation. However, phylloquinone–CviR, together with other metabolites and the reference standard binding of CviR, all had an RMSF value that was less than 3 Å, indicating the relatively good fluctuation of the CviR residue after binding of these ligands.
The RoG measures how complexes are thermodynamically compact over time, so the lower the value, the more compact the complex is [
68]. In this study, the observation that the binding of the top-ranked metabolites and the reference standards to LasR had a negligible effect on the mean RoG values relative to unbound LasR indicates the relative compactness of the complexes. This means that metabolites’ binding had no thermodynamic perturbation effect on LasR. However, it is worth noting the unfolding effect that azithromycin had on LasR between 70 and 80 ns, suggesting lower compactness and stability within this period. As with LasR, the negligible increasing effect on RoG upon CviR binding by the top four metabolites and reference standard might indicate that the thermodynamic geometry of CviR was not perturbed.
The SASA is a key thermodynamic stability metric that examines protein folding and surface area changes over the simulation, with larger SASA values indicating an increase in protein volume [
66]. The physicochemical properties of the amino acid residues that were rearranged or altered determine the degree of variation in the SASA value [
69]. Similar to the RoG results, a higher fluctuation of SASA was observed in the azithromycin-LasR and azithromycin–CviR complex plot over the 100 ns simulation period, suggesting that azithromycin has a larger impact on the surface expansion of LasR and CviR protein. However, the comparable SASA value of the unbound proteins (LasR and CviR) and the top-ranked metabolite complexes shows that the LasR and CviR volume either remains the same or decreases over the course of the simulation, thus suggesting no perturbation in protein following the binding of the top-ranked metabolites and cinnamaldehyde during simulation.
In biochemistry, hydrogen bonds are significant interactions as they are crucial for molecular recognition, structural stability, enzyme catalysis, drug partition, and permeability [
70,
71,
72]. Intramolecular hydrogen bonds and distance are important in the stability of a protein structure and, hence, can be assessed to understand the impact of ligand binding on the stability of a protein during simulation [
64]. However, the membrane partition and permeability of the medication may be negatively impacted by an excess of hydrogen bond donors or acceptors [
73]. In addition to increasing the water desolvation penalty during drug penetration, these polar groups can reduce the attraction for the hydrophobic membrane area [
73]. As observed in this study, before and after the top three compounds and reference standards bindings, the stable fluctuation in the pattern of the number of hydrogen bonds produced in LasR and CviR indicates that, following binding to compounds, LasR and CviR thermodynamic entropy was unaffected [
19]. In LasR, the decrease in the average number of intramolecular hydrogen bonds created by the top three compounds and controls in complexes with LasR could suggest breakage in some intramolecular hydrogen bonds due to ligand binding. This observation is unlike what was reported by Aribisala and Sabiu [
33] and those observed in this study with CviR, where top-ranked metabolites and reference standards binding of CviR caused increased intramolecular hydrogen bonds. The increased intramolecular hydrogen bonds of CviR could be due to the addition of intermolecular hydrogen bonds contributed by ligand binding. However, the high number of intramolecular hydrogen bonds noted with phylloquinone–LasR and phylloquinone–CviR protein corroborates the stability observed with the complex, which implies that there was a high degree of thermodynamic compatibility during the 100 ns simulation duration, which may have caused the higher binding free energy observed with the compound against LasR and CviR.
The top-ranked compounds and the reference standards exhibited different bond lengths and numbers of interactions in the active site of LasR and CviR, which were found to have an impact on the free energy of binding observed in this study. The highest number of interactions and hydrogen bonding contacts observed with the phylloquinone–LasR and the phylloquinone–CviR complexes after 100 ns is consistent with the binding free energy observed with the metabolites against the respective target. This observation indicated that a stronger and more stable ligand–protein complex results from more interactions and hydrogen bonding contacts, suggesting greater inhibition in the phylloquinone–LasR and phylloquinone–CviR complexes observed in this study. This conclusion agrees with the observation of Forli et al. [
74], who showed that the higher the number of interactions, including hydrogen bonding, the higher the inhibitory effect of the inhibitor. Similar observations were noted with the other top-ranked metabolites compared to the reference standards against LasR and CviR, where top-ranked metabolites with higher binding free energy showed a higher number of interactions and hydrogen bonding contacts. In addition to the highest number of hydrogen bonding contacts, phylloquinone–LasR and phylloquinone–CviR complexes had the highest number of important interactions, such as the stacking interaction (stacked), which is one of the most powerful driving forces behind the biological complexation process, and protein folding. Taken together, all of these observations may have contributed to the observed higher affinity and improved stability between phylloquinone and the investigated targets. Since analyzing the number and type of interactions in just one simulation time frame could lead to a false positive conclusion, snapshots at the different time frames of the simulation were taken, and the observation that the consistent number of interactions and some conserved residues exist at 30 ns, 60 ns, and 100 ns during the simulation point to the potential enhanced inhibitory effect on the proteins by the top-ranked metabolites and reference standards during the 100 ns simulation period.
To avoid or reduce the tendency of high failure rate during the preclinical and clinical phases of drug development, evaluation of the pharmacokinetics, drug likeliness, synthetic feasibility, and toxicity characteristics of potential therapeutic agents have been identified as a critical step and a prerequisite, and this was undertaken for the top-ranked metabolites in this study. The rule of 5 (RO5), also known as Lipinski’s rule, provides a useful framework to determine whether a tested molecule will be orally accessible and bioavailable [
75]. It also affirms that molecules exhibit strong absorption or permeation if they have an octanol/water partition coefficient (log P) < 5, molecular weight (MW) < 500 g/mol, number of hydrogen bond donors (n OH, NH) < 5, and number of hydrogen bond acceptors (n O, N) ≤ 10 [
76]. Interestingly, the top-ranked metabolites fulfilling Lipinski’s rule showed their ability to be orally administrable to reach target sites and exert their pharmacological effects, while azithromycin failing the rule suggests its relatively lower tendency to be orally administered and, thus, pinpoints the probable advantage of the top-ranked compounds over azithromycin as drug candidates. However, this does not imply that azithromycin is not a good drug but suggests that it can nonetheless be modified to improve its administration via the oral route, which is mostly preferred [
77]. On the other hand, while the low GIT absorption rate of azithromycin and phylloquinone indicates that they are less likely to be absorbed through the GIT, the high GIT absorption rate of linoleic acid and oleic acid indicates their advantage over azithromycin and phylloquinone. Azithromycin, with the lowest bioavailability score, indicates that it is not preferred as an oral drug over cinnamaldehyde, and the top-ranked compounds with higher bioavailability scores mean a high rate of absorption of a drug and the concentration of unchanged drug that reaches the site of action to exert its pharmacological effect. This observation agrees with the findings of Shode et al. [
78] regarding anti-COVID-19 drug candidates with relatively higher bioavailability scores than the reference standards used. Additionally, phylloquinone and the other top-ranked compounds had bioavailability ratings comparable to cinnamaldehyde, which suggests that they can be used orally and will be bioavailable since cinnamaldehyde is an oral drug. Interestingly, the top-ranked compounds, except phylloquinone, were also water-soluble, suggesting their advantage of being easily transported through the bloodstream compared to azithromycin, which is poorly soluble in water.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}